Abstract
Several new derivatives of adenine, purine, and theophylline containing the (CF3)2CH group connected to a nitrogen atom of the imidazole ring were prepared by the reaction of 2,2,4,4-tetrakis(trifluoromethyl)-1,3-dithietane (1) with the corresponding substrates, resulting in the selective alkylation of one of the nitrogen atoms of the imidazole ring. The reaction proceeds under mild conditions in a polar solvent, giving the alkylated products in 47–78% yield. While for purine and 4- and 5-azabenzimidazole, the reaction led to a mixture of two isomers, the reaction of adenine and the corresponding 2-fluoro derivative was regioselective, resulting in the formation of only one isomer in each case. The alkylation of theophylline led to the formation of a new derivative of caffeine.

Graphical Abstract
Introduction
Despite significant progress in the last 20–30 years, the selective introduction of fluorine and polyfluoroalkyl substituents into organic molecules remains a challenging problem in the synthesis of fluorinated biologically active compounds, especially larger moieties, such as C2F5, CF(CF3)2, and CH(CF3)2. With respect to the hexafluoroisopropyl group, the methods are limited to a relatively short list: the reduction of the OH group of the C(CF3)2OH moiety (this protocol was employed in the synthesis of enantiomerically pure (S)-5,5,5,5',5',5'-hexafluoroleucine [1]); the Wittig reaction of Ph3P=C(O)C(O)OR with hexafluoroacetone, followed by the hydrogenation of the CH=C(CF3)2 unit, and the conversion of the ketoester into an amino acid [2]; and the reaction of aldehydes with either Ph3P/(CF3)2CCl2 [3] or 2,2,4,4-tetrakis(trifluoromethyl)-1,3-dithietane [4,5]. The last method was employed in the synthesis of 16,16,16,17,17,17-hexafluororetinal, as reported by the Haas group [5].
Benzimidazoles are an important class of organic materials, and many derivatives of these group are biologically active [6-9]. The benzimidazole moiety “ […] is isosteric with indole and purine nuclei, which are present in a number of fundamental cellular components and bioactive compounds. Indeed, a number of important drugs used in different therapeutic areas contain the benzimidazole ring […], such as proton pump inhibitors (omeprazole), antihypertensives (candesartan, telmisartan), antihistaminics (astemizole), antihelmintics (albendazole, mebendazole), as well as several other kinds of still investigational therapeutic agents, including antitumorals and antivirals […]” [8].
Interestingly, most fluorine-containing benzimidazoles contain the fluorinated substituent either on the aromatic ring or on a side chain connected to the aromatic ring; examples of benzimidazoles containing a fluoroalkyl group connected to a nitrogen atom of the imidazole segment are relatively rare. Known examples include benzimidazoles carrying NCF3 [10], NCF2H [11-13], NRf (Rf = CF2Cl, CF2Br, CF2CFClH, CF=CFClF) [14,15], and hexafluoroisopropyl groups attached to the nitrogen atom [16]. Recently, we reported a new method for the introduction of a hexafluoroisopropyl group based on the reaction of tetrakis(trifluoromethyl)-1,3-dithietane (1) with the corresponding azoles [17]. Since this methodology is simple and works for a wide variety of imidazoles, including benzimidazole, we decided to explore the applicability of this method to the modification of biologically active imidazoles, such as adenine and purine derivatives. The results of this study are reported in this article.
Results and Discussion
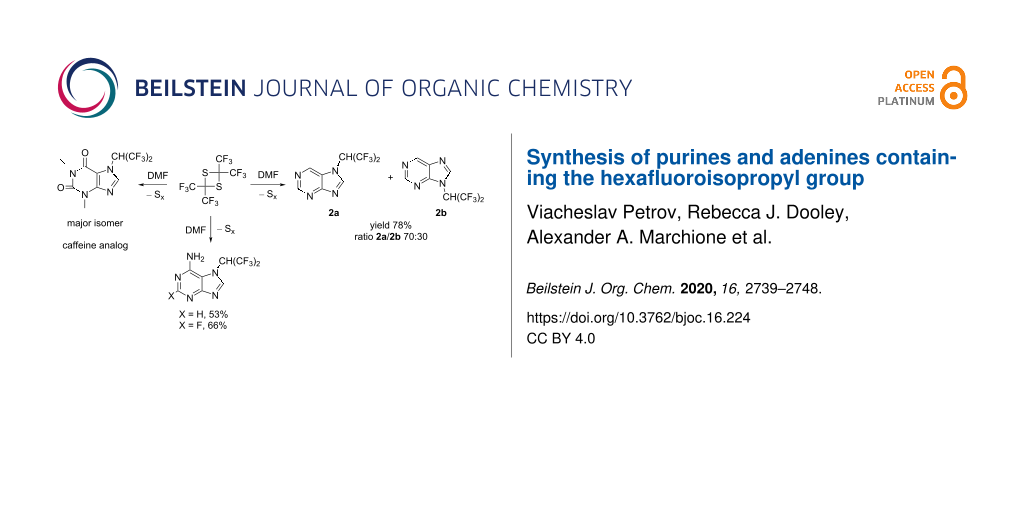
Despite the presence of two electron-withdrawing nitrogen atoms in the aromatic ring, purine (2) was found to react with tetrakis(trifluoromethyl)-1,3-dithietane (1) in a manner similar to benzimidazole [17]. The reaction proceeds without catalyst in DMSO solvent at ambient temperature, leading to the formation of 7- and 9-(hexafluoroisopropyl)purine (2a and 2b), respectively, along with the formation of one equivalent of elemental sulfur (Scheme 1).
![[1860-5397-16-224-i1]](/bjoc/content/inline/1860-5397-16-224-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of purine (2) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
Scheme 1: Reaction of purine (2) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
A crude mixture of 2a and 2b (ratio 70:30) was isolated in 78% yield. Since the minor isomer 2b had a significantly higher solubility in hexane, washing of the crude mixture with hexane resulted in an enrichment of the major isomer 2a (ratio 2a/2b 95:5 after washing with hexane). Pure 2a was isolated after recrystallization from hexane (see Experimental section), and the structure was established by X-ray diffraction (Figure 1).
![[1860-5397-16-224-1]](/bjoc/content/figures/1860-5397-16-224-1.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Crystal structure of 2a, with the thermal ellipsoids drawn at 30% probability.
Figure 1: Crystal structure of 2a, with the thermal ellipsoids drawn at 30% probability.
4-Azabenzimidazole (3) reacted with tetrakis(trifluoromethyl)-1,3-dithietane (1) under similar conditions, with the formation of the two isomeric products 3a and 3b (Scheme 2).
![[1860-5397-16-224-i2]](/bjoc/content/inline/1860-5397-16-224-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of 4-azabenzimidazole (3) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
Scheme 2: Reaction of 4-azabenzimidazole (3) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
The major isomer 3a was isolated in a pure form, and the structure was confirmed by X-ray diffraction. It should be pointed out that the 19F NMR spectra of the two isomers were very different. While the resonance corresponding to the CH(CF3)2 group in the spectrum of 3b was a sharp and well-resolved doublet (δ = −69.95 ppm, JFH = 6.9 Hz in CDCl3), the analogous resonance in the major isomer 3a was significantly broadened (δ = −68.95 ppm, ∆√1/2 = 60Hz, CDCl3), indicating a restricted rotation around the C–N bond, likely due to the steric interaction of the CF3 groups of the hexafluoroisopropyl fragment with the neighboring hydrogen atom of the pyridine ring. A similar phenomenon was previously observed for N-(hexafluoroisopropyl)benzimidazole [17].
5-Azabenzimidazole (4) reacted similarly with tetrakis(trifluoromethyl)-1,3-dithietane (1), which also resulted in the formation of two regioisomers, 4a and 4b (Scheme 3).
![[1860-5397-16-224-i3]](/bjoc/content/inline/1860-5397-16-224-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of 5-azabenzimidazole (4) with 1.
Scheme 3: Reaction of 5-azabenzimidazole (4) with 1.
In this context, the isomers 4a and 4b were separated by column chromatography (see Experimental section), and the structure of both was established by X-ray diffraction. In the 19F NMR spectra of both isomers 4a and 4b, the resonances corresponding to the CH(CF3)2 group were significantly broadened.
While all attempts to involve guanine in the reaction with 1 failed (DMSO, 25–70 °C, 4 d), the reaction of adenine (5) or 2-fluoroadenine (6) with tetrakis(trifluoromethyl)-1,3-dithietane (1) was found to proceed under mild conditions (DMSO, 25 °C, 1 d), leading (unexpectedly) to the formation of only one isomer (5a and 6a, respectively) in each case (Scheme 4).
![[1860-5397-16-224-i4]](/bjoc/content/inline/1860-5397-16-224-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of adenine (5) and 2-fluoroadenine (6) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
Scheme 4: Reaction of adenine (5) and 2-fluoroadenine (6) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
Another surprising feature of these reactions was the selective alkylation of the nitrogen atoms of the imidazole ring rather than the amino group. This is in sharp contrast to the report by Ishikawa and Kitazume that the reaction of anilines with tetrakis(trifluoromethyl)-1,3-dithietane (1) results in the formation of the corresponding imines of hexafluoroacetone [18]. Unfortunately, we were not able to confirm the structures by X-ray diffraction, and the structural assignment for the compounds 5a and 6a relies solely on NMR data. In both cases, the data were consistent with the structures 5a or 6a, carrying NH2 and CH(CF3)2 groups on the same side of the molecule. Indeed, in the 19F NMR spectra of 5a and 6a, the resonance corresponding to CH(CF3)2 was significantly broadened (5a: ∆√1/2 = 150 Hz; 6a: ∆√1/2 = 50 Hz), and in the 1H NMR spectrum of 6a, the 1H nuclei of the NH2 group were observed as two separate resonances, with significantly different chemical shifts (δ = 1.66 (br. s) and 5.98 ppm (1H, br. s) in CDCl3 solvent, while in DMSO-d6, only one broad resonance (δ = 7.69 ppm, 2H, br. s) was observed for the NH2 group. We believe that these observations are consistent with the restricted rotation of the CH(CF3)2 group because of the proximity to the amino group.
Theobromine or caffeine both were found to be inert towards tetrakis(trifluoromethyl)-1,3-dithietane (1) – even at elevated temperature (65 °C, 4–6 d, DMSO or DMF) or in the presence of a catalyst (CsF, DMSO, 25 °C, 2 d), but the structurally similar theophylline (7) reacted with 1 in the presence of CsF catalyst, producing a mixture of the two isomers 7a and 7b, with a significant predominance of 7a (Scheme 5).
![[1860-5397-16-224-i5]](/bjoc/content/inline/1860-5397-16-224-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of theophylline (7) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
Scheme 5: Reaction of theophylline (7) with tetrakis(trifluoromethyl)-1,3-dithietane (1).
The major isomer 7a was isolated by recrystallization from hexane, and the structure was unambiguously established by X-ray diffraction (Figure 2).
![[1860-5397-16-224-2]](/bjoc/content/figures/1860-5397-16-224-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of 7a, with the thermal ellipsoids drawn at 30% probability.
Figure 2: Crystal structure of 7a, with the thermal ellipsoids drawn at 30% probability.
Exchange broadening of the resonances of the (CF3)2CH group was not observed in the 19F NMR spectra of 7a and 7b.
Compound 7a can be viewed as a caffeine analogue, modified by the attachment of two CF3 groups to the methyl group. This is the first example of an introduction of a fluoroalkyl group bigger than CF3 or CF2H into a theophylline (7) molecule, since known examples of fluorinated caffeine derivatives are limited to 7-trifluoromethyl caffeine, claimed in the patent literature [19,20] and 7-difluoromethyl caffeine, prepared by the insertion of difluorocarbene into the N–H bond of theophylline (7) [11,21,22]. Similar to the reaction of tetrakis(trifluoromethyl)-1,3-dithietane (1) and theophylline (7, Scheme 5), this process also led to the formation of two regioisomers, but interestingly, the ratio of the 7-CF2H- and 9-CF2H-isomers varied broadly from 1:2 [22] to 3:2, depending on the reaction conditions and the source of difluorocarbene [11].
Mechanism, regiochemistry, and rotation barriers of the CH(CF3)2 group
The mechanism of the reaction of tetrakis(trifluoromethyl)-1,3-dithietane (1) with the azoles 2–7 is not entirely clear. Although there are two similar reactions reported in the literature (the reaction of 1 with anilines [18] and azoles [17]), no satisfactory mechanism was suggested for these reactions. Recently, it was demonstrated that the majority of the reactions of 1 can involve monomeric hexafluorothioacetone (HFTA) as long as the reaction is carried out in a polar and nucleophilic solvent, such as DMF or DMSO [23]. One can speculate that an alternative mechanism may involve the interaction of monomeric HFTA and imidazole. In this case, the reaction can proceed through the attack of a nitrogen atom of the imidazole ring at the carbon atom of the C=S bond, leading to the formation of the zwitterion B (Scheme 6).
![[1860-5397-16-224-i6]](/bjoc/content/inline/1860-5397-16-224-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Probable mechanism of the reaction of tetrakis(trifluoromethyl)-1,3-dithietane (1) with compounds 2–7.
Scheme 6: Probable mechanism of the reaction of tetrakis(trifluoromethyl)-1,3-dithietane (1) with compounds 2–...
The transfer of a proton from the nitrogen atom to the sulfur atom leads to the formation of thiol C, which can lose sulfur, giving rise to the final product. Because of the unsymmetrical nature of substrate A, the attack on the C=S bond of HFTA proceeds by either nitrogen atom of the imidazole ring, resulting in the formation of two isomers for compounds 2–4 and 7. The highly selective reaction of adenines 5 and 6 may reflect a significantly higher nucleophilicity of the nitrogen atom of the imidazole ring versus that of the NH2 group.
It should be pointed out that the mechanism depicted in Scheme 6 is highly speculative since the nature of the last step (the conversion of intermediate C into the final product, with extrusion of elemental sulfur) is not well understood and may be more complex, involving additional steps. The mechanism clarification of the reaction of tetrakis(trifluoromethyl)-1,3-dithietane (1) with azoles (and anilines) does require further investigation.
As was mentioned above, the broadening of the resonances corresponding to the CF3 group as observed in the 19F NMR spectra of derivatives 3a, 4a, and 4b at ambient temperature is likely to be related to a restricted rotation around the C–N bond as a result of the steric interaction between the fluorine atoms of the (CF3)2CH group and a neighboring hydrogen of an aromatic ring. Indeed, a similar phenomenon was observed for benzimidazole carrying a CH(CF3)2 group at a nitrogen atom of the imidazole ring [17]. Another argument in favor of this explanation is the absence of any evidence for the restricted rotation in the isomer 3b (Scheme 2), which has a nitrogen atom in the β-position relative to the CH(CF3)2 group (see Table 1), while signal broadening was clearly present in the 19F NMR spectra of both isomers 4a and 4b. All these data are consistent with the observed restricted rotation of the (CF3)2CH group in compounds 5a and 6a (see Table 2) because of the presence in the β-position relative to the CH(CF3)2 unit of the amino group, which is substantially bigger than a hydrogen atom.
Table 1: Reaction products, conditions, yield, ratio of isomers, and melting points for the new materials.
| compound | solvent (cat.) | T, °C (time, d) | yield, % | ratio of isomers (crude product) | mp, °C (crude product) | mp, °C (recrystallized from hexane) |
| 2a | DMSO | 25 (1) | 78 | 2a/2b 70:30 | 158–167 | – |
| 2b | 2b, 122–123 | |||||
| 3a,b | DMSO | 25 (1) | 74 | 3a/3b 82:18 | 137–163 | 3a, 164–165 |
| 4a,b | DMSO | 25 (1) | 70 | 4a/4b 67:33 | – |
4a, 157–158
4b, 110–111 |
| 5a | DMSO | 25 (1) | 53 | one isomer |
212–214
(dec.) |
5a, 214–216
(dec.) |
| 6a | DMSO | 25 (1) | 66 | one isomer | – | 6a, 220–222 |
| 7a,b | DMF (CsF) | 25 (4) | 47 | 7a/7b 85:15 | 86–88 | 7a, 95–95.5 (purity 98%) |
Table 2: Enthalpy and entropy of activation for the rotameric interconversion of a subset of hexafluoroisopropyl azoles.
| compound | ΔH‡, major to minor (kcal⋅mol−1) | ΔS‡, major to minor (cal⋅mol−1 K−1) | ΔH‡, minor to major (kcal⋅mol−1) | ΔS‡, minor to major (cal⋅mol−1 K−1) | population of the major rotamer at 298 K |
| 3a | 11.6 | −5.38 | 11.1 | −5.90 | 0.691 |
| 4a | 12.0 | −4.12 | 11.5 | −3.67 | 0.760 |
| 4b | 11.6 | −5.91 | 10.6 | −7.76 | 0.680 |
| 6a | 12.0 | −3.72 | 9.79 | −7.09 | 0.862 |
Dynamic NMR spectroscopy
The broadening of resonances corresponding to the hexafluoroisopropyl moieties in the 19F NMR spectra described above was observed at near-ambient temperatures and at resonance frequencies from 376 to 564 MHz, consistent with a slow exchange between rotameric states on the NMR timescale under these conditions. The activation parameters of the hindered rotation were obtained for a subset of these compounds, 3a, 4a, 4b, and 6a, by NMR experiments similar to those used in recent studies [24]. In brief, solutions of each compound were made in N,N-dimethylformamide-d7. A series of 19F NMR spectra were acquired at subambient temperature: both simple one-dimensional spectra and selective inversion spectra, in which the resonance corresponding to one rotamer was selectively inverted, allowed to interconvert with the other site for a variable period and then converted to observable magnetization with an excitation pulse. The former experiments allowed an assay of the population of each site as a function of the temperature, which enabled an extrapolation of the population differences at an elevated temperature (via the van ‘t Hoff equation). The latter experiments provided information directly on the kinetics of the interconversion between the sites. A series of spectra were then acquired at elevated temperature; here, the kinetics of the interconversion between the sites could be derived directly from the linewidth. Figure 3 illustrates the spectra acquired over a range of temperatures using compound 3a as an example. With the rate constants of the interconversion between the rotamers in hand, an Eyring plot was generated, and the enthalpy and entropy of activation were derived (Table 2).
![[1860-5397-16-224-3]](/bjoc/content/figures/1860-5397-16-224-3.jpg?scale=1.7&max-width=1024&background=FFFFFF)
Figure 3: Top: 19F NMR spectra of 3a acquired over a sample temperature range of 223–373 K. Left: Fitted plot of integrated intensity vs interpulse delay time from the experiment inverting the minor rotamer of 3a at 253 K. Right: Eyring plot of the interconversion of the rotamers of 3a.
Figure 3: Top: 19F NMR spectra of 3a acquired over a sample temperature range of 223–373 K. Left: Fitted plot...
The structural identification of the major and minor rotamers was not attempted by NMR, but in silico investigations of 3a supported the intuitive notion that the rotamer of lower energy was that with the CF3 moieties nearer the five-membered ring of the azabenzimidazole unit, and the rotamer of higher energy was that with the CF3 moieties near the six-membered ring (see Figure 4). Density functional theory calculations predict an enthalpy difference of 0.48 kcal⋅mol−1 between the two rotamers. By comparison, ΔH between the two rotamers was obtained experimentally by fitting the van ‘t Hoff equation to the population differences observed in the 19F NMR spectra between 224 and 264 K. 0.89 kcal⋅mol−1 is in tolerable agreement with the computation.
![[1860-5397-16-224-4]](/bjoc/content/figures/1860-5397-16-224-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: DFT-optimized structures of the two rotamers of 3a. Left: Lower-energy rotamer. Right: Higher-energy rotamer.
Figure 4: DFT-optimized structures of the two rotamers of 3a. Left: Lower-energy rotamer. Right: Higher-energ...
Comparing 3a, 4a, 4b, and 6a in Table 2, the enhanced imbalance in the population of the rotamers of 6a is notable and consistent with the amino moiety imposing a greater steric hindrance. The differences among the other compounds are more modest. The small entropy of activation is consistent with a simple rotation over a steric barrier (with minimal contribution from such forces as hydrogen bonding or solvent interactions), and the relative enthalpy of activation for each compound (major to minor vs minor to major) are roughly similar to the difference in total enthalpy for the two rotamers for each compound.
Experimental
GC and GC–MS analyses were carried out on a HP-6890 instrument using a HP FFAP capillary column and either TCD (GC) or mass-selective detectors (GS–MS), respectively. Purine (2), 4-aza- and 5-azabenzimidazole (3 and 4), adenine (5), theophylline (7), dry DMF, and DMSO were obtained from commercial source (Sigma-Aldrich) and used without further purification. CsF (Sigma-Aldrich) was dried in vacuum and stored and handled inside a glove box. Compound 1 was prepared according to a literature procedure [24], purity 96–98% and was used without further purification. The purity of the new materials, determined by GC and NMR spectroscopy, was at least 99%, unless stated otherwise.
Standard NMR experiments
The NMR data presented in Table 2 and Table 3 were derived from spectra acquired on a Bruker Neo spectrometer with a 9.4 T magnetic field, equipped with a 5 mm 15N,77Se, 31P {19F,1H} nitrogen cryoprobe. Chemical shifts are referenced to internal tetramethylsilane or trichlorofluoromethane. The spectra were acquired at 298 K.
Table 3: NMR and MS data for the compounds 2a,b, 3a,b, 4a,b, 5a, 6a, and 7a,b.
| compound | 1H NMR (in CDCl3, δ, ppm; J, Hz) | 19F NMR (in CDCl3, δ, ppm; J, Hz) | 13C NMR (δ, ppm; J, Hz)a | MS (m/z)b |
| 2a | 5.55 (1H, sept, 6.5), 8.40 (1H, s), 9.08 (1H, s), 9.28 (1H, s) | −68.75 (d, 6.6) | – | 270 (M+, C8H4F6N4+) |
| 2b | 6.08 (1H, sept, 7.0), 8.33 (1H, s), 9.09 (1H, s), 9.30 (1H, s) | −69.75 (d, 7.0) | 55.07 (sept, 33.7), 120.51 (q, 283.8), 133.87, 141.75 (sept, 1.5), 149.83, 151.43, 153.80 | 270 (M+, C8H4F6N4+) |
| 3a | 5.63 (1H, sept, 6.7), 7.40 (1H, dd, 8.4, 4.7), 7.89 (1H, d, 8.4), 8.37 (1H, s), 8.70 (1H, d, 4.7) | −68.95 (br. s, ∆√1/2 = 60 Hz) | 52.50 (br. s), 118.99, 119.61, 121.61 (q, 287.00), 144.00, 146.01, 159.81, 205.22 | 269 (M+, C9H5F6N3+) |
| 3b |
6.18 (1H, sept), 7.40 (1H),
8.25 (1H), 8.37 (1H), 8.50 (1H) |
−69.95 (d, 6.9) | – | – |
| 4a | 5.46 (sept, 6.7), 7.41 (1H, d, 5.7), 8.15 (1H, s), 8.61 (1H, d, 5.7), 9.24 (1H, s) | −69.15 (br. s) | 58.38 (sept, 32.8), 115.74, 120.78 (q, 284.4), 131.28, 132.90, 143.65, 144.00, 148.35 | 269 (M+, C9H5F6N3+) |
| 4b | 5.46 (sept, 6.7), 7.41 (1H, d, 5.7), 8.15 (1H, s), 8.61 (1H, d, 5.7), 9.24 (1H, s) | −69.08 (br. s) | 58.10 (sept, 33.2), 104.64, 120.77 (q, 284.4), 138.78, 139.97, 142.11, 144.06, 144.09 | – |
| 5a |
5.67 (2H, br. s), 5.95 (1H, sept, 7.2), 8.02 (1H, s), 8.48 (1H, s);
DMSO-d6: 7.23 (1H, sept, 7.2 Hz), 7.64 (2H, s), 8.27 (1H, s), 8.42 (1H, s) |
−69.08 (br. s, ∆√1/2 > 150 Hz);
DMSO-d6: −69.88 (br. s, ∆√1/2 > 150 Hz) |
55.03 (sept, 33.0), 117.93, 121.44 (q, 282.0), 138.87, 150.41, 154.27, 156.20c | 285 (M+, C8H5F6N5+) |
| 6a |
1.66 (1H, br. s), 5.81 (1H, sept, 7.2), 5.98 (1H, br. s), 7.98 (1H, s);
DMSO-d6: 6.48 (1H, sept, 7.1), 7.69 (2H, br. s), 7.96 (1H, s) |
−69.84 (6F, br. s, ∆√1/2 = 50 Hz), −49.15 (1F, br. s);
DMSO-d6: −69.4 (6F, br. s, ∆√1/2 = 50 Hz), −50.42 (1F, br. s) |
59.70 (sept, 32.0), 117.21, 121.51 (q, 283.8), 139.27, 151.88 (d, 20.4), 158.55 (d, 21.70), 159.68 (d, 205.9)c | 303 (M+, C8H4F7N5+) |
| 7a | 3.40 (3H, s), 3.60 (3H, s), 6.74 (1H, sept, 7.0), 7.85 (1H, s) | −70.00 (d, 7.0) | – | 330 (M+, C10H8F6N4O2+) |
| 7b | 3.39 (3H, s), 3.59 (3H, s), 4.68 (1H, sept, 7.8), 7.80 (1H, s) | −65.02 (d, 7.8) | – | – |
a13C{1H} in CDCl3, unless stated otherwise. bMixture of isomers, solution in acetone. cIn DMSO-d6 as a solvent.
Dynamic NMR experiments
The dynamics of rotamer interconversion were studied on a Bruker AVIIIHD spectrometer with a 9.4 T magnetic field, equipped with a 10 mm F{H} probe suitable for high- and low-temperature operation. The sample temperature was calibrated against neat ethylene glycol and neat methanol standards. Selective inversion experiments at subambient temperature effected the selective inversion with iBURP shapes [25]. The spin–lattice relaxation time T1 for each site was determined with nonselective inversion recovery experiments. The rate constant for the interconversion between the rotamers at each temperature was determined using numerical integration, minimizing the square of residuals, with the obtained T1 relaxation time as an input to the model. The linewidths at elevated temperature were determined using the peak fitting utility in ACDLabs Spectrus Processor software (v. 2018.1.1).
X-ray diffraction
X-ray data for 7a, 2a, 3a, 4a, and 6a were collected at −100 °C using a Bruker APEX-II CCD system equipped with a sealed tube molybdenum source and a graphite monochromator. The structures were solved and refined using the SHELXTL software package [26], refinement by full-matrix least squares on F2, scattering factors from [25]. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publication nos. CCDC #1998565−1998569.
Computational chemistry
Computational optimization of 8a and 8b structures was performed with Gaussian 9 software. Density functional theory at the B3LYP level, with a 6-311g++ basis set (Co phase) was employed.
Reaction of purine (2) and 1
A mixture of 1.2 g (0.01 mol) of purine (2), 3 g (0.08 mol) of 1, and 15 mL of DMSO was agitated for 24 h at ambient temperature. The precipitated sulfur was filtered and dried on a filter (isolated: 0.25 g, 78%). The filtrate was poured in 300 mL of water, and the mixture was extracted with hexane (50 mL × 2). The hexane layer was dried, and the solvent was removed under vacuum to leave 0.6 g of a white solid, which was found to be the 99% pure minor isomer 2b (mp 122–123 °C after the second crystallization from hexane; this sample was used for X-ray diffraction studies). The rest of the reaction mixture was extracted with DCM (50 mL × 2). The DCM layer was washed with water (200 mL × 3) and dried, and the solvent was removed under vacuum to leave 1.5 g of a crude orange solid, which was found to be a mixture of 2a and 2b in the ratio 70:30 (GC, NMR), mp 158–167 °C. Total yield of 2a,b: 78%.
A small sample of a mixture of 2a,b was washed with cold hexane on a ceramic filter to give a material with a 2a,b ratio of 95:5. Recrystallization of this sample from hexane gave the isomer 2b (purity 99%, by NMR), and this sample was used for an X-ray diffraction experiment. The reaction conditions, mass spectrometry, and NMR data are given in Table 1 and Table 3.
Reaction of 4-azabenzimidazole (3) and 1
A mixture of 1.2 g (0.01 mol) of 4-azabenzimidazole (3), 2 g (0.06 mol) of 1, and 15 mL of DMSO was agitated at ambient temperature for 24 h. The precipitated sulfur was filtered, washed with hexane, and dried on a filter (isolated: 0.25 g, 78%). The reaction mixture was poured into 500 mL of water, and the solids were extracted with a 1:1 mixture of hexane/dichloromethane (50 mL × 2), washed with water (100 mL × 2), dried with MgSO4, and filtered. The solvent was removed under reduced pressure to give 2 g (74%) of the crude product, which was found to be a mixture of two isomers (ratio 82:18, by NMR), mp 137–163 °C.
A small sample of this material was recrystallized from hot hexane to give a sample of the major isomer 3a (purity 99.8%, mp 164–165 °C), which was used for an X-ray diffraction experiment. The reaction conditions, mass spectrometry, and NMR data are given in Table 1 and Table 3.
Reaction of 5-azabenzimidazole (4) and 1
A mixture of 1.2 g (0.01 mol) of 5-azabenzimidazole (4), 2 g (0.06 mol) of 1, and 15 mL of DMSO was agitated at ambient temperature for 24 h. The precipitated sulfur was filtered. The reaction mixture was poured in 500 mL of water, and the solids were filtered off to give 1.9 g (70%) of a crude product, which was found to be a mixture of the isomers 4a and 4b in a ratio of 67:33 (by NMR).
Pure isomers were isolated using column chromatography (Biotage instrument, hexane/ethyl acetate gradient, 100 mL/min, cartridge SNAP 340 g). The structure of each isomer was determined by X-ray diffraction. The reaction conditions, mass spectrometry, and NMR data are given in Table 1 and Table 3.
Reaction of adenine (5) and 2-fluoroadenine (6), respectively, and 1
A mixture of 0.01 mol of the corresponding adenine (1.35 g of 5 or 1.53 g of 6), 2 g (0.06 mol) of 1, and 15 mL of DMSO was agitated at ambient temperature for 24 h. The precipitated sulfur was filtered and dried on a filter (isolated: 0.25 and 0.30 g, 78–94%, respectively). The reaction mixture was poured in 500 mL of water, and the solids were filtered off, washed with water (100 mL × 2), and dried on a filter. The crude material was washed with cold hexane and dried to give 1.51 g of 5a (yield 53%, purity 98%) and 1.99 g of 6a (yield 66%, purity 97%). Pure samples (>99%, by GC and NMR) of 5a and 5b were obtained by recrystallization of small sample from hexane. The reaction conditions, mass spectrometry, and NMR data are given in Table 1 and Table 3.
Reaction of theophylline (7) and 1
A mixture of 1.8 g (0.01 mol) of theophylline (7), 2 g (0.012 mol) of 1, 0.5 g (0.003 mol) of dry CsF, and 15 mL of DMF was agitated for 4 d at ambient temperature. The reaction mixture was filtered to remove precipitated sulfur and the catalyst. The filtrate was poured in 300 mL of water, and the mixture was extracted by a dichloromethane/hexane mixture (3:1, 50 mL × 2). The organic layer was dried with MgSO4, filtered, and the solvent was removed under reduced pressure to give 1.5 g of a slightly yellow solid. This was redissolved in 20 mL of 1:1 mixture of dichloromethane/hexane and kept at −15 °C. A yellow precipitate (0.1 g) was filtered off, and the solvent was removed to leave 1.2 g of the crude product (mp 86–88 °C), which was found to be a mixture of the two isomers 7a and 7b in a ratio of 85:15 (purity 98%, yield 47%). A small sample of this material was recrystallized from hot hexane to give a sample of 7a (mp 95 °C, purity 99%), which was used for an X-ray diffraction experiment. The identity of a single crystal and the bulk of the material was confirmed by powder diffraction data and compared to simulated powder diffraction data from the single crystal diffraction experiment. The reaction conditions, mass spectrometry, and NMR data are given in Table 1 and Table 3.
References
-
Anderson, J. T.; Toogood, P. L.; Marsh, E. N. G. Org. Lett. 2002, 4, 4281–4283. doi:10.1021/ol026922j
Return to citation in text: [1] -
Chiu, H.-P.; Cheng, R. P. Org. Lett. 2007, 9, 5517–5520. doi:10.1021/ol702470j
Return to citation in text: [1] -
Hanack, M.; Korhummel, C. Synthesis 1987, 944–947. doi:10.1055/s-1987-28135
Return to citation in text: [1] -
Burton, D. J.; Inouye, Y. Tetrahedron Lett. 1979, 20, 3397–3400. doi:10.1016/s0040-4039(01)95419-x
Return to citation in text: [1] -
Abele, H.; Haas, A.; Lieb, M.; Zwingenberger, J. Chem. Ber. 1994, 127, 145–149. doi:10.1002/cber.19941270123
Return to citation in text: [1] [2] -
Tonelli, M.; Simone, M.; Tasso, B.; Novelli, F.; Boido, V.; Sparatore, F.; Paglietti, G.; Pricl, S.; Giliberti, G.; Blois, S.; Ibba, C.; Sanna, G.; Loddo, R.; La Colla, P. Bioorg. Med. Chem. 2010, 18, 2937–2953. doi:10.1016/j.bmc.2010.02.037
Return to citation in text: [1] -
Tonelli, M.; Paglietti, G.; Boido, V.; Sparatore, F.; Marongiu, F.; Marongiu, E.; La Colla, P.; Loddo, R. Chem. Biodiversity 2008, 5, 2386–2401. doi:10.1002/cbdv.200890203
Return to citation in text: [1] -
Tonelli, M.; Novelli, F.; Tasso, B.; Vazzana, I.; Sparatore, A.; Boido, V.; Sparatore, F.; La Colla, P.; Sanna, G.; Giliberti, G.; Busonera, B.; Farci, P.; Ibba, C.; Loddo, R. Bioorg. Med. Chem. 2014, 22, 4893–4909. doi:10.1016/j.bmc.2014.06.043
Return to citation in text: [1] [2] -
Akhtar, W.; Khan, M. F.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M. A.; Mehdi, S. H.; Akhter, M.; Alam, M. M. Eur. J. Med. Chem. 2017, 126, 705–753. doi:10.1016/j.ejmech.2016.12.010
Return to citation in text: [1] -
Engl, P. S.; Senn, R.; Otth, E.; Togni, A. Organometallics 2015, 34, 1384–1395. doi:10.1021/acs.organomet.5b00137
Return to citation in text: [1] -
Prakash, G. K. S.; Krishnamoorthy, S.; Ganesh, S. K.; Kulkarni, A.; Haiges, R.; Olah, G. A. Org. Lett. 2014, 16, 54–57. doi:10.1021/ol403007j
Return to citation in text: [1] [2] [3] -
Poludnenko, V. G.; Didinskaya, O. B.; Pozharskii, A. F.; Gil'burd, M. M. Khim. Geterotsikl. Soedin. 1982, 1696.
Return to citation in text: [1] -
Poludnenko, V. G.; Didinskaya, O. B.; Pozharskii, A. F. Khim. Geterotsikl. Soedin. 1984, 520–523.
Return to citation in text: [1] -
Yagupolskii, L. M.; Fedyuk, D. V.; Petko, K. I.; Troitskaya, V. I.; Rudyk, V. I.; Rudyuk, V. V. J. Fluorine Chem. 2000, 106, 181–187. doi:10.1016/s0022-1139(00)00321-3
Return to citation in text: [1] -
Rudyuk, V. V.; Fedyuk, D. V.; Yagupolskii, L. M. J. Fluorine Chem. 2004, 125, 1465–1471. doi:10.1016/j.jfluchem.2004.05.003
Return to citation in text: [1] -
Petko, K. I.; Vorobey, V. M. Ukr. Khim. Zh. (Russ. Ed.) 2015, 81, 106–110.
Return to citation in text: [1] -
Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2013, 156, 262–267. doi:10.1016/j.jfluchem.2013.07.004
Return to citation in text: [1] [2] [3] [4] [5] -
Ishikawa, N.; Kitazume, T. Chem. Lett. 1972, 1, 169–170. doi:10.1246/cl.1972.169
Return to citation in text: [1] [2] -
Huang, W. 2,4-(1H,3H)-Pyrimidinedione derivatives and preparation method therefor. WO Patent WO2015106624A1, July 23, 2015.
Return to citation in text: [1] -
Huang, W. 2,4-(1H,3H)-Pyrimidinedione derivatives and preparation method thereof. Chinese Patent CN103788095A, May 14, 2014.
Return to citation in text: [1] -
Hao, X.; Chen, W.; Chen, M.; Lan, S.; Zhang, J.; Li, W. J. Huazhong Univ. Sci. Technol., Nat. Sci. 2007, 41, 546–548.
Return to citation in text: [1] -
Mehta, V. P.; Greaney, M. F. Org. Lett. 2013, 15, 5036–5039. doi:10.1021/ol402370f
Return to citation in text: [1] [2] -
Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2015, 179, 56–63. doi:10.1016/j.jfluchem.2015.04.003
Return to citation in text: [1] -
Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2010, 131, 1144–1155. doi:10.1016/j.jfluchem.2010.06.011
Return to citation in text: [1] [2] -
International Tables for Crystallography, Volume C, Tables 4.2.6.8 and 6.1.1.4.
Return to citation in text: [1] [2] -
SHELXTL, Version 6.0; Bruker AXS: Madison, WI, USA, 1996.
Return to citation in text: [1]
| 25. | International Tables for Crystallography, Volume C, Tables 4.2.6.8 and 6.1.1.4. |
| 1. | Anderson, J. T.; Toogood, P. L.; Marsh, E. N. G. Org. Lett. 2002, 4, 4281–4283. doi:10.1021/ol026922j |
| 5. | Abele, H.; Haas, A.; Lieb, M.; Zwingenberger, J. Chem. Ber. 1994, 127, 145–149. doi:10.1002/cber.19941270123 |
| 18. | Ishikawa, N.; Kitazume, T. Chem. Lett. 1972, 1, 169–170. doi:10.1246/cl.1972.169 |
| 4. | Burton, D. J.; Inouye, Y. Tetrahedron Lett. 1979, 20, 3397–3400. doi:10.1016/s0040-4039(01)95419-x |
| 5. | Abele, H.; Haas, A.; Lieb, M.; Zwingenberger, J. Chem. Ber. 1994, 127, 145–149. doi:10.1002/cber.19941270123 |
| 19. | Huang, W. 2,4-(1H,3H)-Pyrimidinedione derivatives and preparation method therefor. WO Patent WO2015106624A1, July 23, 2015. |
| 20. | Huang, W. 2,4-(1H,3H)-Pyrimidinedione derivatives and preparation method thereof. Chinese Patent CN103788095A, May 14, 2014. |
| 17. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2013, 156, 262–267. doi:10.1016/j.jfluchem.2013.07.004 |
| 17. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2013, 156, 262–267. doi:10.1016/j.jfluchem.2013.07.004 |
| 11. | Prakash, G. K. S.; Krishnamoorthy, S.; Ganesh, S. K.; Kulkarni, A.; Haiges, R.; Olah, G. A. Org. Lett. 2014, 16, 54–57. doi:10.1021/ol403007j |
| 12. | Poludnenko, V. G.; Didinskaya, O. B.; Pozharskii, A. F.; Gil'burd, M. M. Khim. Geterotsikl. Soedin. 1982, 1696. |
| 13. | Poludnenko, V. G.; Didinskaya, O. B.; Pozharskii, A. F. Khim. Geterotsikl. Soedin. 1984, 520–523. |
| 10. | Engl, P. S.; Senn, R.; Otth, E.; Togni, A. Organometallics 2015, 34, 1384–1395. doi:10.1021/acs.organomet.5b00137 |
| 17. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2013, 156, 262–267. doi:10.1016/j.jfluchem.2013.07.004 |
| 8. | Tonelli, M.; Novelli, F.; Tasso, B.; Vazzana, I.; Sparatore, A.; Boido, V.; Sparatore, F.; La Colla, P.; Sanna, G.; Giliberti, G.; Busonera, B.; Farci, P.; Ibba, C.; Loddo, R. Bioorg. Med. Chem. 2014, 22, 4893–4909. doi:10.1016/j.bmc.2014.06.043 |
| 6. | Tonelli, M.; Simone, M.; Tasso, B.; Novelli, F.; Boido, V.; Sparatore, F.; Paglietti, G.; Pricl, S.; Giliberti, G.; Blois, S.; Ibba, C.; Sanna, G.; Loddo, R.; La Colla, P. Bioorg. Med. Chem. 2010, 18, 2937–2953. doi:10.1016/j.bmc.2010.02.037 |
| 7. | Tonelli, M.; Paglietti, G.; Boido, V.; Sparatore, F.; Marongiu, F.; Marongiu, E.; La Colla, P.; Loddo, R. Chem. Biodiversity 2008, 5, 2386–2401. doi:10.1002/cbdv.200890203 |
| 8. | Tonelli, M.; Novelli, F.; Tasso, B.; Vazzana, I.; Sparatore, A.; Boido, V.; Sparatore, F.; La Colla, P.; Sanna, G.; Giliberti, G.; Busonera, B.; Farci, P.; Ibba, C.; Loddo, R. Bioorg. Med. Chem. 2014, 22, 4893–4909. doi:10.1016/j.bmc.2014.06.043 |
| 9. | Akhtar, W.; Khan, M. F.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M. A.; Mehdi, S. H.; Akhter, M.; Alam, M. M. Eur. J. Med. Chem. 2017, 126, 705–753. doi:10.1016/j.ejmech.2016.12.010 |
| 14. | Yagupolskii, L. M.; Fedyuk, D. V.; Petko, K. I.; Troitskaya, V. I.; Rudyk, V. I.; Rudyuk, V. V. J. Fluorine Chem. 2000, 106, 181–187. doi:10.1016/s0022-1139(00)00321-3 |
| 15. | Rudyuk, V. V.; Fedyuk, D. V.; Yagupolskii, L. M. J. Fluorine Chem. 2004, 125, 1465–1471. doi:10.1016/j.jfluchem.2004.05.003 |
| 11. | Prakash, G. K. S.; Krishnamoorthy, S.; Ganesh, S. K.; Kulkarni, A.; Haiges, R.; Olah, G. A. Org. Lett. 2014, 16, 54–57. doi:10.1021/ol403007j |
| 11. | Prakash, G. K. S.; Krishnamoorthy, S.; Ganesh, S. K.; Kulkarni, A.; Haiges, R.; Olah, G. A. Org. Lett. 2014, 16, 54–57. doi:10.1021/ol403007j |
| 21. | Hao, X.; Chen, W.; Chen, M.; Lan, S.; Zhang, J.; Li, W. J. Huazhong Univ. Sci. Technol., Nat. Sci. 2007, 41, 546–548. |
| 22. | Mehta, V. P.; Greaney, M. F. Org. Lett. 2013, 15, 5036–5039. doi:10.1021/ol402370f |
| 22. | Mehta, V. P.; Greaney, M. F. Org. Lett. 2013, 15, 5036–5039. doi:10.1021/ol402370f |
| 25. | International Tables for Crystallography, Volume C, Tables 4.2.6.8 and 6.1.1.4. |
| 24. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2010, 131, 1144–1155. doi:10.1016/j.jfluchem.2010.06.011 |
| 24. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2010, 131, 1144–1155. doi:10.1016/j.jfluchem.2010.06.011 |
| 23. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2015, 179, 56–63. doi:10.1016/j.jfluchem.2015.04.003 |
| 17. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2013, 156, 262–267. doi:10.1016/j.jfluchem.2013.07.004 |
| 18. | Ishikawa, N.; Kitazume, T. Chem. Lett. 1972, 1, 169–170. doi:10.1246/cl.1972.169 |
| 17. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2013, 156, 262–267. doi:10.1016/j.jfluchem.2013.07.004 |
© 2020 Petrov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)