Abstract
Photochemical reactions of bicyclo[2.2.2]oct-5-en-2-ones having mixed chromophores like a 5,6 dibenzoyl moiety and bulky electron-deficient substituents like phenyl or isopropenyl at the bridgehead position were analyzed for the first time in different solvents and upon irradiation with different wavelengths. In all cases, a regioselective photoinduced 1,5-phenyl migration leading to vinyl ketenes from the more congested site of the molecule to the less congested one has been observed. The ketenes were exceptionally stable both in air and solution. Its stability studies in acetonitrile through time-dependent UV absorption spectra revealed that it remained almost unchanged at least for a couple of weeks.
Graphical Abstract
Introduction
Enones exhibit a rich and diverse photochemistry. The deep-seated photochemical rearrangements found in these systems have attracted numerous mechanistic studies. In this context, the photochemistry of α,β-enones A and β,γ-enones B (Figure 1) has become an actively researched area for more than two decades [1-6], particularly because of their fascinating photochemical rearrangements. In view of their rich photochemistry, it may be expected that incorporation of both enones into the same molecule C would lead to a variety of more interesting reactions. Indeed, this type of mixed enone photochemistry is also an attractive field [7,8] for chemists because the photochemistry of such substrates can feature the intramolecular competition of different photochemical rearrangements.
![[1860-5397-16-190-1]](/bjoc/content/figures/1860-5397-16-190-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
For several years, our laboratory (S. Lahiri et al.) is primarily engaged in synthesizing various types of such multichromophoric systems to compare their photochemistry in detail [9-15]. Since in acyclic enones like C, E/Z-photoisomerization becomes one of the major energy dissipation pathways, we selected rigid bicyclic multichromophoric enones like D to minimize this route. Interestingly such rigid systems may either be considered as α,β- or β,γ-enone with δ-keto conjugation. However, in one of our previous works [16], we have established that the system should be considered as α,β-enone with δ-keto homoconjugation.
Simple β,γ-enone systems show an absorption band at about 210–250 nm characteristic for (π,π*) transition and at around 280–300 nm characteristic for (n,π*) transition. However, enone D shows enhanced intensity of an (n,π*) absorption band near 300 nm (log ε ≈ 3.5), along with an additional weak (n,π*) absorption band near 350 nm (log ε ≈ 2.0). Probably this is due to interactions of these two chromophores [7]. In many cases probable unselective populations of the excited states owing to the presence of these two individual chromophores are expected to give rise to different photoproduct(s) and in some cases distribution of these photoproducts may be wavelength dependent [7].
In order to investigate the photoreaction of such enones, we observed a complete 1,2-acyl shift (1,2-AS) photoproduct formation when 1a–h have been irradiated (Scheme 1) [14,16]. We also observed that such a rearrangement took place very efficiently via a triplet-mediated pathway without use of any external sensitizer. Similar to bicyclic systems, some of the tricyclic system like 1h yielded 1,2-AS product 2h under similar reaction conditions [16].
![[1860-5397-16-190-i1]](/bjoc/content/inline/1860-5397-16-190-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Quantitative photoisomerization of 1 to 2 in all types of solvents.
Scheme 1: Quantitative photoisomerization of 1 to 2 in all types of solvents.
Initially we envisaged two mechanistic pathways: the oxa-di-π-methane (ODPM) path (characteristic of β,γ-enone moiety) and the type-B path (characteristic of an α,β-enone moiety). However, with the help of the photoinduced electron transfer (PET) reaction of 1a,c,d,g–h and photoreaction of the partially olefinated product of 1c,d,h, we have recognized that in a mixed α,β- and β,γ-enone system, the photorearrangement route is selectively from the α,β-enone part and we confirmed that the irradiation follows a type B rearrangement (Scheme 2) [16].
![[1860-5397-16-190-i2]](/bjoc/content/inline/1860-5397-16-190-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Accepted mechanistic pathway for the photochemical transformations of 1.
Scheme 2: Accepted mechanistic pathway for the photochemical transformations of 1.
Another interesting set of competitive 1,5-(C–O) acyl shift rearrangements (formation of vinyl ketene) with that of 1,2-AS were observed when photoreactions of bridged bicyclic systems like 3a–g were carried out [9-13]. These molecules contain a δ-keto-α,β-enone system juxtaposed with a (cis)-dibenzoylalkene moiety. Interestingly, photoreactions of 3a–f were found to follow regioselective photoindiuced 1,5-phenyl migration from the more congested site of the molecule to the less one leading to the formation of vinyl ketenes 4c–e and 5b,d,f along with the formation of the 1,2-AS photoproduct 6a,d (Scheme 3, Table 1). Some of the vinyl ketenes were stable enough to be kept at room temperature up to 24 hours together as verified by time-dependent absorption spectra [13]. Also no external sensitizer was needed for these transformations. So an efficient intersystem crossing (ISC) may be involved in such systems to reach to the triplet state. A further fascinating aspect is that the two competitive pathways viz. 1,2-AS and photoindiuced 1,5-phenyl migration have been found to be subtly dependent on the substitution pattern. For instance, the rate of 1,2-AS in 3b has been found to be much faster than the one of 3e. It is probably due to the extra hindrance of gem-dimethyl substituents at C-7 position in the latter. Moreover, only 1,2-AS photoproduct 6g was obtained when 3g was irradiated [10].
![[1860-5397-16-190-i3]](/bjoc/content/inline/1860-5397-16-190-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Photochemical reactions of 3a–g. Irradiation using a Hanovia medium pressure 450 W lamp with a pyrex filter; a) conditions and yields are given in Table 1.
Scheme 3: Photochemical reactions of 3a–g. Irradiation using a Hanovia medium pressure 450 W lamp with a pyre...
Table 1: Chemical yields of the formation of photoproducts 4, 5 and 6.
| Starting enones | Time (h) | Medium | Photoproduct(s), yield (%) |
| 3a | 5 | acetone | 6a (25)a |
| 3b | ≈35 | suspended in distilled water | 5b (55)b |
| 3c | ≈35 | suspended in distilled water | 4c (55)b |
| 3d | 6 | acetone | 4d (13)a, 5d (17)a, 6d (23)a |
| 3e | ≈35 | suspended in distilled water | 4e (82)b |
| 3f | ≈35 | suspended in distilled water | 5f (85)b |
| 3g |
5
5.5 5 |
acetone
benzene MeOH |
6g (60)a
6g (45)a 6g (55)a |
aIsolated yields; bbased on recovered starting materials.
In all of the molecules, the bridgehead positions contain only electron-donor groups (such as OMe, Me, etc.), but same type of rigid enones having electron deficient groups at the bridgehead positions have not been checked so far. To bridge this gap, we carried out the photoreactions of 5,6-dibenzoyl-4-phenylbicyclo[2.2.2]oct-5-en-2-one (7a) and 5,6-dibenzoyl-4-isopropenylbicyclo-[2.2.2]oct-5-en-2-one (7b) in detail (Figure 2). Both of the molecules contain electron-acceptor groups like phenyl and isopropenyl at its bridgehead position (C-4) and these groups were bulkier than the earlier reported one.
![[1860-5397-16-190-2]](/bjoc/content/figures/1860-5397-16-190-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
5,6-Dibenzoyl-4-phenylbicyclo[2.2.2]oct-5-en-2-one (7a) and 5,6-dibenzoyl-4-isopropenylbicyclo[2.2.2]oct-5-en-2-one (7b) have been prepared through Diels–Alder reaction between substituted silyloxycyclohexa-1,3-dienes 8a and 8b [17,18] and dibenzoyl acetylene (9) followed by hydrolysis of the silyloxy adduct (Scheme 4).
![[1860-5397-16-190-i4]](/bjoc/content/inline/1860-5397-16-190-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Both 7a and 7b showed an enhanced (n,π*) absorption band near 300 nm (log ε ≈ 3.5) along with an additional weak band near 343 nm (log ε ≈ 2.5) in their UV spectra as expected for δ-keto-α,β-enone [7].
Photoreaction of 7a and 7b
Irradiation of a degassed solution of 7a,b in benzene at 254 nm for 3–4 h yielded a pale yellow solid of ketene 10a,b (Scheme 5, Table 2) through regioselective photoinduced 1,5-phenyl migration and we did not obtain any 1,2-AS or any other photoproduct. The same ketenes were isolated in moderate yields in acetone and acetonitrile solvents. However, we did not find an alternative 1,2-AS photoproduct. Even a change of the wavelength had no effect on the reactivity and similar results were obtained at 300 and 350 nm. The results are summarized in Table 2.
![[1860-5397-16-190-i5]](/bjoc/content/inline/1860-5397-16-190-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Photochemical reaction of 7a,b; a) solvent and conditions are given in Table 2.
Scheme 5: Photochemical reaction of 7a,b; a) solvent and conditions are given in Table 2.
Table 2: Result of photoreaction of 7a,b at various wavelengths in different solvents.
| Wavelength (nm) | Solvent | Starting compounda | Irradiation time (h) | Productb (yield/ %) |
| 254 | benzene | 7a | 3 | 10a (38) |
| 7b | 4 | 10b (32) | ||
| acetonitrile | 7a | 3 | 10a (54) | |
| 7b | 4 | 10b (38) | ||
| acetone | 7a | 3 | 10a (44) | |
| 7b | 4 | 10b (33) | ||
| 300 | benzene | 7a | 5 | 10a (46) |
| 7b | 5.5 | 10b (41) | ||
| acetonitrile | 7a | 5 | 10a (41) | |
| 7b | 5.5 | 10b (40) | ||
| acetone | 7a | 5 | 10a (49) | |
| 350 | benzene | 7a | 5 | 10a (43) |
| acetonitrile | 7a | 5 | 10a (49) | |
aPrepared for the first time, bisolated yield.
The structures of ketenes 10a,b were established from their analytical and spectral data. The ketene stetching (γC=C=O) was detected in the IR spectrum near 2090 cm−1 and the ring ketone stetching (γC=O) appeared at 1730 cm−1. The 1H NMR spectra of these compounds were very similar to those of the parent molecules but their 13C NMR data were found to be much more informative (Table 3). In 1H NMR, a doublet and a triplet in the region of δ 6.6–6.9 ppm confirmed the presence of the OPh group while the bridgehead protons of the ketenes appeared at a slightly up-field position with respect to the parent compounds.
Table 3: Comparison of spectral data of ketens 10a,b with parent compounds 7a,b.
| Compound (mp [°C]) | IR [cm−1] | 1H NMR [ppm] | 13C NMR [ppm] | |||||
| γC=C=O | γC=O | δbridgehead | δOPh | δC+O | δ+C+O | δC+C+O | δ+C-OPh | |
| 7a (224–226) | – |
1726
1666 1646 |
3.82
(t, J = 2.5 Hz) |
– |
208.4,
195.4, 193.3 |
– | – | – |
| 7b (144–146) | – |
1722
1663 1649 |
3.73
(t, J = 2.7 Hz) |
– |
208.9,
194.8, 193.6 |
– | – | – |
| 10a (162–164) | 2087 | 1730 | 3.69 (brs) |
6.68
(d, J = 8 Hz, 2H), 6.79 (t, J = 7.4 Hz, 3H) |
209.9 | 200.2 | 48.9 | 155.9 |
| 10b (128–130) | 2094 | 1732 | 3.67 (brs) |
6.77
(d, J = 8 Hz, 2H), 6.91 (t, J = 7.3 Hz, 3H) |
210.3 | 204.6 | 48.9 | 144.3 |
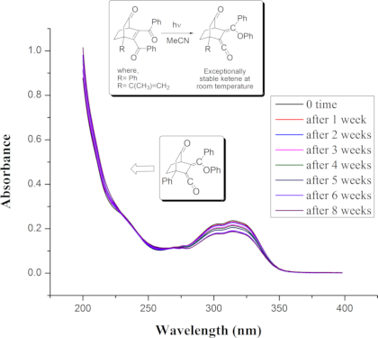
Ketenes 10a,b were exceptionally stable both in air and solution. Their stability studies in acetonitrile through time-dependent absorption spectra (Figure 3) revealed that they remained almost unchanged at least for six to eight weeks.
![[1860-5397-16-190-3]](/bjoc/content/figures/1860-5397-16-190-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Time-dependent absorption spectra of 10a,b in acetonitrile at rt.
Figure 3: Time-dependent absorption spectra of 10a,b in acetonitrile at rt.
Such photoinduced intramolecular 1,5-phenyl migration from carbon to proximate oxygen is a general reaction route for (cis)-dibenzoylalkene chromophores [19-22]. In unsymmetrical derivatives such migrations have been found to take place from the benzoyl group attached to the more crowded olefinic carbon atom to the less crowded one [23]. A similar observation has been reported from our laboratory for bicyclo[2.2.2]octenone systems [13]. Thus, selective formation of 10a,b is expected with the exception that the ketenes 10a,b were unusually stable. This may be due to the imposition of the bulky electron-withdrawing group at the bridgehead position. The non-requirement of any sensitizer in the process indicates that the reaction presumably proceeds via a triplet-mediated pathway. Moreover, the non-appearence of any 1,2-AS product signifies that the β,γ-enone part is inactive. On the other hand (cis)-dibenzoylalkene like α,β-enone part of such multichromophoric bicyclic system actively participates during the photoirradiation.
The structure of 10a was further confirmed by converting it quantitatively to the corresponding methyl esters 11a,b with methanol under reflux conditions (Scheme 6). The isomeric ratio (1:6) of 11a and 11b was confirmed from the NMR spectrum of the crude reaction mixture. Since the polarity of the two methyl esters is very similar, we could not separate them through column chromatography. However, 11a was separated in a trace amount through repeated preparative TLC.
![[1860-5397-16-190-i6]](/bjoc/content/inline/1860-5397-16-190-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Conversion of ketene 10a to its methyl esters 11a,b.
Scheme 6: Conversion of ketene 10a to its methyl esters 11a,b.
Moreover, when a degassed solution of 7a in methanol was irradiated under similar reaction conditions, a mixture of methyl esters 11a and 11b was obtained in the same 1:6 ratio with a moderate yield (60%).
Conclusion
From the above results it was evident that the presence of phenyl or isopropenyl like electron-deficient bulky substituents at the bridgehead position of bicyclo[2.2.2]oct-5-en-2-ones juxtaposed with the (cis)-dibenzoylalkene moiety facilitates the regioselective photo-1,5-phenyl migration from the more congested site of the molecule with the formation of vinyl ketenes presumably via a triplet-mediated pathway. To the best of our knowledge this is the first example of the formation of ketenes which are exceptionally stable for more than 6–8 weeks. Furthermore, we conclude that in a mixed α,β- and β,γ-enone type system, the photorearrangement occures selectively from the α,β-enone part since there was no 1,2-AS or ODPM rearranged photochemical product.
Experimental
The synthetic procedures, photochemical studies and spectra (UV, IR, mass, 1H NMR, 13C-NMR, DEPT-135) of all new compounds can be found in the Supporting Information File 1 and Supporting Information File 2.
References
-
Zimmerman, H. E.; Armesto, D. Chem. Rev. 1996, 96, 3065–3112. doi:10.1021/cr910109c
and references cited therein.
Return to citation in text: [1] -
Hixson, S. S.; Mariano, P. S.; Zimmerman, H. E. Chem. Rev. 1973, 73, 531–551. doi:10.1021/cr60285a005
Return to citation in text: [1] -
Dauben, W. G.; Lodder, G.; Ipaktschi, J. Top. Curr. Chem. 1975, 54, 73–114. doi:10.1007/bfb0046181
Return to citation in text: [1] -
Houk, K. N. Chem. Rev. 1976, 76, 1–74. doi:10.1021/cr60299a001
Return to citation in text: [1] -
Schuster, D. I. Photochemical Rearrangements of Enones. In Rearrangements of Enones in Ground and Excited States; de Mayo, P., Ed.; Organic Chemistry: A Series of Monographs, Vol. 3; Academic Press: New York, NY, USA, 1980; pp 167–279. doi:10.1016/b978-0-12-481303-8.50010-x
Return to citation in text: [1] -
Singh, V. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool, W.; Lenci, F., Eds.; CRC Press: Boca Raton, FL, USA, 2003; 78.1.
Return to citation in text: [1] -
Eichenberger, H.; Tsutsumi, K.; de Weck, G.; Wolf, H. R. Helv. Chim. Acta 1980, 63, 1499–1519. doi:10.1002/hlca.19800630620
Return to citation in text: [1] [2] [3] [4] -
Pfenninger, E.; Poel, D. E.; Berse, C.; Wehrli, H.; Schaffner, K.; Jeger, O. Helv. Chim. Acta 1968, 51, 772–803. doi:10.1002/hlca.660510418
Return to citation in text: [1] -
Maiti, B. C.; Singh, R.; Lahiri, S. J. Chem. Res., Synop. 1993, 500.
Return to citation in text: [1] [2] -
Maiti, B. C.; Singh, R.; Lahiri, S. J. Photochem. Photobiol., A 1995, 91, 27–32. doi:10.1016/1010-6030(95)04105-o
Return to citation in text: [1] [2] [3] -
Maji, D.; Singh, R.; Mostafa, G.; Ray, S.; Lahiri, S. J. Org. Chem. 1996, 61, 5165–5168. doi:10.1021/jo951038a
Return to citation in text: [1] [2] -
Maiti, B. C.; Lahiri, S. Tetrahedron 1998, 54, 9111–9122. doi:10.1016/s0040-4020(98)00548-1
Return to citation in text: [1] [2] -
Chanda, M.; Maji, D.; Lahiri, S. Chem. Commun. 2001, 543–544. doi:10.1039/b009013l
Return to citation in text: [1] [2] [3] [4] -
Yadav, S.; Banerjee, S.; Maji, D.; Lahiri, S. Tetrahedron 2007, 63, 10979–10990. doi:10.1016/j.tet.2007.08.054
Return to citation in text: [1] [2] -
Banerjee, S.; Yadav, S.; Lahiri, S. Org. Lett. 2009, 11, 3494–3497. doi:10.1021/ol901307v
Return to citation in text: [1] -
Sengupta, A.; Chakraborty, I.; Ghosh, A.; Lahiri, S. Tetrahedron 2011, 67, 1689–1695. doi:10.1016/j.tet.2010.12.064
Return to citation in text: [1] [2] [3] [4] -
Ghosh, A.; Chakraborty, I.; Adarsh, N. N.; Lahiri, S. Tetrahedron 2010, 66, 164–171. doi:10.1016/j.tet.2009.11.006
Return to citation in text: [1] -
The O-silyloxydienes were highly moisture sensitive, hence all steps of the reaction were performed following the same procedure as described in [17].
Return to citation in text: [1] -
Griffin, G. W.; O'Connell, E. J. J. Am. Chem. Soc. 1962, 84, 4148–4149. doi:10.1021/ja00880a036
Return to citation in text: [1] -
Zimmerman, H. E.; Dürr, H. G. C.; Lewis, R. G.; Bram, S. J. Am. Chem. Soc. 1962, 84, 4149–4150. doi:10.1021/ja00880a037
Return to citation in text: [1] -
Murty, B. A. R. C.; Kumar, C. V.; Dabral, V.; Das, P. K.; George, M. V. J. Org. Chem. 1984, 49, 4165–4171. doi:10.1021/jo00196a013
Return to citation in text: [1] -
Singh, R.; Sinha, A.; Lahiri, S. J. Chem. Res., Synop. 1992, 372.
Return to citation in text: [1] -
Padwa, A.; Crumrine, D.; Shubber, A. J. Am. Chem. Soc. 1966, 88, 3064–3069. doi:10.1021/ja00965a031
Return to citation in text: [1]
| 13. | Chanda, M.; Maji, D.; Lahiri, S. Chem. Commun. 2001, 543–544. doi:10.1039/b009013l |
| 17. | Ghosh, A.; Chakraborty, I.; Adarsh, N. N.; Lahiri, S. Tetrahedron 2010, 66, 164–171. doi:10.1016/j.tet.2009.11.006 |
| 1. |
Zimmerman, H. E.; Armesto, D. Chem. Rev. 1996, 96, 3065–3112. doi:10.1021/cr910109c
and references cited therein. |
| 2. | Hixson, S. S.; Mariano, P. S.; Zimmerman, H. E. Chem. Rev. 1973, 73, 531–551. doi:10.1021/cr60285a005 |
| 3. | Dauben, W. G.; Lodder, G.; Ipaktschi, J. Top. Curr. Chem. 1975, 54, 73–114. doi:10.1007/bfb0046181 |
| 4. | Houk, K. N. Chem. Rev. 1976, 76, 1–74. doi:10.1021/cr60299a001 |
| 5. | Schuster, D. I. Photochemical Rearrangements of Enones. In Rearrangements of Enones in Ground and Excited States; de Mayo, P., Ed.; Organic Chemistry: A Series of Monographs, Vol. 3; Academic Press: New York, NY, USA, 1980; pp 167–279. doi:10.1016/b978-0-12-481303-8.50010-x |
| 6. | Singh, V. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool, W.; Lenci, F., Eds.; CRC Press: Boca Raton, FL, USA, 2003; 78.1. |
| 7. | Eichenberger, H.; Tsutsumi, K.; de Weck, G.; Wolf, H. R. Helv. Chim. Acta 1980, 63, 1499–1519. doi:10.1002/hlca.19800630620 |
| 19. | Griffin, G. W.; O'Connell, E. J. J. Am. Chem. Soc. 1962, 84, 4148–4149. doi:10.1021/ja00880a036 |
| 20. | Zimmerman, H. E.; Dürr, H. G. C.; Lewis, R. G.; Bram, S. J. Am. Chem. Soc. 1962, 84, 4149–4150. doi:10.1021/ja00880a037 |
| 21. | Murty, B. A. R. C.; Kumar, C. V.; Dabral, V.; Das, P. K.; George, M. V. J. Org. Chem. 1984, 49, 4165–4171. doi:10.1021/jo00196a013 |
| 22. | Singh, R.; Sinha, A.; Lahiri, S. J. Chem. Res., Synop. 1992, 372. |
| 16. | Sengupta, A.; Chakraborty, I.; Ghosh, A.; Lahiri, S. Tetrahedron 2011, 67, 1689–1695. doi:10.1016/j.tet.2010.12.064 |
| 23. | Padwa, A.; Crumrine, D.; Shubber, A. J. Am. Chem. Soc. 1966, 88, 3064–3069. doi:10.1021/ja00965a031 |
| 9. | Maiti, B. C.; Singh, R.; Lahiri, S. J. Chem. Res., Synop. 1993, 500. |
| 10. | Maiti, B. C.; Singh, R.; Lahiri, S. J. Photochem. Photobiol., A 1995, 91, 27–32. doi:10.1016/1010-6030(95)04105-o |
| 11. | Maji, D.; Singh, R.; Mostafa, G.; Ray, S.; Lahiri, S. J. Org. Chem. 1996, 61, 5165–5168. doi:10.1021/jo951038a |
| 12. | Maiti, B. C.; Lahiri, S. Tetrahedron 1998, 54, 9111–9122. doi:10.1016/s0040-4020(98)00548-1 |
| 13. | Chanda, M.; Maji, D.; Lahiri, S. Chem. Commun. 2001, 543–544. doi:10.1039/b009013l |
| 14. | Yadav, S.; Banerjee, S.; Maji, D.; Lahiri, S. Tetrahedron 2007, 63, 10979–10990. doi:10.1016/j.tet.2007.08.054 |
| 15. | Banerjee, S.; Yadav, S.; Lahiri, S. Org. Lett. 2009, 11, 3494–3497. doi:10.1021/ol901307v |
| 17. | Ghosh, A.; Chakraborty, I.; Adarsh, N. N.; Lahiri, S. Tetrahedron 2010, 66, 164–171. doi:10.1016/j.tet.2009.11.006 |
| 18. | The O-silyloxydienes were highly moisture sensitive, hence all steps of the reaction were performed following the same procedure as described in [17]. |
| 7. | Eichenberger, H.; Tsutsumi, K.; de Weck, G.; Wolf, H. R. Helv. Chim. Acta 1980, 63, 1499–1519. doi:10.1002/hlca.19800630620 |
| 8. | Pfenninger, E.; Poel, D. E.; Berse, C.; Wehrli, H.; Schaffner, K.; Jeger, O. Helv. Chim. Acta 1968, 51, 772–803. doi:10.1002/hlca.660510418 |
| 7. | Eichenberger, H.; Tsutsumi, K.; de Weck, G.; Wolf, H. R. Helv. Chim. Acta 1980, 63, 1499–1519. doi:10.1002/hlca.19800630620 |
| 16. | Sengupta, A.; Chakraborty, I.; Ghosh, A.; Lahiri, S. Tetrahedron 2011, 67, 1689–1695. doi:10.1016/j.tet.2010.12.064 |
| 13. | Chanda, M.; Maji, D.; Lahiri, S. Chem. Commun. 2001, 543–544. doi:10.1039/b009013l |
| 16. | Sengupta, A.; Chakraborty, I.; Ghosh, A.; Lahiri, S. Tetrahedron 2011, 67, 1689–1695. doi:10.1016/j.tet.2010.12.064 |
| 10. | Maiti, B. C.; Singh, R.; Lahiri, S. J. Photochem. Photobiol., A 1995, 91, 27–32. doi:10.1016/1010-6030(95)04105-o |
| 14. | Yadav, S.; Banerjee, S.; Maji, D.; Lahiri, S. Tetrahedron 2007, 63, 10979–10990. doi:10.1016/j.tet.2007.08.054 |
| 16. | Sengupta, A.; Chakraborty, I.; Ghosh, A.; Lahiri, S. Tetrahedron 2011, 67, 1689–1695. doi:10.1016/j.tet.2010.12.064 |
| 7. | Eichenberger, H.; Tsutsumi, K.; de Weck, G.; Wolf, H. R. Helv. Chim. Acta 1980, 63, 1499–1519. doi:10.1002/hlca.19800630620 |
| 9. | Maiti, B. C.; Singh, R.; Lahiri, S. J. Chem. Res., Synop. 1993, 500. |
| 10. | Maiti, B. C.; Singh, R.; Lahiri, S. J. Photochem. Photobiol., A 1995, 91, 27–32. doi:10.1016/1010-6030(95)04105-o |
| 11. | Maji, D.; Singh, R.; Mostafa, G.; Ray, S.; Lahiri, S. J. Org. Chem. 1996, 61, 5165–5168. doi:10.1021/jo951038a |
| 12. | Maiti, B. C.; Lahiri, S. Tetrahedron 1998, 54, 9111–9122. doi:10.1016/s0040-4020(98)00548-1 |
| 13. | Chanda, M.; Maji, D.; Lahiri, S. Chem. Commun. 2001, 543–544. doi:10.1039/b009013l |
© 2020 Ghosh; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)