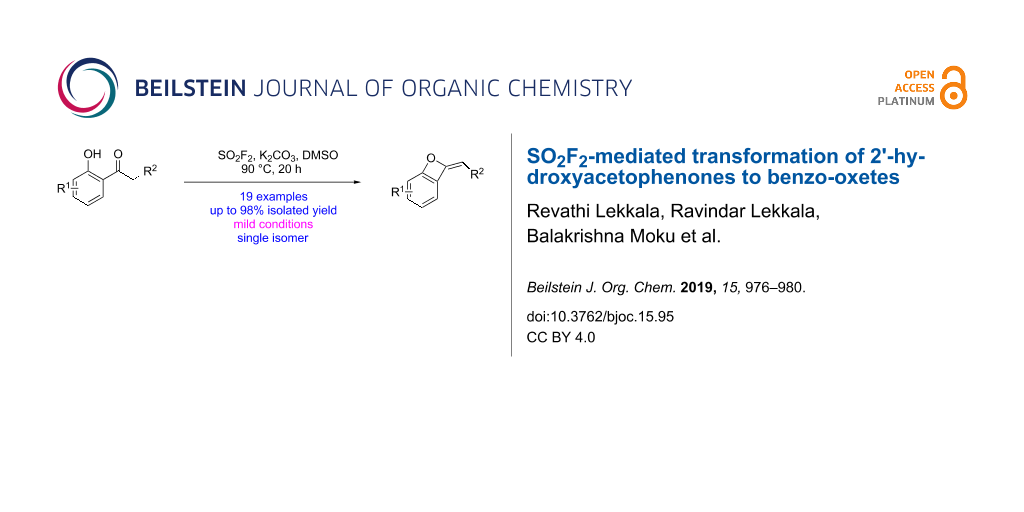
Abstract
A catalyst-free novel and efficient methodology for the challenging synthesis of benzo-oxetes from 2'-hydroxyacetophenones mediated by sulfuryl fluoride (SO2F2) gas has been realized. The combination of 2'-hydroxyacetophenones and SO2F2 furnishes synthetically challenging benzo-oxetanes in moderate to excellent yields. The highlight of this work is the design and synthesis of strained four-membered oxete rings.

Graphical Abstract
Introduction
Oxetanes are versatile elements in drug discovery and synthesis [1,2], and represent important moieties in some natural products and biological active molecules [3]. The synthesis and chemistry of oxetanes have been reviewed [1,2,4,5]. In medicinal chemistry, oxetanes have remained neglected units for many years since the first parent structure has been reported by Reboul in 1878 [6]. As strained cyclic ethers oxetanes exhibit an attractive combination of stable patterns for medicinal chemistry and reactivity for further synthesis [4]. These characteristics formulate them a captivating pattern for a rising scope of applications in the chemical sciences. Recently, oxetanes emerged as small, stable, attractive, and less lipophilic molecular units in drug discovery [7,8]. In view of the broad applicability of oxetanes in materials sciences, organic synthesis, and medicinal chemistry [7-15], the progress of synthetic routes for the formation of a range of oxetanes is highly desirable.
However, the intrinsic ring strain in oxetanes makes cyclization a basic challenge, moreover, the kinetics of cyclization to make four-membered ring ethers are considerably slower than those of six, five, and three-membered ring compounds [16]. Therefore, the formation of oxetanes with satisfactory yields from the functionalized acyclic precursors generally requires good leaving groups and anions. A large number of synthetic methods has been described for the formation of oxetane derivatives and among them, two common strategies are widely applied. The first strategy engages an intramolecular etherification reaction, which is called Williamson ether synthesis (Scheme 1a). This method was first utilized to synthesize oxetanes in 1878 and has remained as general tool in the synthesis of complex oxetane-containing compounds since then [17-20]. The second strategy involves a [2 + 2] cycloaddition that furnishes substituted oxetanes rapidly, for example Paterno–Büchi reaction (Scheme 1b) [21]. However, the recognition of facial selectivity control in [2 + 2] cycloaddition is not an easy task.
![[1860-5397-15-95-i1]](/bjoc/content/inline/1860-5397-15-95-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathways towards oxetanes and benzo-oxetes.
Scheme 1: Synthetic pathways towards oxetanes and benzo-oxetes.
The availability of various methods for the synthesis of oxetanes and their strained nature has afforded opportunities for the discovery of new transformations. Previous reports on the synthesis of substituted oxetanes from ketones described the requirement of chiral reagents or catalysts [11-14,22-24]. Gaseous sulfuryl fluoride (SO2F2) widely has been utilized as a fumigant for more than five decades [25,26], and only recently it has attracted significant attention as an organic synthetic reagent. SO2F2 is a cheap and relatively stable gas (up to 400 °C when dry) and a highly reactive electrophile [27-29]. Under basic conditions, SO2F2 hydrolyzes rapidly into fluoride and fluorosulfate ions. In continuation of our research on the application of SO2F2 for the transformation of organic compounds [30-37], we herein report a novel strategy for the synthesis of benzo-oxete derivatives from ketones using the inexpensive and abundant SO2F2 through a catalyst-free cascade method (Scheme 1c).
Results and Discussion
Initially, we screened various reaction conditions (see Supporting Information File 1) using 1-(2-hydroxyphenyl)ethanone (1a) as a model substrate. As illustrated in Table 1, the desired product 2a was produced in 50–76% yield when different inorganic bases (3 equiv) were used in DMSO at 90 °C under a SO2F2 atmosphere (balloon) for 15 h (Table 1, entries 1–4). The use of K2CO3 provided a significantly higher product yield than the other bases tested (Table 1, entry 5) under the same reaction conditions. When the reaction time was decreased from 15 to 5 h, the yield of the desired product dropped from 90% to 76% (Table 1, entry 6 vs entry 5). On the other hand, increasing the reaction time to 18 h led to an increased product yield (from 90% to 94%, Table 1, entry 7 vs entry 5) and after 20 h reaction time a maximum yield of 98% was achieved (Table 1, entry 8). Next, screening of the reaction temperature revealed that lowering the temperature from 90 °C to 80 °C or further to 70 °C resulted in an obvious decrease of the product yield (from 98% to 91%, and 72%, respectively, Table 1, entries 8, 9 and 10) thus indicating that performing the reaction at 90 °C was essential for high efficiency of the transformation. Additional screening revealed that decreasing the amount of K2CO3 to 2.0 equiv or 1.0 equiv affected the cyclization process significantly to provide benzo-oxetes in 81% and 59% yield, respectively (Table 1, entries 11 and 12).
Table 1: Screening and optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-95-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Base (equiv) | T (°C) | t (h) | Yield (2a, %)b |
| 1 | KOAc (3) | 90 | 15 | 76 |
| 2 | NaOEt (3) | 90 | 15 | 50 |
| 3 | NaOH (3) | 90 | 15 | 76 |
| 4 | NaOAc (3) | 90 | 15 | 50 |
| 5 | K2CO3 (3) | 90 | 15 | 90 |
| 6 | K2CO3 (3) | 90 | 5 | 76 |
| 7 | K2CO3 (3) | 90 | 18 | 94 |
| 8 | K2CO3 (3) | 90 | 20 | 98 |
| 9 | K2CO3 (3) | 80 | 20 | 91 |
| 10 | K2CO3 (3) | 70 | 20 | 72 |
| 11 | K2CO3 (2) | 90 | 20 | 81 |
| 12 | K2CO3 (1) | 90 | 20 | 59 |
aReaction conditions: a mixture of 1-(2-hydroxyphenyl)ethanone (1a, 1.0 mmol, 1.0 equiv), base (X mmol, X equiv) in DMSO (2.0 mL) was stirred at the indicated temperature, under a SO2F2 atmosphere (balloon) for 5–20 h. bIsolated yields.
With the optimized reaction conditions in hand, we next evaluated the generality of the SO2F2-mediated transformation by subjecting diverse 2'-hydroxyacetophenones to the reaction (Scheme 2) [38,39]. Under the optimal reaction conditions, various 2'-hydroxyacetophenones containing both electron-withdrawing and electron-donating groups in the ortho, meta or para-positions of the aryl ring (1b–j) efficiently were transformed to the corresponding benzo-oxetes (2b–j) in 31– 98% yields. Furthermore, also multifunctionalized 2'-hydroxyacetophenones 1k–o were converted smoothly to the corresponding benzo-oxetes (2k–o) affording the products in 31% to 78% yield. In addition, naphthalenones 1p–s also successfully were transformed to their corresponding benzo-oxetes 2p–s in 30–95% yield under identical reaction conditions. For the substituted structure, there is one single isomer obtained, which was proposed to be E-configured. However, the exact configuration was not confirmed [40].
![[1860-5397-15-95-i2]](/bjoc/content/inline/1860-5397-15-95-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of the SO2F2-mediated transformation of 2'-hydroxyacetophenones to benzo-oxetes. Reaction conditions: a mixture of 1-(2-hydroxyphenyl)ethanone (1a, 1.0 mmol, 1.0 equiv), K2CO3 (3.0 mmol, 3.0 equiv) in DMSO (2.0 mL) was stirred at 90 ° C under a SO2F2 atmosphere (balloon) for 20 h. Yields refer to isolated yields. aReaction performed at 50 °C.
Scheme 2: Substrate scope of the SO2F2-mediated transformation of 2'-hydroxyacetophenones to benzo-oxetes. Re...
Two possible reaction mechanisms were proposed for this SO2F2-mediated transformation of 2'-hydroxyacetophenones to benzo-oxetes (Scheme 3). The first mechanism commences with the deprotonation of 2'-hydroxyacetophenone 1 by the base (K2CO3) generating phenoxide A, which subsequently reacts with SO2F2 to give intermediate B with concomitant release of a fluoride anion. The proton of the acetyl group adjacent to the carbonyl moiety in intermediate B is deprotonated by the base (K2CO3) to generate enolated intermediate C, which finally undergoes an intramolecular cyclization furnishing the corresponding benzo-oxete 2. The second mechanism starts with the elimination of the proton from the acetyl group next to the carbonyl moiety of the 2'-hydroxyacetophenone 1 in the presence of the base to afford enol intermediate I. The latter then reacts with SO2F2 to give intermediate II and fluoride anion. The subsequent deprotonation of intermediate B by the base generates phenol anion III, which finally undergoes an intramolecular cyclization to give the corresponding benzo-oxete 2.
![[1860-5397-15-95-i3]](/bjoc/content/inline/1860-5397-15-95-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Two proposed reaction mechanisms. B = base.
Scheme 3: Two proposed reaction mechanisms. B = base.
Conclusion
We have developed a new protocol for the transformation of 2'-hydroxyacetophenones to benzo-oxetes. This SO2F2-mediated cascade reaction showed exceptional functional group tolerance and a broad substrate scope which, to the best of our knowledge, is the first example of employing cheap and abundant 2'-hydroxyacetophenones as starting materials for the synthesis of benzo-oxetes.
Supporting Information
| Supporting Information File 1: Conditions optimization, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 3.4 MB | Download |
References
-
Burkhard, J. A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 9052–9067. doi:10.1002/anie.200907155
Angew. Chem. 2010, 122, 9236–9251. doi:10.1002/ange.200907155
Return to citation in text: [1] [2] -
Wuitschik, G.; Carreira, E. M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. J. Med. Chem. 2010, 53, 3227–3246. doi:10.1021/jm9018788
Return to citation in text: [1] [2] -
Das, B.; Damodar, K. Epoxides and Oxetanes. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, 2011; pp 63–95. doi:10.1002/9783527634880.ch3
Return to citation in text: [1] -
Bull, J. A.; Croft, R. A.; Davis, O. A.; Doran, R.; Morgan, K. F. Chem. Rev. 2016, 116, 12150–12233. doi:10.1021/acs.chemrev.6b00274
Return to citation in text: [1] [2] -
Malapit, C. A.; Howell, A. R. J. Org. Chem. 2015, 80, 8489–8495. doi:10.1021/acs.joc.5b01255
Return to citation in text: [1] -
Reboul, M. Ann. Chim. (Cachan, Fr.) 1878, 14, 495–497.
Return to citation in text: [1] -
Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E. M. Angew. Chem., Int. Ed. 2006, 45, 7736–7739. doi:10.1002/anie.200602343
Angew. Chem. 2006, 118, 7900–7903. doi:10.1002/ange.200602343
Return to citation in text: [1] [2] -
Wuitschik, G.; Rogers-Evans, M.; Buckl, A.; Bernasconi, M.; Märki, M.; Godel, T.; Fischer, H.; Wagner, B.; Parrilla, I.; Schuler, F.; Schneider, J.; Alker, A.; Schweizer, W. B.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4512–4515. doi:10.1002/anie.200800450
Angew. Chem. 2008, 120, 4588–4591. doi:10.1002/ange.200800450
Return to citation in text: [1] [2] -
Permana, Y.; Nakano, K.; Yamashita, M.; Watanabe, D.; Nozaki, K. Chem. – Asian J. 2008, 3, 710–718. doi:10.1002/asia.200700339
Return to citation in text: [1] -
Bouchékif, H.; Philbin, M. I.; Colclough, E.; Amass, A. J. Macromolecules 2008, 41, 1989–1995. doi:10.1021/ma062761d
Return to citation in text: [1] -
Lo, M. M.-C.; Fu, G. C. Tetrahedron 2001, 57, 2621–2634. doi:10.1016/s0040-4020(01)00082-5
Return to citation in text: [1] [2] -
Soai, K.; Niwa, S.; Yamanoi, T.; Hikima, H.; Ishizaki, M. J. Chem. Soc., Chem. Commun. 1986, 1018–1019. doi:10.1039/c39860001018
Return to citation in text: [1] [2] -
Bertolini, F.; Crotti, S.; Di Bussolo, V.; Macchia, F.; Pineschi, M. J. Org. Chem. 2008, 73, 8998–9007. doi:10.1021/jo801568a
Return to citation in text: [1] [2] -
Hintzer, K.; Koppenhoefer, B.; Schurig, V. J. Org. Chem. 1982, 47, 3850–3854. doi:10.1021/jo00141a009
Return to citation in text: [1] [2] -
Dussault, P. H.; Trullinger, T. K.; Noor-e-Ain, F. Org. Lett. 2002, 4, 4591–4593. doi:10.1021/ol0265259
Return to citation in text: [1] -
Di Martino, A.; Galli, C.; Gargano, P.; Mandolini, L. J. Chem. Soc., Perkin Trans. 2 1985, 1345–1349. doi:10.1039/p29850001345
Return to citation in text: [1] -
Picard, P.; Leclercq, D.; Bats, J.-P.; Moulines, J. Synthesis 1981, 550–551. doi:10.1055/s-1981-29523
Return to citation in text: [1] -
Rosowsky, A.; Tarbell, D. S. J. Org. Chem. 1961, 26, 2255–2260. doi:10.1021/jo01351a026
Return to citation in text: [1] -
Balsamo, A.; Ceccarelli, G.; Crotti, P.; Macchia, F. J. Org. Chem. 1975, 40, 473–476. doi:10.1021/jo00892a021
Return to citation in text: [1] -
Berkowitz, P. T.; Baum, K. J. Org. Chem. 1980, 45, 4853–4857. doi:10.1021/jo01312a010
Return to citation in text: [1] -
Bach, T. Synthesis 1998, 683–703. doi:10.1055/s-1998-2054
Return to citation in text: [1] -
Sone, T.; Lu, G.; Matsunaga, S.; Shibasaki, M. Angew. Chem., Int. Ed. 2009, 48, 1677–1680. doi:10.1002/ange.200805473
Angew. Chem. 2009, 121, 1705–1708. doi:10.1002/anie.200805473
Return to citation in text: [1] -
Brown, H. C.; Ramachandran, P. V. Acc. Chem. Res. 1992, 25, 16–24. doi:10.1021/ar00013a003
Return to citation in text: [1] -
Davies, A. T.; Slawin, A. M. Z.; Smith, A. D. Chem. – Eur. J. 2015, 21, 18944–18948. doi:10.1002/chem.201504256
Return to citation in text: [1] -
Kenaga, E. E. J. Econ. Entomol. 1957, 50, 1–6. doi:10.1093/jee/50.1.1
Return to citation in text: [1] -
Sulbaek Andersen, M. P.; Blake, D. R.; Rowland, F. S.; Hurley, M. D.; Wallington, T. J. Environ. Sci. Technol. 2009, 43, 1067–1070. doi:10.1021/es802439f
Return to citation in text: [1] -
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Return to citation in text: [1] -
Revathi, L.; Ravindar, L.; Leng, J.; Rakesh, K. P.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 662–682. doi:10.1002/ajoc.201700591
Return to citation in text: [1] -
Veryser, C.; Demaerel, J.; Bieliunas, V.; Gilles, P.; De Borggraeve, W. M. Org. Lett. 2017, 19, 5244–5247. doi:10.1021/acs.orglett.7b02522
Return to citation in text: [1] -
Fang, W.-Y.; Leng, J.; Qin, H.-L. Chem. – Asian J. 2017, 12, 2323–2331. doi:10.1002/asia.201700891
Return to citation in text: [1] -
Wang, X.-Y.; Leng, J.; Wang, S.-M.; Asiri, A. M.; Marwani, H. M.; Qin, H.-L. Tetrahedron Lett. 2017, 58, 2340–2343. doi:10.1016/j.tetlet.2017.04.070
Return to citation in text: [1] -
Fang, W.-Y.; Huang, Y.-M.; Leng, J.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 751–756. doi:10.1002/ajoc.201800037
Return to citation in text: [1] -
Zhao, C.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2018, 5, 1835–1839. doi:10.1039/c8qo00295a
Return to citation in text: [1] -
Zha, G.-F.; Fang, W.-Y.; Li, Y.-G.; Leng, J.; Chen, X.; Qin, H.-L. J. Am. Chem. Soc. 2018, 140, 17666–17673. doi:10.1021/jacs.8b10069
Return to citation in text: [1] -
Zhang, X.; Rakesh, K. P.; Qin, H.-L. Chem. Commun. 2019, 55, 2845–2848. doi:10.1039/c8cc09693g
Return to citation in text: [1] -
Lekkala, R.; Lekkala, R.; Moku, B.; Qin, H.-L. Org. Chem. Front. 2019, 6, 796–800. doi:10.1039/c8qo01388h
Return to citation in text: [1] -
Zhao, C.; Zha, G.-F.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Eur. J. Org. Chem. 2019, 1801–1807. doi:10.1002/ejoc.201801888
Return to citation in text: [1] -
We tried to open the ring with several reagents, such as NaCN, NaN3, OH, no products were observed.
Return to citation in text: [1] -
The compound 2a did not decompose in toluene at 100 °C for 6 h.
Return to citation in text: [1] -
The attempt to grow crystals was not successful.
Return to citation in text: [1]
| 1. |
Burkhard, J. A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 9052–9067. doi:10.1002/anie.200907155
Angew. Chem. 2010, 122, 9236–9251. doi:10.1002/ange.200907155 |
| 2. | Wuitschik, G.; Carreira, E. M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. J. Med. Chem. 2010, 53, 3227–3246. doi:10.1021/jm9018788 |
| 4. | Bull, J. A.; Croft, R. A.; Davis, O. A.; Doran, R.; Morgan, K. F. Chem. Rev. 2016, 116, 12150–12233. doi:10.1021/acs.chemrev.6b00274 |
| 38. | We tried to open the ring with several reagents, such as NaCN, NaN3, OH, no products were observed. |
| 39. | The compound 2a did not decompose in toluene at 100 °C for 6 h. |
| 1. |
Burkhard, J. A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 9052–9067. doi:10.1002/anie.200907155
Angew. Chem. 2010, 122, 9236–9251. doi:10.1002/ange.200907155 |
| 2. | Wuitschik, G.; Carreira, E. M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. J. Med. Chem. 2010, 53, 3227–3246. doi:10.1021/jm9018788 |
| 4. | Bull, J. A.; Croft, R. A.; Davis, O. A.; Doran, R.; Morgan, K. F. Chem. Rev. 2016, 116, 12150–12233. doi:10.1021/acs.chemrev.6b00274 |
| 5. | Malapit, C. A.; Howell, A. R. J. Org. Chem. 2015, 80, 8489–8495. doi:10.1021/acs.joc.5b01255 |
| 27. | Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399 |
| 28. | Revathi, L.; Ravindar, L.; Leng, J.; Rakesh, K. P.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 662–682. doi:10.1002/ajoc.201700591 |
| 29. | Veryser, C.; Demaerel, J.; Bieliunas, V.; Gilles, P.; De Borggraeve, W. M. Org. Lett. 2017, 19, 5244–5247. doi:10.1021/acs.orglett.7b02522 |
| 3. | Das, B.; Damodar, K. Epoxides and Oxetanes. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, 2011; pp 63–95. doi:10.1002/9783527634880.ch3 |
| 30. | Fang, W.-Y.; Leng, J.; Qin, H.-L. Chem. – Asian J. 2017, 12, 2323–2331. doi:10.1002/asia.201700891 |
| 31. | Wang, X.-Y.; Leng, J.; Wang, S.-M.; Asiri, A. M.; Marwani, H. M.; Qin, H.-L. Tetrahedron Lett. 2017, 58, 2340–2343. doi:10.1016/j.tetlet.2017.04.070 |
| 32. | Fang, W.-Y.; Huang, Y.-M.; Leng, J.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 751–756. doi:10.1002/ajoc.201800037 |
| 33. | Zhao, C.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2018, 5, 1835–1839. doi:10.1039/c8qo00295a |
| 34. | Zha, G.-F.; Fang, W.-Y.; Li, Y.-G.; Leng, J.; Chen, X.; Qin, H.-L. J. Am. Chem. Soc. 2018, 140, 17666–17673. doi:10.1021/jacs.8b10069 |
| 35. | Zhang, X.; Rakesh, K. P.; Qin, H.-L. Chem. Commun. 2019, 55, 2845–2848. doi:10.1039/c8cc09693g |
| 36. | Lekkala, R.; Lekkala, R.; Moku, B.; Qin, H.-L. Org. Chem. Front. 2019, 6, 796–800. doi:10.1039/c8qo01388h |
| 37. | Zhao, C.; Zha, G.-F.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Eur. J. Org. Chem. 2019, 1801–1807. doi:10.1002/ejoc.201801888 |
| 17. | Picard, P.; Leclercq, D.; Bats, J.-P.; Moulines, J. Synthesis 1981, 550–551. doi:10.1055/s-1981-29523 |
| 18. | Rosowsky, A.; Tarbell, D. S. J. Org. Chem. 1961, 26, 2255–2260. doi:10.1021/jo01351a026 |
| 19. | Balsamo, A.; Ceccarelli, G.; Crotti, P.; Macchia, F. J. Org. Chem. 1975, 40, 473–476. doi:10.1021/jo00892a021 |
| 20. | Berkowitz, P. T.; Baum, K. J. Org. Chem. 1980, 45, 4853–4857. doi:10.1021/jo01312a010 |
| 11. | Lo, M. M.-C.; Fu, G. C. Tetrahedron 2001, 57, 2621–2634. doi:10.1016/s0040-4020(01)00082-5 |
| 12. | Soai, K.; Niwa, S.; Yamanoi, T.; Hikima, H.; Ishizaki, M. J. Chem. Soc., Chem. Commun. 1986, 1018–1019. doi:10.1039/c39860001018 |
| 13. | Bertolini, F.; Crotti, S.; Di Bussolo, V.; Macchia, F.; Pineschi, M. J. Org. Chem. 2008, 73, 8998–9007. doi:10.1021/jo801568a |
| 14. | Hintzer, K.; Koppenhoefer, B.; Schurig, V. J. Org. Chem. 1982, 47, 3850–3854. doi:10.1021/jo00141a009 |
| 22. |
Sone, T.; Lu, G.; Matsunaga, S.; Shibasaki, M. Angew. Chem., Int. Ed. 2009, 48, 1677–1680. doi:10.1002/ange.200805473
Angew. Chem. 2009, 121, 1705–1708. doi:10.1002/anie.200805473 |
| 23. | Brown, H. C.; Ramachandran, P. V. Acc. Chem. Res. 1992, 25, 16–24. doi:10.1021/ar00013a003 |
| 24. | Davies, A. T.; Slawin, A. M. Z.; Smith, A. D. Chem. – Eur. J. 2015, 21, 18944–18948. doi:10.1002/chem.201504256 |
| 16. | Di Martino, A.; Galli, C.; Gargano, P.; Mandolini, L. J. Chem. Soc., Perkin Trans. 2 1985, 1345–1349. doi:10.1039/p29850001345 |
| 25. | Kenaga, E. E. J. Econ. Entomol. 1957, 50, 1–6. doi:10.1093/jee/50.1.1 |
| 26. | Sulbaek Andersen, M. P.; Blake, D. R.; Rowland, F. S.; Hurley, M. D.; Wallington, T. J. Environ. Sci. Technol. 2009, 43, 1067–1070. doi:10.1021/es802439f |
| 7. |
Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E. M. Angew. Chem., Int. Ed. 2006, 45, 7736–7739. doi:10.1002/anie.200602343
Angew. Chem. 2006, 118, 7900–7903. doi:10.1002/ange.200602343 |
| 8. |
Wuitschik, G.; Rogers-Evans, M.; Buckl, A.; Bernasconi, M.; Märki, M.; Godel, T.; Fischer, H.; Wagner, B.; Parrilla, I.; Schuler, F.; Schneider, J.; Alker, A.; Schweizer, W. B.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4512–4515. doi:10.1002/anie.200800450
Angew. Chem. 2008, 120, 4588–4591. doi:10.1002/ange.200800450 |
| 9. | Permana, Y.; Nakano, K.; Yamashita, M.; Watanabe, D.; Nozaki, K. Chem. – Asian J. 2008, 3, 710–718. doi:10.1002/asia.200700339 |
| 10. | Bouchékif, H.; Philbin, M. I.; Colclough, E.; Amass, A. J. Macromolecules 2008, 41, 1989–1995. doi:10.1021/ma062761d |
| 11. | Lo, M. M.-C.; Fu, G. C. Tetrahedron 2001, 57, 2621–2634. doi:10.1016/s0040-4020(01)00082-5 |
| 12. | Soai, K.; Niwa, S.; Yamanoi, T.; Hikima, H.; Ishizaki, M. J. Chem. Soc., Chem. Commun. 1986, 1018–1019. doi:10.1039/c39860001018 |
| 13. | Bertolini, F.; Crotti, S.; Di Bussolo, V.; Macchia, F.; Pineschi, M. J. Org. Chem. 2008, 73, 8998–9007. doi:10.1021/jo801568a |
| 14. | Hintzer, K.; Koppenhoefer, B.; Schurig, V. J. Org. Chem. 1982, 47, 3850–3854. doi:10.1021/jo00141a009 |
| 15. | Dussault, P. H.; Trullinger, T. K.; Noor-e-Ain, F. Org. Lett. 2002, 4, 4591–4593. doi:10.1021/ol0265259 |
| 7. |
Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E. M. Angew. Chem., Int. Ed. 2006, 45, 7736–7739. doi:10.1002/anie.200602343
Angew. Chem. 2006, 118, 7900–7903. doi:10.1002/ange.200602343 |
| 8. |
Wuitschik, G.; Rogers-Evans, M.; Buckl, A.; Bernasconi, M.; Märki, M.; Godel, T.; Fischer, H.; Wagner, B.; Parrilla, I.; Schuler, F.; Schneider, J.; Alker, A.; Schweizer, W. B.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4512–4515. doi:10.1002/anie.200800450
Angew. Chem. 2008, 120, 4588–4591. doi:10.1002/ange.200800450 |
© 2019 Lekkala et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)