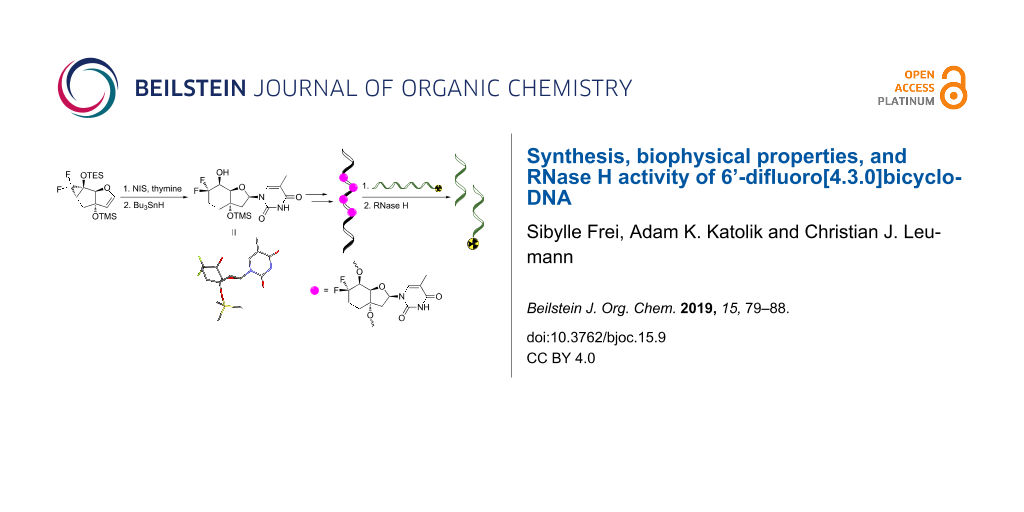
Abstract
Here we present the synthesis, the biophysical properties, and the RNase H profile of 6’-difluorinated [4.3.0]bicyclo-DNA (6’-diF-bc4,3-DNA). The difluorinated thymidine phosphoramidite building block was synthesized starting from an already known gem-difluorinated tricyclic glycal. This tricyclic siloxydifluorocyclopropane was converted into the [4.3.0]bicyclic nucleoside via cyclopropane ring-opening through the addition of an electrophilic iodine during the nucleosidation step followed by reduction. The gem-difluorinated bicyclic nucleoside was then converted into the corresponding phosphoramidite building block which was incorporated into oligonucleotides. Thermal denaturation experiments of these oligonucleotides hybridized to complementary DNA or RNA disclosed a significant destabilization of both duplex types (ΔTm/mod = −1.6 to −5.5 °C). However, in the DNA/RNA hybrid the amount of destabilization could be reduced by multiple insertions of the modified unit. In addition, CD spectroscopy of the oligonucleotides hybridized to RNA showed a similar structure than the natural DNA/RNA duplex. Furthermore, since the structural investigation on the nucleoside level by X-ray crystallography and ab initio calculations pointed to a furanose conformation in the southern region, a RNase H cleavage assay was conducted. This experiment revealed that the oligonucleotide containing five modified units was able to elicit the RNase H-mediated cleavage of the complementary RNA strand.

Graphical Abstract
Introduction
The fluorine atom is a very attractive substituent in medicinal chemistry due to the beneficial biological effects induced by this atom on the overall drug behaviour [1-5]. The positive influences on the drug behaviour is not limited to small molecules but is also valid for antisense oligonucleotides (AONs) [6]. An effective way to tune the properties of antisense oligonucleotides is by the insertion of the fluorine atom in the sugar moiety of the nucleoside. In this way, the sugar pucker can be controlled which ideally results in an increased affinity towards complementary RNA [7]. An improved affinity for RNA as complement can be found in DNA oligonucleotides containing 2’-deoxy-2’-fluoro-RNA (F-RNA) [8] or 2’-deoxy-2’-fluoroarabino nucleic acid (F-ANA, Figure 1) [9]. In the former the sugar pucker adopts a C3’-endo conformation [10] and the duplex formation is entropically stabilized. The reason for the stabilization is an increased strength of the Watson–Crick base pairing and base stacking interactions due to the electronic effects of the axially oriented 2’-fluorine atom [11,12]. Additionally, FC–H···O hydrogen bonds between the 2’-fluorine and the 4’-oxygen or 5’-oxygen of the 3’-adjacent nucleotide are thought to favourably contribute in both F-RNA and F-ANA duplexes [13]. Furthermore, in duplexes of the F-ANA with complementary RNA, internucleosidic C–H···F–C pseudohydrogen bonds are proposed at pyrimidine-purine steps to additionally stabilize the structure [14,15]. The β-orientation of the fluorine substituent in F-ANA leads to a gauche interaction between O4’–C1’–C2’–F2’ favouring the C2’-endo/O4’-endo conformations of the sugar in solution [16,17]. These DNA-like sugar conformations cause that F–ANA is among the few modifications which can trigger the cleavage of the RNA strand of an AON/RNA hybrid structure by the endonuclease RNase H [9,18]. Both, the F–ANA and the F–RNA, are appealing modifications for several oligonucleotide-based silencing applications [8,19-25].
![[1860-5397-15-9-1]](/bjoc/content/figures/1860-5397-15-9-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structure of selected fluorine-modified nucleic acids.
Figure 1: Chemical structure of selected fluorine-modified nucleic acids.
Also evaluated on their antisense properties were the 3’-fluorinated hexitol nucleic acids FHNA and Ara-FHNA (Figure 1) with the fluorine in axial or equatorial orientation, respectively. Both modifications preferentially adopt a chair conformation with the nucleobase in axial orientation which mimics the C3’-endo conformation of the furanose ring. Thermal denaturation experiments with complementary RNA displayed a duplex stabilization for FHNA and a duplex destabilization for Ara-FHNA. The reason for the stabilization of the former was accounted to a combination of the increased rigidity of the six-membered ring and the positioning of the axial oriented fluorine atom pointing into the minor groove. Conversely, in the Ara-FHNA, repulsive electrostatic interactions between the fluorine atom and the 4’-oxygen of the 3’-adjected nucleotide resulted in a partial unstacking of the nucleobases and a destabilizing effect upon duplex formation [26]. Also other fluorinated nucleic acids such as 2’-fluorocyclohexenyl nucleic acid (F-CeNA, Figure 1) [27] and other modifications [28-31] have been analyzed on their antisense properties.
In our own work we already investigated the effect of the fluorine substituent at various positions of the [3.3.0]bicyclo-DNA (bc-DNA) and tricyclo-DNA (tc-DNA) scaffold. All these modifications unveiled an either identical or slightly increased affinity versus complementary RNA compared to their non-fluorinated compounds [32-35]. Interestingly, the 2’F-tc-ANA (Figure 1) exhibited in a sequence- and composition-dependent manner the ability to induce the RNase H cleavage of the complementary RNA strand [35]. In continuation of our work we became interested in the fluorination of [4.3.0]bicyclo-DNA [36]. Consequently, the 6’-position of the [4.3.0]bicyclo-DNA was substituted with a difluoromethylene group, and the structural effect of this functional unit was explored. Herein we report on the synthesis and properties of the 6’-diF-bc4,3-thymidine analog (Figure 1), and the biophysical properties of oligonucleotides containing this modification. Moreover, we investigated the substrate recognition of the 6’-diF-bc4,3-T analog by RNase H. In addition, the RNase H experiment was also performed with the previously report 6’F-bc4,3-DNA (Figure 1) [37].
Results and Discussion
Synthesis of the phosphoramidite building block
In the literature there exist several procedures to construct an α,α-difluoroketone from a corresponding siloxydifluorocyclopropane [38-40]. However, based on previous observations in the nucleoside synthesis of the 6’F-bc4,3-T [37] we thought to construct the 6’-diF-bc4,3 building block in utilizing this methodology. Along this synthesis, the glycal 1 was treated with N-iodosuccinimide (NIS) in the presence of persilylated thymine to produce the iodine intermediates 2α/β (Scheme 1, Table 1, entry 1). These instable intermediates were then directly reduced with tributyltin hydride (Bu3SnH) to yield the tricyclic nucleosides 5α/β as main compounds. However, we observed the occurrence of the gem-difluorinated bicyclic nucleoside 6 as the main side product. Since nucleoside 6 possessed the desired stereochemistry at the 1’- and the 5’-positions, we investigated the mechanism of its formation in more detail to be able to increase its yield. To determine in which of the two steps the formation of the nucleoside 6 took place, they had to be analyzed separately. Therefore, a sample of the iodinated nucleosides 2α/β was purified and subjected to the reduction reaction, where nucleosides 5α/β were formed as single products (Table 1, entry 2). Also, the conversion of the nucleosides 5α/β into the bicyclic derivative 6 could be ruled out (Table 1, entry 3). Consequently, the bicyclic derivative 6 was thought to have its origin in the NIS-mediated nucleosidation step. In analyzing the crude reaction product in more detail, apart from the iodinated nucleosides 2α/β, the presence of an inseparable mixture of two diiodo-substituted products was unveiled. The major product was the bicyclic gem-diol 4 and the minor one the corresponding ketone 3. We hypothesize that the α,α-difluoroketone 3 was formed through a cyclopropane ring-opening followed by the addition of the electrophilic iodine in accordance to what was reported by the Dilman group [40]. The α,α-difluoroketone 3 rapidly underwent hydration in a reversible way to form the gem-diol 4. The stereochemistry of compound 4 could be assessed indicating the selective formation of the β-nucleoside with the addition of the iodine at the 7’-position from the exo-face. Even though we were not able to verify the relative configuration of ketone 3, we expect the same stereochemistry at the 1’-, 2’- and 7’-positions as in nucleoside 4. The confirmation that the diiodo-substituted derivatives 3/4 were the precursors of nucleoside 6 came from an experiment in which 3/4 was treated with Bu3SnH resulting in nucleoside 6 as the only observed product (Table 1, entry 4). The S-configuration at the 5’-position of nucleoside 6 could be explained by a Felkin–Ahn transition state with the hydrogen radical attacking from the less hindered exo-face [41,42]. Optimisation of the nucleosidation conditions to an increased amount of NIS (2 equiv) and prolongation of the reaction time in combination with the adjustment of the Bu3SnH amount to 3 equivalents led to higher yields of the bicyclic nucleoside 6 (Table 1, entry 5). An additional proof for the reaction mechanism came from the outcome of the reaction where a tricyclic sugar was first treated with NIS and then with Bu3SnH resulting as expected in a gem-difluorinated bicyclic sugar (Scheme S1, Supporting Information File 1). This reaction also ruled out the involvement of the nucleobase or the iodine at the 2’-position in the reaction mechanism.
![[1860-5397-15-9-i1]](/bjoc/content/inline/1860-5397-15-9-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the bicyclic nucleoside 6. Reagents and conditions: a) BSA, thymine, NIS, DCM, 0 °C to rt, 26 h; b) Bu3SnH, AIBN, toluene, 90 °C, 30 min, 34% (5α/β), 48% (6) over two steps.
Scheme 1: Synthesis of the bicyclic nucleoside 6. Reagents and conditions: a) BSA, thymine, NIS, DCM, 0 °C to...
Table 1: Evaluation of the reaction mechanism for the production of bicyclic nucleoside 6.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-9-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Starting material | Bu3SnH (equiv) | AIBN (equiv) | Yield 5α/β [%] | Yield 6 [%] |
| 1 | 1a | 1.5 | 0.5 | 70b | 21b |
| 2 | 2α/β | 1.5 | 0.1 | 82 | – |
| 3 | 5α/β | 2.5 | 0.1 | 93 | – |
| 4 | 3/4 | 3.1 | 0.1 | – | 64 |
| 5 | 1c | 3.5 | 0.1 | 34b | 48b |
aFirst treated with: thymine (3 equiv), BSA (4.5 equiv), NIS (1.5 equiv), DCM, 0 °C to rt, 4.5 h. bYield over two steps. cFirst reacted with: thymine (3 equiv), BSA (4.5 equiv), NIS (2 equiv), DCM, 0 °C to rt, 26 h.
Having attained nucleoside 6, the synthesis towards the building block for DNA-synthesis continued by subsequent desilylation of this derivative producing intermediate 7 (Scheme 2). DMTr-protection of compound 7 at the 5’-oxygen with in situ prepared DMTr-OTf [43,44] followed by phosphitylation at the 3’-oxygen afforded the phosphoramidite 9.
![[1860-5397-15-9-i2]](/bjoc/content/inline/1860-5397-15-9-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the thymidine phosphoramidite building block 9. Reagents and conditions: a) HF-pyridine, DCM/pyridine 5:1, 0 °C to rt, 2.5 h, 64%; b) DMTr-OTf, DCM/pyridine 1:2, rt, 22 h, 56%; c) CEP-Cl, DIPEA, THF, rt, 3 h, 73%.
Scheme 2: Synthesis of the thymidine phosphoramidite building block 9. Reagents and conditions: a) HF-pyridin...
X-ray structure and molecular modeling of the 6’-diF-bc4,3 nucleoside
To verify the relative configuration of nucleoside 6, crystals of this compound were subjected to X-ray diffraction analysis. The asymmetric unit of a single crystal of nucleoside 6 contained two independent molecules which differed only in the conformation around the C3’–O3’ bond (Figure 2, Table 2, and Tables S1–S3, Supporting Information File 1) [45]. The sugar pucker of both molecules expressed the C2’-endo conformation with the pseudorotation phase angle P adopting values of 175° (6a) and 181° (6b), respectively. The maximum puckering amplitude νmax was 43° (6a) and 40° (6b). Furthermore, the nucleobase displayed an anti orientation. The carbocyclic ring adopted a chair conformation. As a consequence, the angle γ was aligned in the synclincal range and the 5’-hydroxy group in an axial arrangement. Additionally, the distance between the 5’-oxygen and the equatorial fluorine atom Fa of 2.80 Å (6a) and 2.73 Å (6b) correlated with the sum of their van der Waals radii (Table S1, Supporting Information File 1). Interestingly, the two fluorine atoms had an effect on the C5’–C6’ and the C6’–C7’ bond length which were shorter than other C–C bonds in the cyclohexyl ring (Table S2, Supporting Information File 1). Furthermore, the difluoromethylene unit affected the C5’–C6’–C7’ angle and the F–C6’–F angle. The former was widened and the latter shortened compared to the structure of the non-fluorinated bc4,3-T (Table 2 and Table S3, Supporting Information File 1). This phenomenon was also observed for other difluoromethylene containing compounds [46]. Apart from that, the observed parameters of the 6’-diF-bc4,3-T were very similar to the ones of the bc4,3-T, indicating that at least on the nucleoside level the fluorine atoms seemed to have a minor effect on the overall structure.
![[1860-5397-15-9-2]](/bjoc/content/figures/1860-5397-15-9-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structure of nucleoside 6a (left) and 6b (right).
Figure 2: X-ray structure of nucleoside 6a (left) and 6b (right).
Table 2: Selected parameters from the crystal structure of nucleoside 6 and the standard bc4,3-T.
| Nucleoside | P [°] | νmax [°] | γ [°] | δ [°] | χ [°] | C5’–C6’–C7’ [°] | X–C6’–Xa [°] |
| 6a | 175 | 43 | 74 | 158 | −108 | 114 | 105 |
| 6b | 181 | 40 | 72 | 164 | −82 | 114 | 105 |
| bc4,3-T (a)b | 174 | 42 | 70 | 162 | −105 | 111 | 108 |
| bc4,3-T (b)b | 166 | 43 | 71 | 154 | −120 | 110 | 108 |
a6a/b: X = F, bc4,3-T: X = H. bThe structures a and b were two different molecules in the same unit. Data taken from ref [36].
To further study the preferred sugar pucker of the 6’-diF-bc4,3-T nucleoside, a potential energy profile versus pseudorotation phase angle of nucleoside 7 was calculated using quantum mechanical methods. For the calculations we used the Gaussian 09 software package [47] at the second order Møller–Plesset (MP2) level of theory, the 6-311G* basis set, and the same methodology as for the 6’F-bc4,3- T [37]. The obtained energy profile of nucleoside 7 (Figure 3a) surprisingly showed only one single low energy region in the Southern area of the pseudorotational cycle. The minimal energy conformer of nucleoside 7 adopted a C2’-endo furanose conformation (P = 160°) and a twist-boat orientation of the carbocyclic unit (Figure 3b). Hence, the angle γ took up a synclincal arrangement and the 5’-hydroxy group a pseudoaxial orientation. Again, the spacing between the 5’-oxygen and the equatorial aligned fluorine atom Fa of 2.61 Å corresponded to the sum of their van der Waals radii. Interestingly, this distance was shorter in the minimal conformer of nucleoside 7 than in the obtained crystal structure of derivative 6 (Table S1, Supporting Information File 1). The reason for that can be attributed to the different conformations of the six-membered rings in these two structures. Apart from that, the two structures were very similar.
![[1860-5397-15-9-3]](/bjoc/content/figures/1860-5397-15-9-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: a) Potential energy profile versus pseudorotation phase angle of nucleoside 7 and b) its minimal energy conformer.
Figure 3: a) Potential energy profile versus pseudorotation phase angle of nucleoside 7 and b) its minimal en...
Synthesis of the modified oligonucleotides and their thermal melting profiles
The phosphoramidite building block 9 was incorporated into oligonucleotides and duplexed to complementary DNA and RNA to determine the effect of the 6’-diF-bc4,3-T modification on the helical structure and duplex thermostability. The sequences chosen for the investigation were the same as previously used for the 6’F-bc4,3-DNA (ON1: 5’-d(GGA TGT TCt CGA)-3’, ON2: 5’-d(GGA tGT TCT CGA)-3’, ON3: 5’-d(GGA TGt tCT CGA)-3’, ON4: 5’-d(GCA ttt ttA CCG)-3’) [37]. UV-melting experiments of the modified duplexes were recorded to assess the affinity of the 6’-diF-bc4,3-T modified oligonucleotides towards complementary DNA and RNA (Table 3). A single insertion of a 6’-diF-bc4,3-T into DNA strands led to a remarkable duplex destabilization versus both complements (ΔTm/mod = −1.6 to −5.5 °C) in a sequence specific manner but with a slight lesser degree of destabilization towards complementary DNA. However, the degree of destabilization was further minimized with two consecutive insertions of the 6’-diF-bc4,3-T (ΔTm/mod = −4.2 °C and −3.4 °C for complementary DNA and RNA, respectively), whereas five consecutive 6’-diF-bc4,3-T units depressed the duplex stability of both structures again (ΔTm/mod = −5.4 °C and −4.5 °C for complementary DNA and RNA, respectively). Interestingly, the DNA/RNA hybrid structure better accommodated multiple 6’-diF-bc4,3-Ts than a single insertion. Additionally, the hybrid structure also better tolerated multiple 6’-diF-bc4,3-T units than the DNA/DNA duplex. The reason for the destabilizing nature of the 6’-diF-bc4,3-T versus both complements might be found in repulsive electrostatic interactions between the fluorine atoms and the 5’-oxygen. In the case of multiple insertions it might be possible that these interactions might be reduced or maybe compensated by other more favourable effects. The fact, that the two fluorine atoms of the 6’-diF-bc4,3-T have a negative influence on the duplex stability was reflected in the higher Tm values of ONs containing the non-fluorinated bc4,3-T unit paired to DNA or RNA (Table 3). However, the Tm difference between duplexes containing the 6’-diF-bc4,3-T or the 6’F-bc4,3-T modifications are harder to be interpreted and may reflect different structural preferences between the cyclohexyl to the cylcohexenyl ring. But in general, the duplex instability in the DNA/DNA structures was more pronounced in the 6’-diF-bc4,3-T series, whereas to complementary RNA the modified oligonucleotides exhibited a similar degree of destabilization in both series.
Table 3: Tm and ΔTm/mod data from UV-melting curves (260 nm) of ONs containing 6’-diF-bc4,3-T, 6’F-bc4,3-T, or bc4,3-T residues in the DNA backbone hybridized to complementary DNA and RNA.
| Entry | Sequence (5’ → 3’)a | Tm [°C] vs DNA (ΔTm/mod [°C]) | Tm [°C] vs RNA (ΔTm/mod [°C]) | ||||
| X = | 6’-diF-bc4,3-T | 6’F-bc4,3-T | bc4,3-Tb | 6’-diF-bc4,3-T | 6’F-bc4,3-T | bc4,3-Tb | |
| 1 | d(GGA TGT TCX CGA) | 43.5 (−5.2) | 46.0 (−2.7) | 47.3 (−0.2) | 44.5 (−5.5) | 46.0 (−4.0) | 48.9 (−0.8) |
| 2 | d(GGA XGT TCT CGA) | 47.1 (−1.6) | 47.2 (−1.5) | 46.1 (−1.4) | 47.9 (−2.1) | 47.6 (−2.4) | 47.4 (−2.3) |
| 3 | d(GGA TGX XCT CGA) | 40.3 (−4.2) | 41.3 (−3.7) | 47.0 (−0.3) | 43.3 (−3.4) | 42.0 (−4.0) | 51.0 (+0.7) |
| 4 | d(GCA XXX XXA CCG) | 20.2 (−5.4) | 30.3 (−3.4) | – | 22.0 (−4.5) | 22.4 (−4.4) | – |
aTotal strand conc. 2 μM in 10 mM NaH2PO4, 150 mM NaCl, pH 7.0. Tm values of unmodified duplexes: DNA1/DNA = 48.7 °C, DNA1/RNA = 50.0 °C, DNA2/DNA = 47.4 °C, DNA2/RNA = 44.4 °C; DNA1 = 5’-d(GGA TGT TCT CGA)-3’, DNA2 = 5’-d(GCA TTT TTA CCG)-3’. bData taken from ref [36].
An interesting behaviour was observed for the sequence of Table 3, entry 2. There, the ON of the three bc4,3-T analogs not only showed almost identical Tm values when paired to DNA but also to complementary RNA. This means that at least in this sequence context the fluorine atoms had no impact on the duplex stability and the destabilization arose only from the bicyclic scaffold. Whether this finding is a general behaviour or a consequence of the A–X–G nearest neighbour interactions cannot be stated at present.
The base pairing selectivity of the 6’-diF-bc4,3-T was evaluated by UV-melting experiment of ON1 by inserting one of the three possible mismatches (G, C, or T) opposite the modified unit in the otherwise complementary DNA strand (Table 4). As anticipated, the mispairing led to a strong decrease of the melting temperature (Tm = −8.5 to −13.8 °C) with the GT-Wobble mispair exhibiting the least destabilizing effect. Comparing these values to the ones of the natural system revealed that the modification discriminated the mismatches more efficiently. Consequently, this finding indicated a higher tendency for mismatch discrimination of the 6’-diF-bc4,3-T over dT.
Table 4: Tm data [°C] from UV-melting curves (260 nm) of ON1 in duplex with complementary mismatched DNA.
| Entry | Sequencea | X = A | X = T | X = G | X = C |
| DNA1 | 5’-d(GGA TGT TCT CGA)-3’ | 48.7 | 39.0 | 40.2 | 37.3 |
| DNA | 5’-d(TCG XGA ACA TCC)-3’ | (−9.7) | (−8.5) | (−11.4) | |
| ON1 | 5’-d(GGA TGT TCt CGA)-3’ | 43.5 | 29.7 | 35.0 | 30.7 |
| DNA | 5’-d(TCG XGA ACA TCC)-3’ | (−13.8) | (−8.5) | (−12.8) | |
aLowercase letters: modified nucleotide, capital letters: natural DNA. Total strand conc. 2 μM in 10 mM NaH2PO4, 150 mM NaCl, pH 7.0.
To gain more information about the helical structure of ON1–4 hybridized to complementary DNA or RNA, CD spectra of these duplexes were recorded (Figure 4). The duplexes of the four ONs paired to DNA still showed the overall shape of a B-type helix [48], although some duplexes exhibited slight distortions. Modest changes of the ellipticity amplitude maxima in the ON3/DNA duplex existed compared to the natural system. Also, some deviation from the natural structure displayed the ON4/DNA duplex. There, the band at 245 nm was depressed and the two positive ellipticities (≈220 nm and ≈280 nm) expressed increased intensities. Besides this, the ellipticity at 280 nm was blue-shifted (≈6 nm). All four modified DNA/RNA hybrids exhibited the characteristic shape of an intermediate A/B-type helix [48]. Again, some slight deviations from the natural system could be observed for the 6’-diF-bc4,3-T containing duplexes. The most distinct deviations occurred in the case of the ON4/RNA duplex. There the band at 225 nm was blue-shifted (≈5 nm) and the one at 245 slight red-shifted (≈2 nm) and both intensities were changed, too. Furthermore, the intensity of the 210 nm peak was reduced.
![[1860-5397-15-9-4]](/bjoc/content/figures/1860-5397-15-9-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: CD spectra of ON1–4 with complementary a) DNA, and b) RNA. Total strand conc. 2 μM in 10 mM NaH2PO4, 150 mM NaCl, pH 7.0.
Figure 4: CD spectra of ON1–4 with complementary a) DNA, and b) RNA. Total strand conc. 2 μM in 10 mM NaH2PO4...
RNase H cleavage assay
The most important requirement for an antisense oligonucleotide to induce RNase H activity lies in its DNA-like sugar conformations [49]. This is generally fulfilled by the 6’-diF-bc4,3-DNA as well as the 6’F-bc4,3-DNA. Furthermore, duplexes of a 6’F-bc4,3-modified strand paired to RNA unveiled in the MD simulations a flexible minor groove distance [37]. This flexibility is thought to play a crucial role for the fitting of the duplex into the DNA-binding channel and the phosphate-binding pocket of the enzyme. Furthermore, the phosphate-binding pocket requires a large distortion of the backbone angle α in order that the phosphate group of the AON can be positioned in it [50,51]. The 6’F-bc4,3-DNA containing strand also complied with this requirement according to the MD simulations [37]. Therefore, we examined the ability of the 6’F-bc4,3-DNA and the 6’-diF-bc4,3-DNA to induce the RNase H-mediated cleavage of the complementary RNA strand by utilizing the sequence of ON4 (5’-d(GCA ttt ttA CCG)-3’) and a chimeric sense strand. The sense strand consisted of five consecutive ribo-A units placed opposite the modified part and 2’-O-methyl RNA flanks. The same construct was previously applied for the evaluation of 2’F-tc-ANA [35] and a similar one for CeNA [52]. For the assay E. coli RNase H was used due to its commercial availability and its similarity to the human enzyme [22].
The cleavage pattern of the RNase H is presented in Figure 5. In the DNA/RNA positive control, the RNA strand was completely cleaved as expected (lane 1). In the negative controls C1 (no antisense strand) and C2 (no enzyme) no degradation of RNA could be observed (lanes 9 and 10). Acceptable substrates for the RNase H were duplexes containing both the 6’F-bc4,3-DNA (lanes 3–5) and the 6’-diF-bc4,3-DNA (lanes 6–8). The latter was able to induce a more efficient cleavage of the complementary RNA strand than the former. Nevertheless, both modifications recruited the RNase H to a lower extent than natural DNA. The 2’F-tc-ANA modification that has previously been shown to induce RNase H cleavage in a different sequence context was only modestly active (lane 2). Overall, the two new fluorinated bc4,3-analogs are among the few modifications which are able to activate RNase H to some extent. These promising results indicate that the 6’-diF-bc4,3-DNA and the 6’F-bc4,3-DNA may find application in therapeutic gapmer oligonucleotides.
![[1860-5397-15-9-5]](/bjoc/content/figures/1860-5397-15-9-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Hydrolysis products of the RNase H activation assay. The DNA served as positive control, whereas C1 (no antisense strand) and C2 (no enzyme) were negative controls.
Figure 5: Hydrolysis products of the RNase H activation assay. The DNA served as positive control, whereas C1...
Conclusion
In this report, we presented the successful synthesis of the 6’-diF-bc4,3-T building block where the gem-difluorinated bicyclic unit was formed starting from a previously described tricyclic siloxydifluorocyclopropane. The reaction of this tricyclic sugar under NIS-mediated nucleosidation produced two diiodo-substituted intermediates which were reduced by Bu3SnH yielding the β-nucleoside 6 as the only diastereoisomer. The crystal structure of nucleoside 6 exhibited the C2’-endo conformation of the furanose ring and the gauche orientation of the torsions angle γ. Conversion of this nucleoside into the corresponding phosphoramidite building block and its incorporation into oligonucleotides was then successfully achieved. Thermal melting experiment of the modified oligonucleotides paired to complementary DNA or RNA revealed a prominent duplex destabilization for both duplex types (ΔTm/mod = −1.6 to −5.5 °C). A lesser degree of destabilization was observed for oligonucleotides containing several consecutive modifications hybridized to complementary RNA. The reason for the destabilization might be accounted to repulsive electrostatic interactions between the equatorial fluorine atom and the 5’-oxygen. CD spectroscopy of the duplexes disclosed that the helical structure of the modified oligonucleotides paired to complementary DNA was still of a B-type, whereas an intermediate A/B-type helix was observed for RNA as complement. Furthermore, the RNase H assay of the oligonucleotide containing either five consecutive 6’-diF-bc4,3-Ts or 6’F-bc4,3-Ts paired to complementary RNA revealed that both modifications were able to recruit this enzyme. In both cases the RNase H cleaved the complementary RNA strand less efficiently as compared to the natural DNA/RNA duplex. This is a promising finding which points to a possible application for both modifications in therapeutic gapmer oligonucleotides.
Supporting Information
| Supporting Information File 1: Additional information, experimental procedures, NMR spectra, and crystallographic data. | ||
| Format: PDF | Size: 3.5 MB | Download |
Acknowledgements
We gratefully acknowledge the financial support by the Swiss National Science Foundation (grant-no: 200020_165778). We thank the group of Chemical Crystallography of the University of Bern (Prof. Dr. P. Macci) for the X-ray structure and the Swiss National Science Foundation (R’equip project 206021_128724) for co-funding of the single crystal X-ray diffractometer at the Department of Chemistry and Biochemistry of the University of Bern.
References
-
Swallow, S. Prog. Med. Chem. 2015, 54, 65–133. doi:10.1016/bs.pmch.2014.11.001
Return to citation in text: [1] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] -
Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f
Return to citation in text: [1] -
Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Guo, F.; Li, Q.; Zhou, C. Org. Biomol. Chem. 2017, 15, 9552–9565. doi:10.1039/c7ob02094e
Return to citation in text: [1] -
Østergaard, M. E.; Dwight, T.; Berdeja, A.; Swayze, E. E.; Jung, M. E.; Seth, P. P. J. Org. Chem. 2014, 79, 8877–8881. doi:10.1021/jo501381q
Return to citation in text: [1] -
Kawasaki, A. M.; Casper, M. D.; Freier, S. M.; Lesnik, E. A.; Zounes, M. C.; Cummins, L. L.; Gonzalez, C.; Cook, P. D. J. Med. Chem. 1993, 36, 831–841. doi:10.1021/jm00059a007
Return to citation in text: [1] [2] -
Damha, M. J.; Wilds, C. J.; Noronha, A.; Brukner, I.; Borkow, G.; Arion, D.; Parniak, M. A. J. Am. Chem. Soc. 1998, 120, 12976–12977. doi:10.1021/ja982325+
Return to citation in text: [1] [2] -
Ikeda, H.; Fernandez, R.; Barchi, J. J., Jr.; Huang, X.; Marquez, V. E.; Wilk, A. Nucleic Acids Res. 1998, 26, 2237–2244. doi:10.1093/nar/26.9.2237
Return to citation in text: [1] -
Patra, A.; Paolillo, M.; Charisse, K.; Manoharan, M.; Rozners, E.; Egli, M. Angew. Chem. 2012, 124, 12033–12036. doi:10.1002/ange.201204946
Angew. Chem., Int. Ed. 2012, 51, 11863–11866. doi:10.1002/anie.201204946
Return to citation in text: [1] -
Pallan, P. S.; Greene, E. M.; Jicman, P. A.; Pandey, R. K.; Manoharan, M.; Rozners, E.; Egli, M. Nucleic Acids Res. 2011, 39, 3482–3495. doi:10.1093/nar/gkq1270
Return to citation in text: [1] -
Martin-Pintado, N.; Deleavey, G. F.; Portella, G.; Campos-Olivas, R.; Orozco, M.; Damha, M. J.; González, C. Angew. Chem. 2013, 125, 12287–12290. doi:10.1002/ange.201305710
Angew. Chem., Int. Ed. 2013, 52, 12065–12068. doi:10.1002/anie.201305710
Return to citation in text: [1] -
Anzahaee, M. Y.; Watts, J. K.; Alla, N. R.; Nicholson, A. W.; Damha, M. J. J. Am. Chem. Soc. 2011, 133, 728–731. doi:10.1021/ja109817p
Return to citation in text: [1] -
Watts, J. K.; Martín-Pintado, N.; Gómez-Pinto, I.; Schwartzentruber, J.; Portella, G.; Orozco, M.; González, C.; Damha, M. J. Nucleic Acids Res. 2010, 38, 2498–2511. doi:10.1093/nar/gkp1225
Return to citation in text: [1] -
Wilds, C. J.; Damha, M. Nucleic Acids Res. 2000, 28, 3625–3635. doi:10.1093/nar/28.18.3625
Return to citation in text: [1] -
Berger, I.; Tereshko, V.; Ikeda, H.; Marquez, V. E.; Egli, M. Nucleic Acids Res. 1998, 26, 2473–2480. doi:10.1093/nar/26.10.2473
Return to citation in text: [1] -
Li, F.; Sarkhel, S.; Wilds, C. J.; Wawrzak, Z.; Prakash, T. P.; Manoharan, M.; Egli, M. Biochemistry 2006, 45, 4141–4152. doi:10.1021/bi052322r
Return to citation in text: [1] -
Rigo, F.; Hua, Y.; Chun, S. J.; Prakash, T. P.; Krainer, A. R.; Bennett, C. F. Nat. Chem. Biol. 2012, 8, 555–561. doi:10.1038/nchembio.939
Return to citation in text: [1] -
Kalota, A.; Karabon, L.; Swider, C. R.; Viazovkina, E.; Elzagheid, M.; Damha, M. J.; Gewirtz, A. M. Nucleic Acids Res. 2006, 34, 451–461. doi:10.1093/nar/gkj455
Return to citation in text: [1] -
Souleimanian, N.; Deleavey, G. F.; Soifer, H.; Wang, S.; Tiemann, K.; Damha, M. J.; Stein, C. A. Mol. Ther.–Nucleic Acids 2012, 1, e43. doi:10.1038/mtna.2012.35
Return to citation in text: [1] -
Monia, B. P.; Lesnik, E. A.; Gonzalez, C.; Lima, W. F.; McGee, D.; Guinosso, C. J.; Kawasaki, A. M.; Cook, P. D.; Freier, S. M. J. Biol. Chem. 1993, 268, 14514–14522.
Return to citation in text: [1] [2] -
Manoharan, M.; Akinc, A.; Pandey, R. K.; Qin, J.; Hadwiger, P.; John, M.; Mills, K.; Charisse, K.; Maier, M. A.; Nechev, L.; Greene, E. M.; Pallan, P. S.; Rozners, E.; Rajeev, K. G.; Egli, M. Angew. Chem. 2011, 123, 2332–2336. doi:10.1002/ange.201006519
Angew. Chem., Int. Ed. 2011, 50, 2284–2288. doi:10.1002/anie.201006519
Return to citation in text: [1] -
Dowler, T.; Bergeron, D.; Tedeschi, A.-L.; Paquet, L.; Ferrari, N.; Damha, M. J. Nucleic Acids Res. 2006, 34, 1669–1675. doi:10.1093/nar/gkl033
Return to citation in text: [1] -
Davis, S.; Propp, S.; Freier, S. M.; Jones, L. E.; Serra, M. J.; Kinberger, G.; Bhat, B.; Swayze, E. E.; Bennett, C. F.; Esau, C. Nucleic Acids Res. 2009, 37, 70–77. doi:10.1093/nar/gkn904
Return to citation in text: [1] -
Egli, M.; Pallan, P. S.; Allerson, C. R.; Prakash, T. P.; Berdeja, A.; Yu, J.; Lee, S.; Watt, A.; Gaus, H.; Bhat, B.; Swayze, E. E.; Seth, P. P. J. Am. Chem. Soc. 2011, 133, 16642–16649. doi:10.1021/ja207086x
Return to citation in text: [1] -
Seth, P. P.; Yu, J.; Jazayeri, A.; Pallan, P. S.; Allerson, C. R.; Østergaard, M. E.; Liu, F.; Herdewijn, P.; Egli, M.; Swayze, E. E. J. Org. Chem. 2012, 77, 5074–5085. doi:10.1021/jo300594b
Return to citation in text: [1] -
Seth, P. P.; Pallan, P. S.; Swayze, E. E.; Egli, M. ChemBioChem 2013, 14, 58–62. doi:10.1002/cbic.201200669
Return to citation in text: [1] -
Jung, M. E.; Dwight, T. A.; Vigant, F.; Østergaard, M. E.; Swayze, E. E.; Seth, P. P. Angew. Chem. 2014, 126, 10051–10055. doi:10.1002/ange.201405283
Angew. Chem., Int. Ed. 2014, 53, 9893–9897. doi:10.1002/anie.201405283
Return to citation in text: [1] -
Martínez-Montero, S.; Deleavey, G. F.; Martín-Pintado, N.; Fakhoury, J. F.; González, C.; Damha, M. J. ACS Chem. Biol. 2015, 10, 2016–2023. doi:10.1021/acschembio.5b00218
Return to citation in text: [1] -
Martínez-Montero, S.; Deleavey, G. F.; Dierker-Viik, A.; Lindovska, P.; Ilina, T.; Portella, G.; Orozco, M.; Parniak, M. A.; González, C.; Damha, M. J. J. Org. Chem. 2015, 80, 3083–3091. doi:10.1021/jo502948t
Return to citation in text: [1] -
Dugovic, B.; Leumann, C. J. J. Org. Chem. 2014, 79, 1271–1279. doi:10.1021/jo402690j
Return to citation in text: [1] -
Medvecky, M.; Istrate, A.; Leumann, C. J. J. Org. Chem. 2015, 80, 3556–3565. doi:10.1021/acs.joc.5b00184
Return to citation in text: [1] -
Istrate, A.; Medvecky, M.; Leumann, C. J. Org. Lett. 2015, 17, 1950–1953. doi:10.1021/acs.orglett.5b00662
Return to citation in text: [1] -
Istrate, A.; Katolik, A.; Istrate, A.; Leumann, C. J. Chem. – Eur. J. 2017, 23, 10310–10318. doi:10.1002/chem.201701476
Return to citation in text: [1] [2] [3] -
Stauffiger, A.; Leumann, C. J. Eur. J. Org. Chem. 2009, 1153–1162. doi:10.1002/ejoc.200801034
Return to citation in text: [1] [2] [3] -
Frei, S.; Leumann, C. J. Beilstein J. Org. Chem. 2018, 14, 3088–3097. doi:10.3762/bjoc.14.288
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Kageshima, Y.; Suzuki, C.; Oshiro, K.; Amii, H. Synlett 2015, 26, 63–66. doi:10.1055/s-0034-1379599
Return to citation in text: [1] -
Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2015, 17, 760–763. doi:10.1021/acs.orglett.5b00097
Return to citation in text: [1] -
Fedorov, O. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 5870–5876. doi:10.1021/acs.joc.5b00904
Return to citation in text: [1] [2] -
Quintard, J.-P.; Pereyre, M. J. Organomet. Chem. 1974, 82, 103–111. doi:10.1016/s0022-328x(00)80725-1
Return to citation in text: [1] -
Curran, D. P.; Sun, S. Tetrahedron Lett. 1993, 34, 6181–6184. doi:10.1016/s0040-4039(00)73704-x
Return to citation in text: [1] -
Tarköy, M.; Bolli, M.; Leumann, C. Helv. Chim. Acta 1994, 77, 716–744. doi:10.1002/hlca.19940770315
Return to citation in text: [1] -
Sekiguchi, M.; Obika, S.; Harada, Y.; Osaki, T.; Somjing, R.; Mitsuoka, Y.; Shibata, N.; Masaki, M.; Imanishi, T. J. Org. Chem. 2006, 71, 1306–1316. doi:10.1021/jo051187l
Return to citation in text: [1] -
CCDC 1872241 contains the supplementary crystalographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre.
Return to citation in text: [1] -
O'Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/pac-con-11-09-26
Return to citation in text: [1] -
Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Vorlíčková, M.; Kejnovská, I.; Bednářová, K.; Renčiuk, D.; Kypr, J. Chirality 2012, 24, 691–698. doi:10.1002/chir.22064
Return to citation in text: [1] [2] -
Crooke, S. T. Antisense Drug Technology Principles, Strategies, and Applications, 2nd ed.; CRC Press, Inc.: Boca Raton, 2008.
Return to citation in text: [1] -
Lima, W. F.; Nichols, J. G.; Wu, H.; Prakash, T. P.; Migawa, M. T.; Wyrzykiewicz, T. K.; Bhat, B.; Crooke, S. T. J. Biol. Chem. 2004, 279, 36317–36326. doi:10.1074/jbc.m405035200
Return to citation in text: [1] -
Nowotny, M.; Gaidamakov, S. A.; Ghirlando, R.; Cerritelli, S. M.; Crouch, R. J.; Yang, W. Mol. Cell 2007, 28, 264–276. doi:10.1016/j.molcel.2007.08.015
Return to citation in text: [1] -
Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, C.; Rosemeyer, H.; Seela, F.; Van Aerschot, A.; Herdewijn, P. J. Am. Chem. Soc. 2000, 122, 8595–8602. doi:10.1021/ja000018+
Return to citation in text: [1]
| 37. | Frei, S.; Leumann, C. J. Beilstein J. Org. Chem. 2018, 14, 3088–3097. doi:10.3762/bjoc.14.288 |
| 37. | Frei, S.; Leumann, C. J. Beilstein J. Org. Chem. 2018, 14, 3088–3097. doi:10.3762/bjoc.14.288 |
| 1. | Swallow, S. Prog. Med. Chem. 2015, 54, 65–133. doi:10.1016/bs.pmch.2014.11.001 |
| 2. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 3. | Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f |
| 4. | Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023 |
| 5. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 9. | Damha, M. J.; Wilds, C. J.; Noronha, A.; Brukner, I.; Borkow, G.; Arion, D.; Parniak, M. A. J. Am. Chem. Soc. 1998, 120, 12976–12977. doi:10.1021/ja982325+ |
| 28. | Seth, P. P.; Pallan, P. S.; Swayze, E. E.; Egli, M. ChemBioChem 2013, 14, 58–62. doi:10.1002/cbic.201200669 |
| 29. |
Jung, M. E.; Dwight, T. A.; Vigant, F.; Østergaard, M. E.; Swayze, E. E.; Seth, P. P. Angew. Chem. 2014, 126, 10051–10055. doi:10.1002/ange.201405283
Angew. Chem., Int. Ed. 2014, 53, 9893–9897. doi:10.1002/anie.201405283 |
| 30. | Martínez-Montero, S.; Deleavey, G. F.; Martín-Pintado, N.; Fakhoury, J. F.; González, C.; Damha, M. J. ACS Chem. Biol. 2015, 10, 2016–2023. doi:10.1021/acschembio.5b00218 |
| 31. | Martínez-Montero, S.; Deleavey, G. F.; Dierker-Viik, A.; Lindovska, P.; Ilina, T.; Portella, G.; Orozco, M.; Parniak, M. A.; González, C.; Damha, M. J. J. Org. Chem. 2015, 80, 3083–3091. doi:10.1021/jo502948t |
| 37. | Frei, S.; Leumann, C. J. Beilstein J. Org. Chem. 2018, 14, 3088–3097. doi:10.3762/bjoc.14.288 |
| 8. | Kawasaki, A. M.; Casper, M. D.; Freier, S. M.; Lesnik, E. A.; Zounes, M. C.; Cummins, L. L.; Gonzalez, C.; Cook, P. D. J. Med. Chem. 1993, 36, 831–841. doi:10.1021/jm00059a007 |
| 32. | Dugovic, B.; Leumann, C. J. J. Org. Chem. 2014, 79, 1271–1279. doi:10.1021/jo402690j |
| 33. | Medvecky, M.; Istrate, A.; Leumann, C. J. J. Org. Chem. 2015, 80, 3556–3565. doi:10.1021/acs.joc.5b00184 |
| 34. | Istrate, A.; Medvecky, M.; Leumann, C. J. Org. Lett. 2015, 17, 1950–1953. doi:10.1021/acs.orglett.5b00662 |
| 35. | Istrate, A.; Katolik, A.; Istrate, A.; Leumann, C. J. Chem. – Eur. J. 2017, 23, 10310–10318. doi:10.1002/chem.201701476 |
| 35. | Istrate, A.; Katolik, A.; Istrate, A.; Leumann, C. J. Chem. – Eur. J. 2017, 23, 10310–10318. doi:10.1002/chem.201701476 |
| 7. | Østergaard, M. E.; Dwight, T.; Berdeja, A.; Swayze, E. E.; Jung, M. E.; Seth, P. P. J. Org. Chem. 2014, 79, 8877–8881. doi:10.1021/jo501381q |
| 26. | Egli, M.; Pallan, P. S.; Allerson, C. R.; Prakash, T. P.; Berdeja, A.; Yu, J.; Lee, S.; Watt, A.; Gaus, H.; Bhat, B.; Swayze, E. E.; Seth, P. P. J. Am. Chem. Soc. 2011, 133, 16642–16649. doi:10.1021/ja207086x |
| 37. | Frei, S.; Leumann, C. J. Beilstein J. Org. Chem. 2018, 14, 3088–3097. doi:10.3762/bjoc.14.288 |
| 6. | Guo, F.; Li, Q.; Zhou, C. Org. Biomol. Chem. 2017, 15, 9552–9565. doi:10.1039/c7ob02094e |
| 27. | Seth, P. P.; Yu, J.; Jazayeri, A.; Pallan, P. S.; Allerson, C. R.; Østergaard, M. E.; Liu, F.; Herdewijn, P.; Egli, M.; Swayze, E. E. J. Org. Chem. 2012, 77, 5074–5085. doi:10.1021/jo300594b |
| 50. | Lima, W. F.; Nichols, J. G.; Wu, H.; Prakash, T. P.; Migawa, M. T.; Wyrzykiewicz, T. K.; Bhat, B.; Crooke, S. T. J. Biol. Chem. 2004, 279, 36317–36326. doi:10.1074/jbc.m405035200 |
| 51. | Nowotny, M.; Gaidamakov, S. A.; Ghirlando, R.; Cerritelli, S. M.; Crouch, R. J.; Yang, W. Mol. Cell 2007, 28, 264–276. doi:10.1016/j.molcel.2007.08.015 |
| 14. | Anzahaee, M. Y.; Watts, J. K.; Alla, N. R.; Nicholson, A. W.; Damha, M. J. J. Am. Chem. Soc. 2011, 133, 728–731. doi:10.1021/ja109817p |
| 15. | Watts, J. K.; Martín-Pintado, N.; Gómez-Pinto, I.; Schwartzentruber, J.; Portella, G.; Orozco, M.; González, C.; Damha, M. J. Nucleic Acids Res. 2010, 38, 2498–2511. doi:10.1093/nar/gkp1225 |
| 9. | Damha, M. J.; Wilds, C. J.; Noronha, A.; Brukner, I.; Borkow, G.; Arion, D.; Parniak, M. A. J. Am. Chem. Soc. 1998, 120, 12976–12977. doi:10.1021/ja982325+ |
| 18. | Li, F.; Sarkhel, S.; Wilds, C. J.; Wawrzak, Z.; Prakash, T. P.; Manoharan, M.; Egli, M. Biochemistry 2006, 45, 4141–4152. doi:10.1021/bi052322r |
| 48. | Vorlíčková, M.; Kejnovská, I.; Bednářová, K.; Renčiuk, D.; Kypr, J. Chirality 2012, 24, 691–698. doi:10.1002/chir.22064 |
| 13. |
Martin-Pintado, N.; Deleavey, G. F.; Portella, G.; Campos-Olivas, R.; Orozco, M.; Damha, M. J.; González, C. Angew. Chem. 2013, 125, 12287–12290. doi:10.1002/ange.201305710
Angew. Chem., Int. Ed. 2013, 52, 12065–12068. doi:10.1002/anie.201305710 |
| 8. | Kawasaki, A. M.; Casper, M. D.; Freier, S. M.; Lesnik, E. A.; Zounes, M. C.; Cummins, L. L.; Gonzalez, C.; Cook, P. D. J. Med. Chem. 1993, 36, 831–841. doi:10.1021/jm00059a007 |
| 19. | Rigo, F.; Hua, Y.; Chun, S. J.; Prakash, T. P.; Krainer, A. R.; Bennett, C. F. Nat. Chem. Biol. 2012, 8, 555–561. doi:10.1038/nchembio.939 |
| 20. | Kalota, A.; Karabon, L.; Swider, C. R.; Viazovkina, E.; Elzagheid, M.; Damha, M. J.; Gewirtz, A. M. Nucleic Acids Res. 2006, 34, 451–461. doi:10.1093/nar/gkj455 |
| 21. | Souleimanian, N.; Deleavey, G. F.; Soifer, H.; Wang, S.; Tiemann, K.; Damha, M. J.; Stein, C. A. Mol. Ther.–Nucleic Acids 2012, 1, e43. doi:10.1038/mtna.2012.35 |
| 22. | Monia, B. P.; Lesnik, E. A.; Gonzalez, C.; Lima, W. F.; McGee, D.; Guinosso, C. J.; Kawasaki, A. M.; Cook, P. D.; Freier, S. M. J. Biol. Chem. 1993, 268, 14514–14522. |
| 23. |
Manoharan, M.; Akinc, A.; Pandey, R. K.; Qin, J.; Hadwiger, P.; John, M.; Mills, K.; Charisse, K.; Maier, M. A.; Nechev, L.; Greene, E. M.; Pallan, P. S.; Rozners, E.; Rajeev, K. G.; Egli, M. Angew. Chem. 2011, 123, 2332–2336. doi:10.1002/ange.201006519
Angew. Chem., Int. Ed. 2011, 50, 2284–2288. doi:10.1002/anie.201006519 |
| 24. | Dowler, T.; Bergeron, D.; Tedeschi, A.-L.; Paquet, L.; Ferrari, N.; Damha, M. J. Nucleic Acids Res. 2006, 34, 1669–1675. doi:10.1093/nar/gkl033 |
| 25. | Davis, S.; Propp, S.; Freier, S. M.; Jones, L. E.; Serra, M. J.; Kinberger, G.; Bhat, B.; Swayze, E. E.; Bennett, C. F.; Esau, C. Nucleic Acids Res. 2009, 37, 70–77. doi:10.1093/nar/gkn904 |
| 49. | Crooke, S. T. Antisense Drug Technology Principles, Strategies, and Applications, 2nd ed.; CRC Press, Inc.: Boca Raton, 2008. |
| 11. |
Patra, A.; Paolillo, M.; Charisse, K.; Manoharan, M.; Rozners, E.; Egli, M. Angew. Chem. 2012, 124, 12033–12036. doi:10.1002/ange.201204946
Angew. Chem., Int. Ed. 2012, 51, 11863–11866. doi:10.1002/anie.201204946 |
| 12. | Pallan, P. S.; Greene, E. M.; Jicman, P. A.; Pandey, R. K.; Manoharan, M.; Rozners, E.; Egli, M. Nucleic Acids Res. 2011, 39, 3482–3495. doi:10.1093/nar/gkq1270 |
| 36. | Stauffiger, A.; Leumann, C. J. Eur. J. Org. Chem. 2009, 1153–1162. doi:10.1002/ejoc.200801034 |
| 10. | Ikeda, H.; Fernandez, R.; Barchi, J. J., Jr.; Huang, X.; Marquez, V. E.; Wilk, A. Nucleic Acids Res. 1998, 26, 2237–2244. doi:10.1093/nar/26.9.2237 |
| 16. | Wilds, C. J.; Damha, M. Nucleic Acids Res. 2000, 28, 3625–3635. doi:10.1093/nar/28.18.3625 |
| 17. | Berger, I.; Tereshko, V.; Ikeda, H.; Marquez, V. E.; Egli, M. Nucleic Acids Res. 1998, 26, 2473–2480. doi:10.1093/nar/26.10.2473 |
| 48. | Vorlíčková, M.; Kejnovská, I.; Bednářová, K.; Renčiuk, D.; Kypr, J. Chirality 2012, 24, 691–698. doi:10.1002/chir.22064 |
| 37. | Frei, S.; Leumann, C. J. Beilstein J. Org. Chem. 2018, 14, 3088–3097. doi:10.3762/bjoc.14.288 |
| 35. | Istrate, A.; Katolik, A.; Istrate, A.; Leumann, C. J. Chem. – Eur. J. 2017, 23, 10310–10318. doi:10.1002/chem.201701476 |
| 52. | Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, C.; Rosemeyer, H.; Seela, F.; Van Aerschot, A.; Herdewijn, P. J. Am. Chem. Soc. 2000, 122, 8595–8602. doi:10.1021/ja000018+ |
| 36. | Stauffiger, A.; Leumann, C. J. Eur. J. Org. Chem. 2009, 1153–1162. doi:10.1002/ejoc.200801034 |
| 22. | Monia, B. P.; Lesnik, E. A.; Gonzalez, C.; Lima, W. F.; McGee, D.; Guinosso, C. J.; Kawasaki, A. M.; Cook, P. D.; Freier, S. M. J. Biol. Chem. 1993, 268, 14514–14522. |
| 46. | O'Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/pac-con-11-09-26 |
| 36. | Stauffiger, A.; Leumann, C. J. Eur. J. Org. Chem. 2009, 1153–1162. doi:10.1002/ejoc.200801034 |
| 43. | Tarköy, M.; Bolli, M.; Leumann, C. Helv. Chim. Acta 1994, 77, 716–744. doi:10.1002/hlca.19940770315 |
| 44. | Sekiguchi, M.; Obika, S.; Harada, Y.; Osaki, T.; Somjing, R.; Mitsuoka, Y.; Shibata, N.; Masaki, M.; Imanishi, T. J. Org. Chem. 2006, 71, 1306–1316. doi:10.1021/jo051187l |
| 45. | CCDC 1872241 contains the supplementary crystalographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre. |
| 40. | Fedorov, O. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 5870–5876. doi:10.1021/acs.joc.5b00904 |
| 41. | Quintard, J.-P.; Pereyre, M. J. Organomet. Chem. 1974, 82, 103–111. doi:10.1016/s0022-328x(00)80725-1 |
| 42. | Curran, D. P.; Sun, S. Tetrahedron Lett. 1993, 34, 6181–6184. doi:10.1016/s0040-4039(00)73704-x |
| 38. | Kageshima, Y.; Suzuki, C.; Oshiro, K.; Amii, H. Synlett 2015, 26, 63–66. doi:10.1055/s-0034-1379599 |
| 39. | Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2015, 17, 760–763. doi:10.1021/acs.orglett.5b00097 |
| 40. | Fedorov, O. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 5870–5876. doi:10.1021/acs.joc.5b00904 |
| 37. | Frei, S.; Leumann, C. J. Beilstein J. Org. Chem. 2018, 14, 3088–3097. doi:10.3762/bjoc.14.288 |
© 2019 Frei et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)