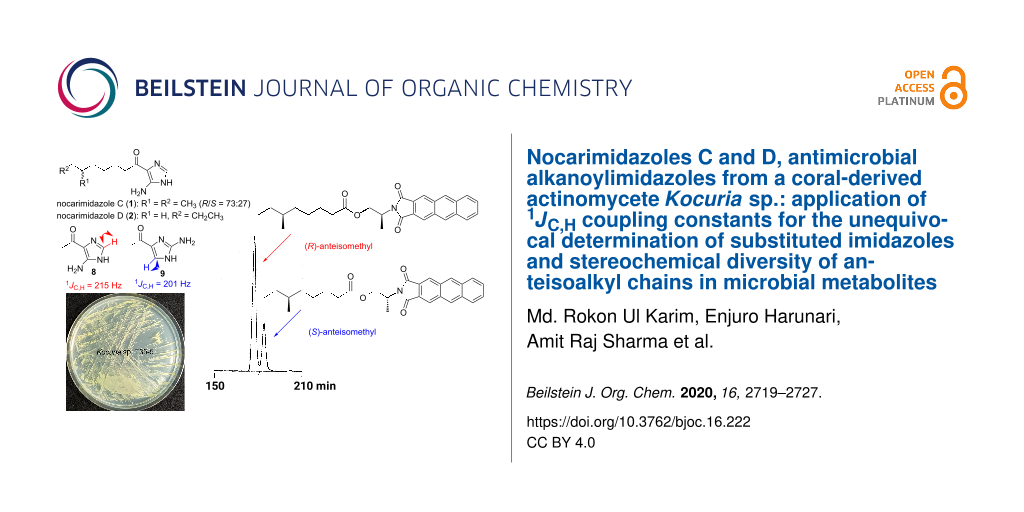
Nocarimidazoles C and D, antimicrobial alkanoylimidazoles from a coral-derived actinomycete Kocuria sp.: application of 1JC,H coupling constants for the unequivocal determination of substituted imidazoles and stereochemical diversity of anteisoalkyl chains in microbial metabolites
1,
1,
1,
1,
2,
1,
3 and
1
Md. Rokon Ul Karim
Biotechnology Research Center and Department of Biotechnology, Toyama Prefectural University, 5180 Kurokawa, Imizu, Toyama 939-0398, Japan
1Biotechnology Research Center and Department of Biotechnology, Toyama Prefectural University, 5180 Kurokawa, Imizu, Toyama 939-0398, Japan
2Shokei Gakuin University, 4-10-1 Yurigaoka, Natori, Miyagi 981-1295, Japan
3Department of Marine Science, Faculty of Fisheries and Marine Science, Diponegoro University, Semarang, Central Java 50275, Indonesia
Corresponding author email
Associate Editor: J. S. Dickschat Beilstein J. Org. Chem.2020,16, 2719–2727.https://doi.org/10.3762/bjoc.16.222 Received 12 Aug 2020,
Accepted 20 Oct 2020,
Published 05 Nov 2020
Chemical investigation of secondary metabolites from a marine-derived actinomycete strain of the genus Kocuria, isolated from a stony coral Mycedium sp., led to the identification of two new alkanoylimidazoles, nocarimidazoles C (1) and D (2) as well as three known congeners, nocarimidazoles A (3) and B (4) and bulbimidazole A (5). Structure analysis of 1 and 2 by NMR and MS revealed that both are 4-alkanoyl-5-aminoimidazoles with a 6-methyloctanoyl or decanoyl chain, respectively. Two possible positions of the amino group on the imidazole rings (C-2 and C-5) posed a challenge in the structure study, which was settled by the measurement of 1JC,H coupling constants for comparison with those of synthetically prepared model imidazoles. The absolute configurations of the anteisoalkanoyl group present in 1, 4, and 5 were determined by low-temperature HPLC analysis of the degradation products labeled with a chiral anthracene reagent, which revealed that 1 is a mixture of the R- and S-enantiomers with a ratio of 73:27, 4 is the pure (S)-enantiomer, and 5 is the (S)-enantiomer with 98% ee. The present study illustrates the diversity in the stereochemistry of anteiso branching in bacterial metabolites. Compounds 1−4 were moderately antimicrobial against Gram-positive bacteria and fungi, with MIC ranges of 6.25–25 μg/mL.
The phylum Actinobacteria contains bacterial genera most prolific as producers of novel natural products with high structural diversity, unique modes of action, and potent bioactivities [1]. More than 10,000 secondary metabolites have been isolated from actinomycetes, accounting for almost 45% of all known microbial secondary metabolites. Particularly, 70% of them were isolated from the genus Streptomyces, the dominant genus commonly found in terrestrial environments [2]. The number of new bioactive compounds from actinomycetes, especially those from terrestrial sources, is likely reaching a plateau after intensive screening activities over several decades [3]. Actinomycetes also inhabit marine environments, including seashores, coastal waters, and bottom sediments or can be found in association with marine organisms such as invertebrates and plants [4,5]. Marine actinomycetes show unique physiological adaptations distinct from their terrestrial counterparts in terms of pressure, salinity, or low-temperature tolerance, which might affect their metabolic ability in their habitat [6,7]. Their secondary metabolite machinery is activated in the sea, as indicated by the isolation of enediyne antitumor antibiotics from marine invertebrates. Namenamicin [8] and the shishijimicins [9], the chalicheamicin-type enediyne polyketides, were isolated from a colonial tunicate. These compounds are considered to be the metabolites of bacterial endosymbionts that internalized biosynthetic genes, likely to have evolved in an actinomycete or an ancestral bacterium in the same lineage. Further exploration of marine habitats has been disclosing a number of unprecedented molecules of actinomycete origin. For example, salinosporamide A, an antitumor drug candidate in clinical trials, is a proteasome inhibitor with an unusual γ-lactam-β-lactone bicyclic core produced by marine Salinospora tropica[10]. Abyssomicin, another example of uncommon polycyclic frameworks, is an antibacterial metabolite of marine Verrucosispora, effective against methicillin-resistant Staphylococcus aureus (MRSA) and Mycobacterium tuberculosis[11,12].
The genus Kocuria, formerly categorized in the genus Micrococcus, is a Gram-positive unicellular coccus belonging to the family Micrococcaceae[13]. Members of the genus Kocuria have been isolated from diverse marine environments such as seawater [14,15], sediments [16], and deep-sea hydrothermal plumes [17]. The genome size of the genus Kocuria is around 2.7 to 3.0 Mbp in average; which is one of the smallest among actinomycetes. However, the latest genomic information suggests the presence of biosynthetic genes for nonribosomal peptide synthetase and type III polyketide synthase in some Kocuria strains [18], which leaves a hope for new secondary metabolites. At present, only a few limited structural types of metabolites, including polyamine-derived siderophores and modified peptides, are known from Kocuria and Micrococcus[19,20].
In our continuing investigation on secondary metabolites from marine bacteria, five alkanoylimidazoles were obtained from the culture extract of a Kocuria strain isolated from a stony coral. Alkanoylimidazoles are a new and rare class of natural products, first described in 2015 by Fenical et al. from marine Nocardiopsis[21] and were recently found from a marine obligate bacterium Microbulbifer by our group [22]. We herein report the isolation, structure determination, and biological activities of two new alkanoylimidazoles, nocarimidazoles C (1) and D (2), along with the identification of three known related compounds, nocarimidazoles A (3) and B (4) as well as bulbimidazole A (5). We also discuss the stereochemical diversity of the anteisoalkanoyl group in these compounds.
Results and Discussion
Strain T35-5 was isolated from the scleractinian coral of the genus Mycedium, collected near the coast of Karimunjawa, Central Java, Indonesia. Analysis of the 16S rRNA gene sequence identified the producing strain as a member of the genus Kocuria. The whole culture broth of Kocuria sp. T35-5, cultured in A11M seawater medium at 30 °C for five days, was extracted with 1-butanol, and the extract was subjected to chromatographic purification, yielding two new alkanoylimidazoles, nocarimidazole C (1) and D (2), along with three known congeners, nocarimidazole A (3) and B (4) as well as bulbimidazole A (5, Figure 1).
Figure 1:
Structure of the nocarimidazoles 1–4 and the bulbimidazoles 5–7.
Figure 1:
Structure of the nocarimidazoles 1–4 and the bulbimidazoles 5–7.
Nocarimidazole C (1) was obtained as a pale yellow amorphous solid. The molecular formula was determined as C12H21N3O based on a protonated molecular ion [M + H]+ at m/z 224.1763 (Δ = +0.6 mmu). The four degrees of unsaturation, calculated from the molecular formula, and the UV absorption band around 296 nm indicated the presence of a conjugated system in this molecule. The IR absorption bands at 3127 and 1664 cm−1 implied the presence of OH/NH and carbonyl groups, respectively. The 1H NMR spectrum was rather simple, displaying only 6 signals: a deshielded proton singlet resonance, three isolated aliphatic methylene unit resonances, a methylene envelope signal, and a doublet and a triplet methyl group resonance overlapping. The 13C NMR spectrum only exhibited several sp3 carbon signals at 10–40 ppm, lacking those of sp2 carbon atoms in CDCl3, CD3OD, or DMSO-d6 (data not shown). The same phenomenon was observed during the study of bulbimidazole A (5), which did not show sp2 carbon signals in neutral solutions due to the presence of multiple resonance structures for the imidazole moiety [22]. We envisaged that due to the presence of an imidazole ring, the UV spectra of 1 and 5 would obviously be different, and as expected, supplementation of a trace amount of trifluoroacetic acid (TFA) to DMSO-d6, combined with a longer relaxation delay (d1 = 30 s) for the 13C NMR experiment, greatly improved the detectability of sp2 carbon resonances. The 13C and HSQC spectra collected in this solvent mixture allowed the assignment of 12 carbon signals to one deshielded carbonyl carbon atom (δC 189.4), two nonprotonated sp2 carbon atoms (δC 109.7, 144.6), one sp2 methine unit (δC 130.9), one sp3 methine carbon atom, five sp3 methylene units, and two methyl moieties (Table 1).
Table 1:1H and 13C NMR spectroscopic data for nocarimidazoles C (1) and D (2) in DMSO-d6 with TFA.
1
2
no.
δCa
δH mult (J in Hz)b
HMBCb,c
δCa
δH mult (J in Hz)b
HMBCb,c
2
130.9, CH
8.62, s
4, 5, 6
130.9, CH
8.59, s
4, 5, 6
4
109.7, C
109.7, C
5
144.6, C
144.7, C
6
189.4, C
189.3, C
7
38.3, CH2
2.66, t (7.4)
6, 8, 9
38.3, CH2
2.66, t (7.4)
4, 6, 8, 9
8
23.9, CH2
1.55, quint (7.2)
6, 10
23.6, CH2
1.54, quint (7.3)
6, 7, 9, 10
9
26.2, CH2
1.27d
11
28.76, CH2
1.22–1.26d
7
10
35.9, CH2
1.07, m
9, 11
29.1, CH2
1.22–1.25d
8
1.27d
9, 11
11
33.8, CH
1.27d
9, 13, 14
29.0,e CH2
1.22–1.25d
9, 10, 12
12
29.0, CH2
1.08, m
11, 13, 14
28.83,e CH2
1.22–1.26d
1.26d
11, 13, 14
13
11.3, CH3
0.80, t (7.4)
11, 12
31.4, CH2
1.21,d m
14
14
19.2, CH3
0.79, d (6.7)
10, 11, 12
22.3, CH2
1.21–1.25d
13, 15
15
14.0, CH3
0.83, t (6.8)
13, 14
aRecorded at 125 MHz (reference δC 39.5). bRecorded at 500 MHz (reference δH 2.49). cFrom the proton stated to the indicated carbon atom(s). dOverlapping signals. eAssignment may be interchangeable.
Three small fragments (H-7/H-8/H-9, H-11/H-14, H-12/H-13) were defined by the analysis of the COSY spectrum (Figure 2). Meanwhile, HMBC correlations from the two methyl protons (H-13 and H-14) to well-resolved C-11, and C-12, along with a correlation from H-14 to C-10, allowed to assemble an anteisomethyl terminus from C-10 to C-14. The connectivity between C-9 and C-10 was established by HMBC correlations from H-8 to C-10 and H-10 to C-9. Furthermore, HMBC correlations from H-7 and H-8 to a deshielded carbonyl carbon atom at δC 189.4 (C-6) supported a 6-methyloctanoyl substructure. The remaining structural unit has the composition formula C3H4N3 with three double bond equivalents, composed in part by the two nonprotonated sp2 carbon atoms (C-4 and C-5) and an sp2 methine unit (CH-2) and exhibited HMBC correlations from H-2 to C-4 and C-5. These requirements were only satisfied by an amino-substituted imidazole ring. Indeed, a four-bond correlation from H-2 to C-6 established the linkage between the chain part and the imidazole ring (Figure 2).
Figure 2:
COSY and key HMBC correlations for 1 and 2.
Figure 2:
COSY and key HMBC correlations for 1 and 2.
The remaining question was whether the amino group is bound to C-2 or C-5 in the imidazole ring. A literature survey suggested a diagnostic use of 1JC,H coupling constants [23]. In imidazole and ʟ-histidine, the 1JC,H values for H-2/C-2 (208 to ≈222 Hz) were found to be always larger by at least 15 Hz than those for H-4/C-4 (188 to ≈208 Hz) at any pH condition below 11 [24]. Because the sp2 methine group in 1 exhibited 1JC,H = 221 Hz in a coupled HSQC experiment, this was assignable to the imidazole 2-position based on this criterion, and hence a C-5 amino substitution. Additionally, 1JC,H measurements of bulbimidazole A (5) gave 221 Hz for H-2/C-2 and 204 Hz for H-4/C-4 (H-5/C-5 in the numbering system for 5), which corroborated the assignment. To finally settle this issue, two aminoimidazoles 8 and 9, possessing 4-acetyl and 5- or 2-amino substitutions, respectively, were synthesized for comparison, according to the reported procedures (Scheme 1) [25-27]. Compound 8 is known as a photolysis product of 6-methylpurine 1-oxide [28], but we prepared it by the Grignard reaction of a commercially available imidazole derivative, 4-isocyano-1H-imidazol-5-amine, with MeMgBr. On the other hand, 9 was synthesized in three steps from pyrimidin-2-amine. Imidation of the starting material with 1,1-dimethoxy-N,N-dimethylmethylamine gave an N,N- dimethylformimidamide derivative, which was cyclized with 1-chloropropan-2-one to give 3-acetylimidazo[1,2-a]pyrimidine, which, following degradation of the pyridimine ring with hydrazine, yielded 9. The coupled HSQC experiments measured 1JC,H = 215 Hz for the H-2/C-2 pair in 8 (Figure S26, Supporting Information File 1) and 201 Hz for H-5/C-5 in 9 (Figure S31, Supporting Information File 1). Thus, the amino substitution at C-5 in 1 was unequivocally established (Figure 3).
Scheme 1:
Synthesis of the model compounds 8 and 9.
Scheme 1:
Synthesis of the model compounds 8 and 9.
The absolute configuration at C-11 in the anteisoalkanoyl chain was determined by the Ohrui–Akasaka method [29]. The imidazole ring was degraded by oxidation using ruthenium tetraoxide and sodium periodate in a biphasic solvent mixture (CCl4/MeCN/H2O), which gave 6-methyloctanoic acid (nat-10). The authentic (S)-6-methyloctanoic acid and (S)-8-methyldecanoic acid were synthesized in our previous studies [22,30]. These anteiso fatty acids were derivatized with a chiral anthracene reagent to yield the esters of (R)- or (S)-2-(anthracene-2,3-dicarboximido)propanol (nat-10-(R)-2A1P, (S)-10-(R)-2A1P, and (S)-10-(S)-2A1P), which were subjected to HPLC analysis for comparison (Figure 4). The retention times of the standard samples were 177 min for (S)-10-(S)-2A1P (chromatographically equivalent to (R)-10-(R)-2A1P) and 184 min for (S)-10-(R)-2A1P, and nat-10-(R)-2A1P gave both peaks with the area ratio of 72.9:27.1 (Figure 4a). Therefore, 1 was confirmed as an enantiomeric mixture comprising 73% of the R- and 27% of the S-enantiomer. We also analyzed the absolute configuration of nocarimidazole B (4) produced by strain TK35-5. The same compound was originally isolated from marine Nocadiopsis, but the absolute configuration was not elucidated. The conversion of 4 into the derivative nat-11-(R)-2A1P, followed by HPLC analysis, revealed that 4 is an enantiomerically pure S-enantiomer (Figure 4b). Additionally, the same chiral analysis with bulbimidazole A (5) obtained in this study proved the enantiomeric ratio as R/S = 1.4:98.6 (Figure 4c).
Figure 4:
Determination of the absolute configuration of 1 (a), 4 (b), and 5 (c) by the Ohrui–Akasaka method.
Figure 4:
Determination of the absolute configuration of 1 (a), 4 (b), and 5 (c) by the Ohrui–Akasaka method.
Nocarimidazole D (2) was isolated as a pale yellow amorphous solid. The molecular formula of 2 was deduced as C13H23N3O, 14 amu (corresponding to CH2) larger than 1, based on the HRESITOFMS data ([M + H]+ at m/z 238.1915, Δ = +0.2 mmu). The UV and IR spectra showed almost the same features as those for 1. The interpretation of the 1H, 13C, and HSQC spectra of 2 in comparison to 1 revealed three additional methylene groups and the absence of one doublet methyl and one methine signal. Two aliphatic fragments, H-7/H-8/H-9 and H-14/H-15, were identified from COSY correlation data. These fragments were then joined into a nonbranching linear alkyl chain by HMBC correlations from H-15 to C-13 and H-8 to C-10, though H-11/C-11 and H-12/C-12 were not completely assignable due to a severe signal overlapping. This alkyl chain was further linked to the imidazole ring through a carbonyl carbon atom C-6 on the basis of HMBC correlations from H-7 and H-2 to C-6 (Figure 2). 1H and 13C NMR chemical shifts for the imidazole part of 2 were almost the same as those for 1 (Table 1).
The antimicrobial activity of 1–4 was tested against Gram-positive bacteria Kocuria rhizophila and Staphylococcus aureus, Gram-negative bacteria Escherichia coli and Rhizobium radiobacter, a yeast Candida albicans, and two fungi Glomerella cingulata and Trichophyton rubrum (Table 2). All compounds exhibited moderate activity against Gram-positive bacteria with MICs of 6.25–12.5 μg/mL but were inactive against Gram-negative bacteria. The compounds 1–4 were also active against the yeast and fungi, with MIC values ranging from 6.25–25 μg/mL. In addition, the compounds 1 and 4 exhibited weak cytotoxicity against P388 murine leukemia cells, with an IC50 of 38 and 33 μM, respectively.
Table 2:
Antimicrobial activity of the nocarimidazoles 1–4.
MIC (μg/mL)
microoroganisms
1
2
3
4
Kocuria rhizophila ATCC9341
6.25
12.5
6.25
6.25
Staphylococcus aureus FDA209P JC-1
12.5
25
12.5
25
Escherichia coli NIHJ JC-2
>100
>100
>100
>100
Rhizobium radiobacter NBRC14554
>100
>100
>100
>100
Candida albicans NBRC0197
25
25
12.5
12.5
Glomerella cingulata NBRC5907
12.5
12.5
25
25
Trichophyton rubrum NBRC5467
6.25
6.25
25
6.25
Conclusion
Alkanoylimidazoles, 4-acylated imidazoles of varying chain length and terminal branching, with occasional substitution at C-5 by an amino group, are an emerging class of marine-derived natural products: nocarimidazoles A (3) and B (4), the first two members in this class, were discovered from a marine actinomycete Nocardiopsis[21]; bulbimidazoles A–C (5–7), on the other hand, were isolated from a marine gammaproteobacterium Microbulbifer[22]. In this study, additional members, nocarimidazoles C (1) and D (2), were obtained from a marine-derived actinomycete of the genus Kocuria. The exclusive origin of these metabolites from marine bacteria, as well as the distribution among phylogenetically distinct taxa imply their potential function in the adaptation of the producing organisms to the marine ecosystem.
Similarly to nocarimidazole B (4) and bulbimidazole A (5), 1 possesses the anteiso-branched alkanoyl chain. The absolute configurations of the anteiso-branched metabolites are in general expected to be S in an association to bacterial anteiso fatty acids, which are biosynthesized from ʟ-isoleucine [31,32]. However, we have previously shown that both the anteiso-branched secondary metabolites of marine bacteria, nocapyrone L [30] and bulbimidazole A (5) [22], are 2:3 and 9:91 mixtures of the R- and S-enantiomers, respectively. Again, we encountered enantiomerically mixed anteiso chains in 1 and 5 but at the same time found purely S-configured 4 (Figure 5), demonstrating the varied enantiomerical purity and chirality of anteiso-branched bacterial metabolites. Intriguingly, the same chemistry is seen among related compounds from a single organism (e.g., 1, 4, and 5) and even with a compound (in terms of having the common planar structure) from different organisms (e.g., 5). This lesson not only warrants the insufficiency of assigning the stereochemistry of the synthesized anteiso-chained natural products only by comparison of the optical rotation values but provides a new insight into the structural/biosynthetic diversity in microbial secondary metabolites. Finally, it should be noted that the R-enantiomer-rich anteisoalkyl group is extremely rare: only two ceramides, each from the dinoflagellate Coolia monotis[33] and the sponge Ephydatia syriaca[34], preceed 1.
Figure 5:
Stereochemical diversity of the anteiso-chain chirality in microbial metabolites.
Figure 5:
Stereochemical diversity of the anteiso-chain chirality in microbial metabolites.
The specific rotations were measured on a JASCO P-1030 polarimeter. UV and IR spectra were recorded on a Shimadzu UV-1800 spectrophotometer and a PerkinElmer Spectrum 100 spectrophotometer, respectively. NMR spectra were obtained on a Bruker AVANCE II 500 MHz NMR spectrometer in DMSO-d6 supplemented with a trace amount of trifluoroacetic acid using the signals of the residual solvent protons (δH 2.49) and carbon atoms (δC 39.5) as internal standards for compounds 1–5, or in CDCl3 using the signals of the residual solvent protons (δH 7.27) and carbon atoms (δC 77.0) as internal standards for other compounds. HRESITOFMS spectra were recorded on a Bruker micrOTOF focus mass spectrometer. An Agilent HP1200 system equipped with a diode array detector was used for analysis and purification.
Microorganism
The coral sample Mycedium sp. was collected at −15 to −20 m from Tanjung Gelam, Karimunjawa National Park, Jepara, Central Java, Indonesia with a permission number of 1096/T.34/TU/SIMAKSI/7/2017. The strain T35-5 was isolated according to the method described previously [35] and identified as a member of the genus Kocuria on the basis of 100.0% similarity in the 16S rRNA gene sequence (1381 nucleotides; DDBJ accession number LC556325) to Kocuria palustris DSM 11925T (accession number Y16263).
Fermentation
In a similar manner as described in [22], the strain T35-5 was maintained on Marine Agar 2216 (Difco). A loopful of the strain T35-5 was inoculated into a 500 mL K-1 flask containing 100 mL of Marine Broth 2216 (Difco) as a seed culture. The seed culture was incubated at 30 °C on a rotary shaker at 200 rpm for 2 days. Three mL each of the seed culture were inoculated into 500 mL K-1 flasks containing 100 mL of A11M production medium, which consists of 0.2% glucose, 2.5% soluble starch, 0.5% yeast extract, 0.5% polypeptone (Wako Pure Chemical Industries, Ltd.), 0.5% NZ-amine (Wako Pure Chemical Industries, Ltd.), 0.3% CaCO3, and 1% Diaion HP-20 (Mitsubishi Chemical Co.) in natural seawater (collected from Toyama Bay, Japan). The pH value of the medium was adjusted to 7.0 before sterilization. The inoculated flasks were incubated at 30 °C for 5 days, with rotational shaking at 200 rpm.
Extraction and isolation
In a similar manner as described in [22], after fermentation, 100 mL of 1-butanol was added to each flask, and the flasks were shaken for 1 h. The emulsified mixture was centrifuged at 6000 rpm for 10 min, and the organic layer was separated from the aqueous layer. Evaporation of the organic solvent gave approximately 3.8 g of extract from 3 L of culture. The extract (3.8 g) was chromatographed over a silica gel column using a mixture solvent of CHCl3/MeOH (1:0, 20:1, 10:1, 4:1, 2:1, 1:1, and 0:1, v/v). Fraction 3 (10:1) was concentrated to yield 0.38 g of a brown oil, which was further fractionated by ODS column chromatography with a stepwise gradient of a MeCN/0.1% HCO2H aqueous solution (2:8, 3:7, 4:6, 5:5, 6:4, 7:3, and 8:2, v/v). Fractions 4 (5:5) and 5 (6:4) were separately concentrated in vacuo, and the remaining aqueous layer was extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to give 40 mg and 45 mg of a semipure material. Final purification was achieved by preparative HPLC (Cosmosil AR-II, Nacalai Tesque Inc., 10 × 250 mm, 4 mL/min, UV detection at 254 nm) with an isocratic elution of MeCN/0.1% HCO2H (42:58) to afford nocarimidazole C (1, 2.4 mg, tR 8.45 min), nocarimidazole D (2, 3.2 mg, tR 14.9 min), nocarimidazole A (3, 3.8 mg, tR 13.5 min), nocarimidazole B (4, 8.0 mg, tR 17.3 min) from fraction 4, and bulbimidazole A (5, 3.2 mg, tR 11.2 min) from fraction 5.
Bioassays
The antimicrobial activity was evaluated in a similar manner as previously reported [22]. The cytotoxicity against P388 murine leukemia cells was examined according to a protocol described in [22].
Supporting Information
Supporting Information File 1:
Copies of the NMR spectra for compounds 1 and 2.
P388 cells were obtained from the JCRB Cell Bank under the accession code JCRB0017 (Lot. 06252002).
Funding
This work was supported by a TPU Young Investigator Overseas Training Expense for E. H. and JSPS KAKENHI Grants JP19K05848 to Y. I., JP18K05827 to N. O., and JP16H06213 to D. U.
References
Bull, A. T.; Stach, J. E. M. Trends Microbiol.2007,15, 491–499. doi:10.1016/j.tim.2007.10.004
Return to citation in text:
[1]
Girão, M.; Ribeiro, I.; Ribeiro, T.; Azevedo, I. C.; Pereira, F.; Urbatzka, R.; Leão, P. N.; Carvalho, M. F. Front. Microbiol.2019,10, 683. doi:10.3389/fmicb.2019.00683
Return to citation in text:
[1]
Almeida, E. L.; Carrillo Rincón, A. F.; Jackson, S. A.; Dobson, A. D. W. Front. Microbiol.2019,10, 1713. doi:10.3389/fmicb.2019.01713
Return to citation in text:
[1]
Seipke, R. F.; Kaltenpoth, M.; Hutchings, M. I. FEMS Microbiol. Rev.2012,36, 862–876. doi:10.1111/j.1574-6976.2011.00313.x
Return to citation in text:
[1]
Undabarrena, A.; Beltrametti, F.; Claverías, F. P.; González, M.; Moore, E. R. B.; Seeger, M.; Cámara, B. Front. Microbiol.2016,7, 1135. doi:10.3389/fmicb.2016.01135
Return to citation in text:
[1]
Abdelmohsen, U. R.; Bayer, K.; Hentschel, U. Nat. Prod. Rep.2014,31, 381–399. doi:10.1039/c3np70111e
Return to citation in text:
[1]
McDonald, L. A.; Capson, T. L.; Krishnamurthy, G.; Ding, W.-D.; Ellestad, G. A.; Bernan, V. S.; Maiese, W. M.; Lassota, P.; Discafani, C.; Kramer, R. A.; Ireland, C. M. J. Am. Chem. Soc.1996,118, 10898–10899. doi:10.1021/ja961122n
Return to citation in text:
[1]
Oku, N.; Matsunaga, S.; Fusetani, N. J. Am. Chem. Soc.2003,125, 2044–2045. doi:10.1021/ja0296780
Return to citation in text:
[1]
Feling, R. H.; Buchanan, G. O.; Mincer, T. J.; Kauffman, C. A.; Jensen, P. R.; Fenical, W. Angew. Chem., Int. Ed.2003,42, 355–357. doi:10.1002/anie.200390115
Return to citation in text:
[1]
Bister, B.; Bischoff, D.; Ströbele, M.; Riedlinger, J.; Reicke, A.; Wolter, F.; Bull, A. T.; Zähner, H.; Fiedler, H.-P.; Süssmuth, R. D. Angew. Chem., Int. Ed.2004,43, 2574–2576. doi:10.1002/anie.200353160
Return to citation in text:
[1]
Freundlich, J. S.; Lalgondar, M.; Wei, J.-R.; Swanson, S.; Sorensen, E. J.; Rubin, E. J.; Sacchettini, J. C. Tuberculosis2010,90, 298–300. doi:10.1016/j.tube.2010.08.002
Return to citation in text:
[1]
Tang, J. S.; Gillevet, P. M. Int. J. Syst. Evol. Microbiol.2003,53, 995–997. doi:10.1099/ijs.0.02372-0
Return to citation in text:
[1]
Seo, Y. B.; Kim, D.-E.; Kim, G.-D.; Kim, H.-W.; Nam, S.-W.; Kim, Y. T.; Lee, J. H. Int. J. Syst. Evol. Microbiol.2009,59, 2769–2772. doi:10.1099/ijs.0.008482-0
Return to citation in text:
[1]
Li, J.; Zhang, S. Int. J. Syst. Evol. Microbiol.2020,70, 785–789. doi:10.1099/ijsem.0.003825
Return to citation in text:
[1]
Kim, S. B.; Nedashkovskaya, O. I.; Mikhailov, V. V.; Han, S. K.; Kim, K.-O.; Rhee, M.-S.; Bae, K. S. Int. J. Syst. Evol. Microbiol.2004,54, 1617–1620. doi:10.1099/ijs.0.02742-0
Return to citation in text:
[1]
Zhang, L.; Xi, L.; Ruan, J.; Huang, Y. Int. J. Syst. Evol. Microbiol.2017,67, 164–169. doi:10.1099/ijsem.0.001599
Return to citation in text:
[1]
Palomo, S.; González, I.; de la Cruz, M.; Martín, J.; Tormo, J. R.; Anderson, M.; Hill, R. T.; Vicente, F.; Reyes, F.; Genilloud, O. Mar. Drugs2013,11, 1071–1086. doi:10.3390/md11041071
Return to citation in text:
[1]
Leutou, A. S.; Yang, I.; Kang, H.; Seo, E. K.; Nam, S.-J.; Fenical, W. J. Nat. Prod.2015,78, 2846–2849. doi:10.1021/acs.jnatprod.5b00746
Return to citation in text:
[1]
[2]
Karim, M. R. U.; Harunari, E.; Oku, N.; Akasaka, K.; Igarashi, Y. J. Nat. Prod.2020,83, 1295–1299. doi:10.1021/acs.jnatprod.0c00082
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
Wasylishen, R. E.; Tomlinson, G. Biochem. J.1975,147, 605–607. doi:10.1042/bj1470605
Return to citation in text:
[1]
Kick, E. K.; Bodas, M.; Mohan, R.; Valente, M.; Wurtz, N.; Patil, S. LXR modulators. PCT Int. Pat. Appl. WO2014144037 A1, Sept 18, 2014.
Return to citation in text:
[1]
Rasapalli, S.; Dhawane, A.; Rees, C.; Golen, J. A.; Singh, B. R.; Cai, S.; Jasinski, J.; Kwasny, S. M.; Moir, D. T.; Opperman, T. J.; Bowlin, T. L. MedChemComm2013,4, 1467. doi:10.1039/c3md00143a
Return to citation in text:
[1]
Love, C.; Van Wauwe, J. P. F.; De Brabander, M.; Cooymans, L.; Vandermaesen, N. 2,4-Disubstituted thiazolyl derivatives. PCT. Int. Pat. Appl. WO200164674 A1, Sept 7, 2001.
Return to citation in text:
[1]
Lam, F. L.; Parham, J. C. J. Am. Chem. Soc.1975,97, 2839–2844. doi:10.1021/ja00843a038
Return to citation in text:
[1]
Akasaka, K.; Meguro, H.; Ohrui, H. Tetrahedron Lett.1997,38, 6853–6856. doi:10.1016/s0040-4039(97)01616-x
Return to citation in text:
[1]
Kim, Y.; Ogura, H.; Akasaka, K.; Oikawa, T.; Matsuura, N.; Imada, C.; Yasuda, H.; Igarashi, Y. Mar. Drugs2014,12, 4110–4125. doi:10.3390/md12074110
Return to citation in text:
[1]
[2]
Akasaka, K.; Shichijyukari, S.; Matsuoka, S.; Murata, M.; Meguro, H.; Ohrui, H. Biosci., Biotechnol., Biochem.2000,64, 1842–1846. doi:10.1271/bbb.64.1842
Return to citation in text:
[1]
Řezanka, T.; Sigler, K.; Dembitsky, V. M. Tetrahedron2006,62, 5937–5943. doi:10.1016/j.tet.2006.04.019
Return to citation in text:
[1]
Sharma, A. R.; Zhou, T.; Harunari, E.; Oku, N.; Trianto, A.; Igarashi, Y. J. Antibiot.2019,72, 634–639. doi:10.1038/s41429-019-0192-x
Return to citation in text:
[1]
Reference 30
30.
Kim, Y.; Ogura, H.; Akasaka, K.; Oikawa, T.; Matsuura, N.; Imada, C.; Yasuda, H.; Igarashi, Y. Mar. Drugs2014,12, 4110–4125. doi:10.3390/md12074110
Undabarrena, A.; Beltrametti, F.; Claverías, F. P.; González, M.; Moore, E. R. B.; Seeger, M.; Cámara, B. Front. Microbiol.2016,7, 1135. doi:10.3389/fmicb.2016.01135
7.
Abdelmohsen, U. R.; Bayer, K.; Hentschel, U. Nat. Prod. Rep.2014,31, 381–399. doi:10.1039/c3np70111e
Palomo, S.; González, I.; de la Cruz, M.; Martín, J.; Tormo, J. R.; Anderson, M.; Hill, R. T.; Vicente, F.; Reyes, F.; Genilloud, O. Mar. Drugs2013,11, 1071–1086. doi:10.3390/md11041071
Girão, M.; Ribeiro, I.; Ribeiro, T.; Azevedo, I. C.; Pereira, F.; Urbatzka, R.; Leão, P. N.; Carvalho, M. F. Front. Microbiol.2019,10, 683. doi:10.3389/fmicb.2019.00683
Bister, B.; Bischoff, D.; Ströbele, M.; Riedlinger, J.; Reicke, A.; Wolter, F.; Bull, A. T.; Zähner, H.; Fiedler, H.-P.; Süssmuth, R. D. Angew. Chem., Int. Ed.2004,43, 2574–2576. doi:10.1002/anie.200353160
12.
Freundlich, J. S.; Lalgondar, M.; Wei, J.-R.; Swanson, S.; Sorensen, E. J.; Rubin, E. J.; Sacchettini, J. C. Tuberculosis2010,90, 298–300. doi:10.1016/j.tube.2010.08.002
Seo, Y. B.; Kim, D.-E.; Kim, G.-D.; Kim, H.-W.; Nam, S.-W.; Kim, Y. T.; Lee, J. H. Int. J. Syst. Evol. Microbiol.2009,59, 2769–2772. doi:10.1099/ijs.0.008482-0
Feling, R. H.; Buchanan, G. O.; Mincer, T. J.; Kauffman, C. A.; Jensen, P. R.; Fenical, W. Angew. Chem., Int. Ed.2003,42, 355–357. doi:10.1002/anie.200390115
Kim, S. B.; Nedashkovskaya, O. I.; Mikhailov, V. V.; Han, S. K.; Kim, K.-O.; Rhee, M.-S.; Bae, K. S. Int. J. Syst. Evol. Microbiol.2004,54, 1617–1620. doi:10.1099/ijs.0.02742-0
McDonald, L. A.; Capson, T. L.; Krishnamurthy, G.; Ding, W.-D.; Ellestad, G. A.; Bernan, V. S.; Maiese, W. M.; Lassota, P.; Discafani, C.; Kramer, R. A.; Ireland, C. M. J. Am. Chem. Soc.1996,118, 10898–10899. doi:10.1021/ja961122n
Kick, E. K.; Bodas, M.; Mohan, R.; Valente, M.; Wurtz, N.; Patil, S. LXR modulators. PCT Int. Pat. Appl. WO2014144037 A1, Sept 18, 2014.
26.
Rasapalli, S.; Dhawane, A.; Rees, C.; Golen, J. A.; Singh, B. R.; Cai, S.; Jasinski, J.; Kwasny, S. M.; Moir, D. T.; Opperman, T. J.; Bowlin, T. L. MedChemComm2013,4, 1467. doi:10.1039/c3md00143a
27.
Love, C.; Van Wauwe, J. P. F.; De Brabander, M.; Cooymans, L.; Vandermaesen, N. 2,4-Disubstituted thiazolyl derivatives. PCT. Int. Pat. Appl. WO200164674 A1, Sept 7, 2001.



![[1860-5397-16-222-1]](/bjoc/content/figures/1860-5397-16-222-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-222-2]](/bjoc/content/figures/1860-5397-16-222-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-222-i1]](/bjoc/content/inline/1860-5397-16-222-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-222-3]](/bjoc/content/figures/1860-5397-16-222-3.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-222-4]](/bjoc/content/figures/1860-5397-16-222-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-222-5]](/bjoc/content/figures/1860-5397-16-222-5.svg?scale=2.0&max-width=1024&background=FFFFFF)