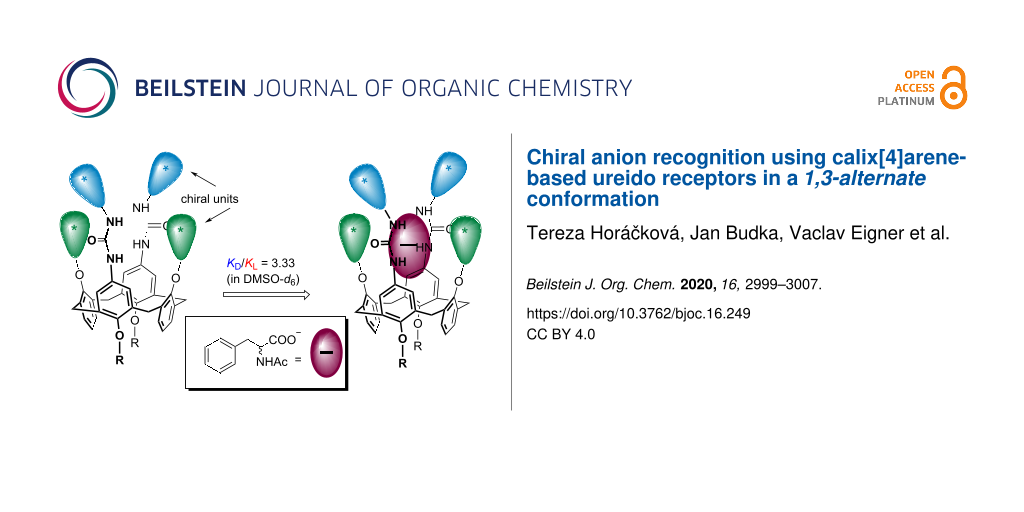
Abstract
The introduction of chiral alkyl substituents into the lower rim of calix[4]arene immobilised in the 1,3-alternate conformation led to a system possessing a preorganised ureido cavity hemmed with chiral alkyl units in the near proximity. As shown by the 1H NMR titration experiments, these compounds can be used as receptors for chiral anions in DMSO-d6. The chiral recognition ability can be further strengthened by the introduction of another chiral moiety directly onto the urea N atoms. The systems with double chiral units being located around the binding ureido cavity showed better stereodiscrimination, with the highest selectivity factor being 3.33 (KL/KD) achieved for N-acetyl-ʟ-phenylalaninate. The structures of some receptors were confirmed by single crystal X-ray analysis.

Graphical Abstract
Introduction
The recognition and complexation of anions has become undoubtedly one of the most important branches of modern supramolecular chemistry, as can easily be demonstrated by an immense number of recent reviews [1-6] and books [7-11] devoted to this topic. Due to the omnipresence of anions in biological systems, their irreplaceable roles in cell functioning have gradually been revealed and are well recognised to date. Consequently, given the importance of anions in many areas of everyday life, including, e.g., biology, medicine, environmental pollution issues, or industrial processes, the design and development of novel artificial receptors/sensors for anions is becoming more and more significant [12-14].
There are many strategies aiming at anion recognition in the literature. Most of the receptors, however, rely on electrostatic interactions. These systems are represented by positively charged molecules, such as quaternary N-, S-, and P-containing onium salts, protonated or alkylated aza-crown ethers and azacryptands, amidinium and guanidinium cations, etc. [15-18]. Due to the low directionality of the Coulomb force, the successful application of purely ionic interactions in the design of selective anion receptors is rather limited. The shapes and geometries of anions are widely different, and therefore the design of corresponding tailor-made receptors is based mostly on more directional interactions, such as hydrogen bonds. Indeed, an incredible number of neutral receptors bearing amide, sulfonamide, urea, thiourea, pyrrole, or triazole functional groups (to name at least some of them) has appeared during the last two decades [19-21].
Due to well-established functionalisation approaches, calix[4]arenes [22-24] are frequently used as molecular platform in the design of more complex receptor systems. The existence of four basic conformations (cone, partial cone, 1,3-alternate, and 1,2-alternate) offers the combination of a precisely defined 3D structure, with functional groups being introduced at exactly defined mutual positions. This makes calix[4]arenes an ideal molecular scaffold [25,26] for the construction of highly sophisticated molecules, including anionic receptors [27-39].
During our ongoing research on anion complexation, we have reported various calix[4]arene receptors based mainly on amide, urea, or thiourea groups [40,41], some of which are available in different conformations. Although the overwhelming majority of calixarene-based receptors makes use of the cone conformer A (Figure 1), the corresponding diureidocalix[4]arenes in the 1,3-alternate conformation B showed [42,43] surprisingly good complexation abilities towards selected anions. Especially for chiral anion recognition, contrary to the cone receptor, a design based on the 1,3-alternate conformer enables the introduction of chiral units into the phenolic functions of the inverted aromatic moieties nearby the ureido cavity responsible for the binding, as in C. This design can be exemplified by our previously published receptors C1 based on a calix[4]arene moiety or by C2 using thiacalix[4]arene as the core scaffold [44,45]. Moreover, the introduction of the tert-butyl groups into the 1,3-alternate conformer should lead to the overall increase rigidity of the molecule, possibly enhancing the interactions within the binding cavity. In this context, we realised that further strengthening of the chiral induction can be reached via the synchronous application of chiral units on the ureido moieties as well, as in D. In this paper, we report the preparation and complexation study of the latter type of receptor, bearing double chiral units in the immediate proximity to the preorganised ureido cavity.
![[1860-5397-16-249-1]](/bjoc/content/figures/1860-5397-16-249-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Design of chiral calix[4]arene-based receptors for anions.
Figure 1: Design of chiral calix[4]arene-based receptors for anions.
Results and Discussion
The introduction of the chiral alkyl moiety based on (S)-2-methylbutan-1-ol into the starting calix[4]arene 1 was carried out using recently described Mitsunobu reaction conditions [44]. Refluxing the reaction mixture of PPh3, DIAD, and toluene for two days provided the distally dialkylated calixarene 2 in 64% yield (Scheme 1). Compound 2 was regioselectively ipso-nitrated with 30 equiv of 65% aq HNO3 in an AcOH and CH2Cl2 mixture, making use of the higher reactivity of the nonalkylated phenolic moieties [45]. The fast reaction afforded the corresponding dinitro derivative 3 within a few minutes with 70% yield.
![[1860-5397-16-249-i1]](/bjoc/content/inline/1860-5397-16-249-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the calix[4]arene-based chiral anionic receptors 7 and 8.
Scheme 1: Synthesis of the calix[4]arene-based chiral anionic receptors 7 and 8.
As we found in our previous attempts [43,45], the alkylation of dinitrocalixarenes to form the 1,3-alternate conformation is a synthetic challenge. In fact, irrespective of the base or solvent used, the alkylation with appropriate n-propyl or n-hexyl halides always led to the partial cone conformation as the main product. To overcome this problematic step, we used the conditions described by Böhmer et al. [46] for a similar system bearing propyl groups on the lower rim. Indeed, one week of stirring 3 with allyl bromide in the presence of Cs2CO3 provided the expected 1,3-alternate conformer 4a in 40% yield accompanied by a small amount of the partial cone conformer 4b (10%).
The unequivocal proof of the structure of the isomer 4a was provided by single crystal X-ray analysis. The compound crystallised in a tetragonal system, space group P41212 as a 1:1 complex with methanol used as a solvent for crystallisation. As shown in Figure 2, the calixarene clearly adopts the 1,3-alternate conformation with an almost ideal tetragonal shape of the cavity. The lengths of the two main diagonals (the distances between opposite bridging CH2 moieties) are essentially identical (7.183 Å and 7.206 Å). If the main plane of the molecule is defined by the four bridging C atoms, all phenolic subunits are almost perpendicular to this plane, with the aromatic parts being slightly tilted out of the cavity. The corresponding interplanar angles Φ with the aromatic subunits are 81.55°, 80.78°, 77.08°, and 80.57°, respectively, starting counterclockwise from the upper subunit bearing a nitro group (Figure 2a).
![[1860-5397-16-249-2]](/bjoc/content/figures/1860-5397-16-249-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structure of 4a: (a) Top view into the cavity. (b) Side view of the same cavity.
Figure 2: X-ray structure of 4a: (a) Top view into the cavity. (b) Side view of the same cavity.
The 1,3-alternate isomer 4a represents quite an interesting synthetic intermediate as the presence of the allyl groups immobilizes the required conformation and at the same time enables the potential incorporation of the macrocycle into a polymeric matrix. Consequently, the subsequent reduction step was carried out in two alternative ways: (i) exclusive reduction of the nitro groups or (ii) concomitant reduction of the nitro groups and the allyl moieties. Thus, the reaction of 4a with SnCl2∙2H2O in ethanol gave the corresponding amine 5 in 57% yield after column chromatography on alumina. On the other hand, the four-day stirring of 4a with Pd/C under a H2 atmosphere (5 atm) in an autoclave at room temperature provided compound 6 in 91% yield.
The starting compound 5 was then reacted with commercially available isocyanates comprising p-nitrophenyl isocyanate, p-n-butylphenyl isocyanate, (S)-α-methylbenzyl isocyanate, and (R)-α-methylbenzyl isocyanate. The reactions were carried out at room temperature in anhydrous dichloromethane, and the products 7a–d were isolated in 40–60% yields. Similarly, the propyl-substituted analogues 8a and 8b were obtained from the reaction of 6 with the corresponding isocyanates in 38% and 45% yield, respectively.
The structures of final receptors 7a–d and 8a,b were confirmed by means of HRMS and NMR techniques. Thus, the HRESIMS (positive mode) analysis of 8 showed signals at m/z = 1141.59784 and 1157.57129, which is in a good agreement with the [M + Na]+ (1141.59846 Da) and [M + K]+ (1157.57240 Da) ions predicted for the product. The splitting pattern and multiplicity of the signals in the 1H NMR spectrum fully corresponded to the expected 1,3-alternate conformation (see Supporting Information File 1). Thus, the two doublets with typical ortho substituent coupling constants (J = 7.3 Hz) at 7.64 and 8.14 ppm support the presence of p-nitrophenyl groups. At the same time, the singlets at 8.36 and 9.37 ppm reflected the ureido NH protons (DMSO-d6, 400 MHz, 298 K).
The structures of the selected receptors 7a and 7d were further proven by single crystal X-ray studies. The calixarene 7a crystallised in a monoclinic system, space group C2, and the unit cell contained two receptors with four molecules of DMSO used as the crystallisation solvent (7·2DMSO complex). Both calixarene molecules exhibited an almost ideal square shape of the cavity, with the lengths of the main diagonals being 7.322 Å × 7.122 Å and 7.323 Å × 7.137 Å, respectively. Every ureido group held one molecule of DMSO via synchronous hydrogen bonding interactions between the two NH protons and a sulfoxide oxygen atom (Figure 3a). The S=O···H–N distances were 1.995, 2.285, 2.033, and 2.328 Å, indicating strong interactions in the solid state. At the same time, the carbonyl groups from the neighbouring receptor urea moieties interacted with the C–H bonds of the DMSO methyl group (the C=O···H–C distances were 2.486 and 2.452 Å), thus forming the calixarene dimer with a head-to-tail mutual orientation. The overall supramolecular binding motif was completed by the close contacts between carbonyl oxygen atoms (of the urea group) and S atoms (of DMSO), indicating possible chalcogen interactions [47,48], and the C=O···S=O distances were 3.269 and 3.308 Å (Figure 3a). The molecular packing was further strengthened by the π–π interactions of the p-nitrophenyl moieties, exhibiting several close CAr···CAr contacts at a 3.373 Å distance (Figure 3b).
![[1860-5397-16-249-3]](/bjoc/content/figures/1860-5397-16-249-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray structure of 7a: (a) Hydrogen bonding interactions (black) in a dimeric motif, chalcogen interactions are shown in green. (b) π–π interactions in the dimeric motif.
Figure 3: X-ray structure of 7a: (a) Hydrogen bonding interactions (black) in a dimeric motif, chalcogen inte...
The receptor 7d crystallised in a triclinic system, space group P1, as a 1:3 complex with acetone (used as solvent for crystallisation). The main packing motive (see Figure 4) was represented by an infinite chain of calixarene molecules joined together by intermolecular hydrogen bonds between the ureido groups (the C=O···H–N distances were 2.293 and 2.048 Å).
One of the ureido functions in the macrocycle also held acetone via a C=O···H–N hydrogen bond (2.085 Å) and via a C=O···H–C bond (2.670 Å) from the meta-position of the adjacent aromatic moiety (Figure 4).
![[1860-5397-16-249-4]](/bjoc/content/figures/1860-5397-16-249-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray structure of 7d, showing hydrogen bonds between the ureido units (green) and hydrogen bonding of acetone molecules (black). One alkyl group in each calixarene was removed for better clarity.
Figure 4: X-ray structure of 7d, showing hydrogen bonds between the ureido units (green) and hydrogen bonding...
The complexation ability of the novel receptors towards selected chiral anions was studied using standard 1H NMR titration experiments. There are several reasons why DMSO-d6 was selected as the solvent for the complexation studies: (i) it dissolves all anionic species tested, (ii) prevents the receptor molecule from self-association, and (iii) reduces the complexation constants to values easily measurable by 1H NMR titrations.
A solution of an anion was gradually added to a solution of 7 or 8 in DMSO-d6 to obtain various calixarene/anion ratios of 1:0.3–1:15. Upon the addition of anions in the form of TBA salts, significant downfield shifts of the ureido NH signals were observed, indicating the complexation under fast exchange conditions [49]. The corresponding complexation constants were calculated based on the analysis of the binding isotherms obtained from the complexation-induced chemical shift (CIS) values of urea NH protons or aromatic CH signals (Figure 5) [50-52]. The nonlinear curve-fitting of the experimental data was performed using the freely available software Bindfit [53]. The stoichiometry of the complexes was determined based on the Bindfit output, where the 1:1 model provided the best fit among all tested stoichiometries (1:1, 1:2, and 2:1).
![[1860-5397-16-249-5]](/bjoc/content/figures/1860-5397-16-249-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: 1H NMR titration of 7c with N-acetyl-ᴅ-phenylalaninate and N-acetyl-ʟ-phenylalaninate (as TBA salts, 400 MHz, 298 K, DMSO-d6, the aromatic signal of the calixarene moiety was used).
Figure 5: 1H NMR titration of 7c with N-acetyl-ᴅ-phenylalaninate and N-acetyl-ʟ-phenylalaninate (as TBA salts...
The results summarised in Table 1 and Table 2 revealed that the complexation properties of the receptors do not depend significantly on the lower-rim substitution of the calixarene. Comparing the complexation constants for the otherwise identical receptors (7a vs 8a or 7b vs 8b), it is obvious that the presence of allyl vs propyl substituents does not impose much difference in terms of absolute K values and selectivity.
Table 1: Binding constants of the receptors 7a and 8a towards selected anions (1H NMR titration, 400 MHz, DMSO-d6, 298 K).
| runa | anion | K (M−1) of 7a | sb | K (M−1) of 8a | sb |
| 1 | N-acetyl-ᴅ-phenylalaninate | 279 |
1.18
(for ʟ) |
290 |
1.10
(for ʟ) |
| 2 | N-acetyl-ʟ-phenylalaninate | 330 | 320 | ||
| 3 | N-acetyl-ᴅ-leucinate | 140 |
1.33
(for ʟ) |
150 |
1.46
(for ʟ) |
| 4 | N-acetyl-ʟ-leucinate | 190 | 220 | ||
| 5 | ᴅ-phenylalaninate | 660 |
1.06
(for ʟ) |
580 |
1.05
(for ʟ) |
| 6 | ʟ-phenylalaninate | 700 | 610 | ||
| 7 | ᴅ-leucinate | 480 |
1.02
(for ʟ) |
320 |
1.12
(for ʟ) |
| 8 | ʟ-leucinate | 490 | 360 | ||
| 9 | (R)-mandelate | 260 |
1.03
(for ʟ) |
250 |
1.04
(for ʟ) |
| 10 | (S)-mandelate | 270 | 260 | ||
aRuns 1–10: tetrabutylammonium (TBA) salts. bSelectivity factor: s = KD/KL or KL/KD to obtain s ≥ 1.
Table 2: Binding constants of the receptors 7b and 8b towards selected anions (1H NMR titration, 400 MHz, DMSO-d6, 298 K).
| runa | anion | K (M−1) of 7b | sb | K (M−1) of 8b | sb |
| 11 | N-acetyl-ᴅ-phenylalaninate | 180 |
1.11
(for ʟ) |
200 |
1.10
(for ʟ) |
| 12 | N-acetyl-ʟ-phenylalaninate | 200 | 220 | ||
| 13 | N-acetyl-ᴅ-leucinate | 20 |
2.00
(for ʟ) |
16 |
1.87
(for ʟ) |
| 14 | N-acetyl-ʟ-leucinate | 40 | 30 | ||
| 15 | ᴅ-phenylalaninate | 90 |
1.11
(for ʟ) |
105 |
1.04
(for ʟ) |
| 16 | ʟ-phenylalaninate | 100 | 110 | ||
| 17 | ᴅ-leucinate | 90 |
1.20
(for ʟ) |
95 |
1.10
(for ʟ) |
| 18 | ʟ-leucinate | 108 | 105 | ||
| 19 | (R)-mandelate | 160 |
1.06
(for ʟ) |
140 |
1.14
(for ʟ) |
| 20 | (S)-mandelate | 170 | 160 | ||
aRuns 11–20: TBA salts. bSelectivity factor: s = KD/KL or KL/KD to obtain s ≥ 1.
As expected, the nitro-substituted receptors 7a and 8a exhibited complexation constants higher than the butyl-substituted analogues 7b and 8b for all anions – compare 7a (Table 1, run 5, K = 660 M−1) vs 7b (Table 2, run 15, K = 90 M−1). On the other hand, despite the differences in the K values, the enantioselectivity remained almost the same in both receptor series. Thus, the selectivity factor s = KL/KD for phenylalaninate was 1.06 for 7a and 1.11 for 7b, and similar values were also obtained for 8a (1.05) and 8b (1.04), using the ʟ-enantiomer in each case. The highest chiral discrimination (for an ʟ-enantiomer) was achieved for N-acetyl-ʟ-leucinate, with a selectivity factor of s = 2.0 and 1.87 for 7b and 8b, respectively.
The above-mentioned results indicate that the binding cavity composed of two preorganised ureido groups and chiral alkyl substituents in the near proximity possesses some ability of enantioselective recognition. In fact, this assumption was already established by our previous receptors C1 and C2 (Figure 1) [44,45], although the direct comparison with our novel results is rather difficult due to the different types of derivatives (C1 has a cavity without tert-butyl groups, and C2 is based on thiacalix[4]arene). Nevertheless, the simultaneous introduction of chiral isocyanate to form chiral urea moieties has not been accomplished yet.
The formation of another chiral centre within the derivatives 7c and 7d led to the expected decrease [42] of the complexation constant values (by approximately one order of magnitude) due to the presence of alkyl instead of aryl urea receptors (Table 3). Thus, going from 7a to 7c, the complexation constant K for ᴅ-leucinate decreased from 480 to 50 (run 7, Table 1 vs run 27, Table 3), and the appropriate values for ᴅ-phenylalaninate are 660 vs 40 (run 5, Table 1 vs run 25, Table 3).
Table 3: Binding constants of the receptors 7c and 7d towards selected anions (1H NMR titration, 400 MHz, DMSO-d6, 298 K).
| runa | anion | K (M−1) of 7c | sb | K (M−1) of 7d | sb) |
| 21 | N-acetyl-ᴅ-phenylalaninate | 15 |
3.33
(for ʟ) |
35 |
1.75
(for ᴅ) |
| 22 | N-acetyl-ʟ-phenylalaninate | 50 | 20 | ||
| 23 | N-acetyl-ᴅ-leucinate | 20 |
2.50
(for ʟ) |
31 |
1.34
(for ᴅ) |
| 24 | N-acetyl-ʟ-leucinate | 50 | 23 | ||
| 25 | ᴅ-phenylalaninate | 40 |
1.37
(for ʟ) |
25 |
1.28
(for ʟ) |
| 26 | ʟ-phenylalaninate | 55 | 32 | ||
| 27 | ᴅ-leucinate | 50 |
1.20
(for ʟ) |
27 |
1.11
(for ʟ) |
| 28 | ʟ-leucinate | 60 | 30 | ||
| 29 | (R)-mandelate | 20 |
1.75
(for ʟ) |
11 |
1.37
(for ʟ) |
| 30 | (S)-mandelate | 35 | 8 | ||
aRuns 21–30: TBA salts. bSelectivity factor: s = KD/KL or KL/KD to obtain s ≥ 1.
On the other hand, the chiral recognition properties of the receptors 7c and 7d are accentuated compared to 7a and 8a or 7b and 8b. The introduction of another chiral centre leads to higher selectivity factors in almost all the cases. The stereodiscrimination of 7c (bearing an (S)-α-methylbenzyl moiety on the urea group) for N-acetylphenylalaninates represents the maximum value achieved (s = 3.33 for ʟ). Interestingly, the diastereomeric isomer 7d, possessing an (R)-chiral centre, prefers the ᴅ-isomer, with a selectivity factor of s = 1.75, and the same holds for N-acetyl-ᴅ-leucinate (s = 1.34). These results indicate that both chiral moieties (the alkyl group on the calixarene and the chiral centre on the urea moiety) function synergistically, and a proper choice of both substituents can lead to an even better stereoselectivity.
Conclusion
In conclusion, the introduction of chiral alkyl groups into the lower rim of calix[4]arene immobilised in the 1,3-alternate conformation resulted in a macrocycle with a preorganised ureido cavity bearing chiral alkyl substituents in the near proximity. As shown by 1H NMR titration experiments, these compounds function as receptors for chiral anions in DMSO-d6. The chiral recognition ability can further be strengthened by the introduction of another chiral moiety directly to the urea nitrogen atoms. The systems with double chiral units located around the binding ureido cavity showed a better stereodiscrimination, with the highest selectivity factor being 3.33 (for ʟ) achieved for N-acetylphenylalaninate.
Supporting Information
| Supporting Information File 1: Experimental details and characterisation data (including X-ray data for 4a, 7a, and 7d, NMR, IR, and HRMS) as well as NMR titration data. | ||
| Format: PDF | Size: 6.8 MB | Download |
References
-
Molina, P.; Zapata, F.; Caballero, A. Chem. Rev. 2017, 117, 9907–9972. doi:10.1021/acs.chemrev.6b00814
Return to citation in text: [1] -
Eytel, L. M.; Fargher, H. A.; Haley, M. M.; Johnson, D. W. Chem. Commun. 2019, 55, 5195–5206. doi:10.1039/c9cc01460h
Return to citation in text: [1] -
Jia, C.; Zuo, W.; Zhang, D.; Yang, X.-J.; Wu, B. Chem. Commun. 2016, 52, 9614–9627. doi:10.1039/c6cc03761e
Return to citation in text: [1] -
Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P. A. Chem. Rev. 2015, 115, 8038–8155. doi:10.1021/acs.chemrev.5b00099
Return to citation in text: [1] -
Zhao, J.; Yang, D.; Yang, X.-J.; Wu, B. Coord. Chem. Rev. 2019, 378, 415–444. doi:10.1016/j.ccr.2018.01.002
Return to citation in text: [1] -
Cai, J.; Sessler, J. L. Chem. Soc. Rev. 2014, 43, 6198–6213. doi:10.1039/c4cs00115j
Return to citation in text: [1] -
Bowman-James, K.; Bianchi, A.; García-España, E., Eds. Anion Coordination Chemistry; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527639502
Return to citation in text: [1] -
Anion Recognition in Supramolecular Chemistry. Gale, P. A.; Dehaen, W., Eds.; Topics in Heterocyclic Chemistry, Vol. 24; Springer Berlin: Berlin, Germany, 2010. doi:10.1007/978-3-642-15444-7
Return to citation in text: [1] -
Recognition of Anions. In Structure and Bonding; Vilar, R., Ed.; Springer: Berlin, Heidelberg, Germany, 2008; Vol. 129. doi:10.1007/978-3-540-79092-1
Return to citation in text: [1] -
Sessler, J. L.; Gale, P. A.; Cho, W. S. In Anion Receptor Chemistry; Stoddart, J. F., Ed.; RSC Publishing: Cambridge, U.K., 2006. doi:10.1039/9781847552471
Return to citation in text: [1] -
Stibor, I., Ed. Anion Sensing; Topics in Current Chemistry, Vol. 255; Springer: Berlin, Heidelberg, Germany, 2005. doi:10.1007/b101055
Return to citation in text: [1] -
Ngo, H. T.; Liu, X.; Jolliffe, K. A. Chem. Soc. Rev. 2012, 41, 4928–4965. doi:10.1039/c2cs35087d
Return to citation in text: [1] -
Kubik, S. Chem. Soc. Rev. 2010, 39, 3648–3663. doi:10.1039/b926166b
Return to citation in text: [1] -
Hein, R.; Beer, P. D.; Davis, J. J. Chem. Rev. 2020, 120, 1888–1935. doi:10.1021/acs.chemrev.9b00624
Return to citation in text: [1] -
Custelcean, R. Chem. Commun. 2020, 56, 10272–10280. doi:10.1039/d0cc04332j
Return to citation in text: [1] -
White, N. G. Dalton Trans. 2019, 48, 7062–7068. doi:10.1039/c8dt05030a
Return to citation in text: [1] -
Mateus, P.; Lima, L. M. P.; Delgado, R. Polyhedron 2013, 52, 25–42. doi:10.1016/j.poly.2012.07.073
Return to citation in text: [1] -
Llinares, J. M.; Powell, D.; Bowman-James, K. Coord. Chem. Rev. 2003, 240, 57–75. doi:10.1016/s0010-8545(03)00019-5
Return to citation in text: [1] -
Dey, S. K.; Basu, A.; Chutia, R.; Das, G. RSC Adv. 2016, 6, 26568–26589. doi:10.1039/c6ra00268d
Return to citation in text: [1] -
Blažek Bregović, V.; Basarić, N.; Mlinarić-Majerski, K. Coord. Chem. Rev. 2015, 295, 80–124. doi:10.1016/j.ccr.2015.03.011
Return to citation in text: [1] -
Dydio, P.; Lichosyt, D.; Jurczak, J. Chem. Soc. Rev. 2011, 40, 2971–2985. doi:10.1039/c1cs15006e
Return to citation in text: [1] -
Neri, P.; Sessler, J. L.; Wang, M.-X., Eds. Calixarenes and Beyond; Springer International Publishing: Basel, Switzerland, 2016. doi:10.1007/978-3-319-31867-7
Return to citation in text: [1] -
Gutsche, C. D. Calixarenes: An Introduction; RSC Publishing: Cambridge, U.K., 2008. doi:10.1039/9781847558190
Return to citation in text: [1] -
Asfari, Z.; Böhmer, V.; Harrowfield, J.; Vicens, J.; Saadioui, M., Eds. Calixarenes 2001; Kluwer Academic Publishers: Dordrecht, Netherlands, 2002. doi:10.1007/0-306-47522-7
Return to citation in text: [1] -
Vicens, J.; Harrowfield, J.; Backlouti, L., Eds. Calixarenes in the Nanoworld; Springer: Dordrecht, Netherlands, 2007. doi:10.1007/978-1-4020-5022-4
Return to citation in text: [1] -
Mandolini, L.; Ungaro, R., Eds. Calixarenes in Action; Imperial College Press: London, U.K., 2000. doi:10.1142/p168
Return to citation in text: [1] -
Edwards, N. Y.; Possanza, A. L. Supramol. Chem. 2013, 25, 446–463. doi:10.1080/10610278.2013.794277
Return to citation in text: [1] -
Joseph, R.; Rao, C. P. Chem. Rev. 2011, 111, 4658–4702. doi:10.1021/cr1004524
Return to citation in text: [1] -
Shokova, E. A.; Kovalev, V. V. Russ. J. Org. Chem. 2009, 45, 1275–1314. doi:10.1134/s1070428009090012
Return to citation in text: [1] -
Matthews, S. E.; Beer, P. D. Supramol. Chem. 2005, 17, 411–435. doi:10.1080/10610270500127089
Return to citation in text: [1] -
Van Rossom, W.; Caers, J.; Robeyns, K.; Van Meervelt, L.; Maes, W.; Dehaen, W. J. Org. Chem. 2012, 77, 2791–2797. doi:10.1021/jo300004p
Return to citation in text: [1] -
Rudzevich, Y.; Cao, Y.; Rudzevich, V.; Böhmer, V. Chem. – Eur. J. 2008, 14, 3346–3354. doi:10.1002/chem.200701694
Return to citation in text: [1] -
Vysotsky, M. O.; Bolte, M.; Thondorf, I.; Böhmer, V. Chem. – Eur. J. 2003, 9, 3375–3382. doi:10.1002/chem.200304912
Return to citation in text: [1] -
Pelizzi, N.; Casnati, A.; Friggeri, A.; Ungaro, R. J. Chem. Soc., Perkin Trans. 2 1998, 1307–1312. doi:10.1039/a801762j
Return to citation in text: [1] -
Staffilani, M.; Hancock, K. S. B.; Steed, J. W.; Holman, K. T.; Atwood, J. L.; Juneja, R. K.; Burkhalter, R. S. J. Am. Chem. Soc. 1997, 119, 6324–6335. doi:10.1021/ja9702172
Return to citation in text: [1] -
Scheerder, J.; Vreekamp, R. H.; Engbersen, J. F. J.; Verboom, W.; van Duynhoven, J. P. M.; Reinhoudt, D. N. J. Org. Chem. 1996, 61, 3476–3481. doi:10.1021/jo9600262
Return to citation in text: [1] -
Rincón, A. M.; Prados, P.; de Mendoza, J. J. Am. Chem. Soc. 2001, 123, 3493–3498. doi:10.1021/ja0036054
Return to citation in text: [1] -
Cho, Y. L.; Rudkevich, D. M.; Rebek, J., Jr. J. Am. Chem. Soc. 2000, 122, 9868–9869. doi:10.1021/ja002345n
Return to citation in text: [1] -
Brody, M. S.; Schalley, C. A.; Rudkevich, D. M.; Rebek, J., Jr. Angew. Chem., Int. Ed. 1999, 38, 1640–1644. doi:10.1002/(sici)1521-3773(19990601)38:11<1640::aid-anie1640>3.0.co;2-y
Return to citation in text: [1] -
Řezanková, M.; Budka, J.; Mikšátko, J.; Eigner, V.; Císařová, I.; Cuřínová, P.; Lhoták, P. Tetrahedron 2017, 73, 742–749. doi:10.1016/j.tet.2016.12.054
Return to citation in text: [1] -
Klejch, T.; Slavíček, J.; Hudeček, O.; Eigner, V.; Gutierrez, N. A.; Cuřínová, P.; Lhoták, P. New J. Chem. 2016, 40, 7935–7942. doi:10.1039/c6nj01271j
Return to citation in text: [1] -
Stibor, I.; Budka, J.; Michlová, V.; Tkadlecová, M.; Pojarová, M.; Cuřínová, P.; Lhoták, P. New J. Chem. 2008, 32, 1597–1607. doi:10.1039/b802871k
Return to citation in text: [1] [2] -
Cuřínová, P.; Stibor, I.; Budka, J.; Sýkora, J.; Lang, K.; Lhoták, P. New J. Chem. 2009, 33, 612–619. doi:10.1039/b816790g
Return to citation in text: [1] [2] -
Mačková, M.; Mikšátko, J.; Budka, J.; Eigner, V.; Cuřínová, P.; Lhoták, P. New J. Chem. 2015, 39, 1382–1389. doi:10.1039/c4nj01956c
Return to citation in text: [1] [2] [3] -
Botha, F.; Budka, J.; Eigner, V.; Hudeček, O.; Vrzal, L.; Císařová, I.; Lhoták, P. Tetrahedron 2014, 70, 477–483. doi:10.1016/j.tet.2013.11.030
Return to citation in text: [1] [2] [3] [4] -
Danila, C.; Bolte, M.; Böhmer, V. Org. Biomol. Chem. 2005, 3, 172–184. doi:10.1039/b414173c
Return to citation in text: [1] -
Gleiter, R.; Haberhauer, G.; Werz, D. B.; Rominger, F.; Bleiholder, C. Chem. Rev. 2018, 118, 2010–2041. doi:10.1021/acs.chemrev.7b00449
Return to citation in text: [1] -
Vogel, L.; Wonner, P.; Huber, S. M. Angew. Chem., Int. Ed. 2019, 58, 1880–1891. doi:10.1002/anie.201809432
Return to citation in text: [1] -
Depending on the anion used, the complexation-induced chemical shift (CIS) values of the NH protons were up to 500 Hz, while at the same time, the maximum CIS values for aromatic CH signals of the calixarene skeleton were less than 80 Hz. Unfortunately, in most cases, the signals of the NH protons became extremely diffused and finally almost invisible upon the addition of an anion.
Return to citation in text: [1] -
Hibbert, D. B.; Thordarson, P. Chem. Commun. 2016, 52, 12792–12805. doi:10.1039/c6cc03888c
Return to citation in text: [1] -
Ulatowski, F.; Dąbrowa, K.; Bałakier, T.; Jurczak, J. J. Org. Chem. 2016, 81, 1746–1756. doi:10.1021/acs.joc.5b02909
Return to citation in text: [1] -
Thordarson, P. Chem. Soc. Rev. 2011, 40, 1305–1323. doi:10.1039/c0cs00062k
Return to citation in text: [1] -
The binding constants were calculated using the Bindfit application, freely available at http://supramolecular.org.
Return to citation in text: [1]
| 53. | The binding constants were calculated using the Bindfit application, freely available at http://supramolecular.org. |
| 49. | Depending on the anion used, the complexation-induced chemical shift (CIS) values of the NH protons were up to 500 Hz, while at the same time, the maximum CIS values for aromatic CH signals of the calixarene skeleton were less than 80 Hz. Unfortunately, in most cases, the signals of the NH protons became extremely diffused and finally almost invisible upon the addition of an anion. |
| 50. | Hibbert, D. B.; Thordarson, P. Chem. Commun. 2016, 52, 12792–12805. doi:10.1039/c6cc03888c |
| 51. | Ulatowski, F.; Dąbrowa, K.; Bałakier, T.; Jurczak, J. J. Org. Chem. 2016, 81, 1746–1756. doi:10.1021/acs.joc.5b02909 |
| 52. | Thordarson, P. Chem. Soc. Rev. 2011, 40, 1305–1323. doi:10.1039/c0cs00062k |
| 1. | Molina, P.; Zapata, F.; Caballero, A. Chem. Rev. 2017, 117, 9907–9972. doi:10.1021/acs.chemrev.6b00814 |
| 2. | Eytel, L. M.; Fargher, H. A.; Haley, M. M.; Johnson, D. W. Chem. Commun. 2019, 55, 5195–5206. doi:10.1039/c9cc01460h |
| 3. | Jia, C.; Zuo, W.; Zhang, D.; Yang, X.-J.; Wu, B. Chem. Commun. 2016, 52, 9614–9627. doi:10.1039/c6cc03761e |
| 4. | Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P. A. Chem. Rev. 2015, 115, 8038–8155. doi:10.1021/acs.chemrev.5b00099 |
| 5. | Zhao, J.; Yang, D.; Yang, X.-J.; Wu, B. Coord. Chem. Rev. 2019, 378, 415–444. doi:10.1016/j.ccr.2018.01.002 |
| 6. | Cai, J.; Sessler, J. L. Chem. Soc. Rev. 2014, 43, 6198–6213. doi:10.1039/c4cs00115j |
| 19. | Dey, S. K.; Basu, A.; Chutia, R.; Das, G. RSC Adv. 2016, 6, 26568–26589. doi:10.1039/c6ra00268d |
| 20. | Blažek Bregović, V.; Basarić, N.; Mlinarić-Majerski, K. Coord. Chem. Rev. 2015, 295, 80–124. doi:10.1016/j.ccr.2015.03.011 |
| 21. | Dydio, P.; Lichosyt, D.; Jurczak, J. Chem. Soc. Rev. 2011, 40, 2971–2985. doi:10.1039/c1cs15006e |
| 46. | Danila, C.; Bolte, M.; Böhmer, V. Org. Biomol. Chem. 2005, 3, 172–184. doi:10.1039/b414173c |
| 15. | Custelcean, R. Chem. Commun. 2020, 56, 10272–10280. doi:10.1039/d0cc04332j |
| 16. | White, N. G. Dalton Trans. 2019, 48, 7062–7068. doi:10.1039/c8dt05030a |
| 17. | Mateus, P.; Lima, L. M. P.; Delgado, R. Polyhedron 2013, 52, 25–42. doi:10.1016/j.poly.2012.07.073 |
| 18. | Llinares, J. M.; Powell, D.; Bowman-James, K. Coord. Chem. Rev. 2003, 240, 57–75. doi:10.1016/s0010-8545(03)00019-5 |
| 47. | Gleiter, R.; Haberhauer, G.; Werz, D. B.; Rominger, F.; Bleiholder, C. Chem. Rev. 2018, 118, 2010–2041. doi:10.1021/acs.chemrev.7b00449 |
| 48. | Vogel, L.; Wonner, P.; Huber, S. M. Angew. Chem., Int. Ed. 2019, 58, 1880–1891. doi:10.1002/anie.201809432 |
| 12. | Ngo, H. T.; Liu, X.; Jolliffe, K. A. Chem. Soc. Rev. 2012, 41, 4928–4965. doi:10.1039/c2cs35087d |
| 13. | Kubik, S. Chem. Soc. Rev. 2010, 39, 3648–3663. doi:10.1039/b926166b |
| 14. | Hein, R.; Beer, P. D.; Davis, J. J. Chem. Rev. 2020, 120, 1888–1935. doi:10.1021/acs.chemrev.9b00624 |
| 45. | Botha, F.; Budka, J.; Eigner, V.; Hudeček, O.; Vrzal, L.; Císařová, I.; Lhoták, P. Tetrahedron 2014, 70, 477–483. doi:10.1016/j.tet.2013.11.030 |
| 7. | Bowman-James, K.; Bianchi, A.; García-España, E., Eds. Anion Coordination Chemistry; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527639502 |
| 8. | Anion Recognition in Supramolecular Chemistry. Gale, P. A.; Dehaen, W., Eds.; Topics in Heterocyclic Chemistry, Vol. 24; Springer Berlin: Berlin, Germany, 2010. doi:10.1007/978-3-642-15444-7 |
| 9. | Recognition of Anions. In Structure and Bonding; Vilar, R., Ed.; Springer: Berlin, Heidelberg, Germany, 2008; Vol. 129. doi:10.1007/978-3-540-79092-1 |
| 10. | Sessler, J. L.; Gale, P. A.; Cho, W. S. In Anion Receptor Chemistry; Stoddart, J. F., Ed.; RSC Publishing: Cambridge, U.K., 2006. doi:10.1039/9781847552471 |
| 11. | Stibor, I., Ed. Anion Sensing; Topics in Current Chemistry, Vol. 255; Springer: Berlin, Heidelberg, Germany, 2005. doi:10.1007/b101055 |
| 43. | Cuřínová, P.; Stibor, I.; Budka, J.; Sýkora, J.; Lang, K.; Lhoták, P. New J. Chem. 2009, 33, 612–619. doi:10.1039/b816790g |
| 45. | Botha, F.; Budka, J.; Eigner, V.; Hudeček, O.; Vrzal, L.; Císařová, I.; Lhoták, P. Tetrahedron 2014, 70, 477–483. doi:10.1016/j.tet.2013.11.030 |
| 40. | Řezanková, M.; Budka, J.; Mikšátko, J.; Eigner, V.; Císařová, I.; Cuřínová, P.; Lhoták, P. Tetrahedron 2017, 73, 742–749. doi:10.1016/j.tet.2016.12.054 |
| 41. | Klejch, T.; Slavíček, J.; Hudeček, O.; Eigner, V.; Gutierrez, N. A.; Cuřínová, P.; Lhoták, P. New J. Chem. 2016, 40, 7935–7942. doi:10.1039/c6nj01271j |
| 44. | Mačková, M.; Mikšátko, J.; Budka, J.; Eigner, V.; Cuřínová, P.; Lhoták, P. New J. Chem. 2015, 39, 1382–1389. doi:10.1039/c4nj01956c |
| 45. | Botha, F.; Budka, J.; Eigner, V.; Hudeček, O.; Vrzal, L.; Císařová, I.; Lhoták, P. Tetrahedron 2014, 70, 477–483. doi:10.1016/j.tet.2013.11.030 |
| 27. | Edwards, N. Y.; Possanza, A. L. Supramol. Chem. 2013, 25, 446–463. doi:10.1080/10610278.2013.794277 |
| 28. | Joseph, R.; Rao, C. P. Chem. Rev. 2011, 111, 4658–4702. doi:10.1021/cr1004524 |
| 29. | Shokova, E. A.; Kovalev, V. V. Russ. J. Org. Chem. 2009, 45, 1275–1314. doi:10.1134/s1070428009090012 |
| 30. | Matthews, S. E.; Beer, P. D. Supramol. Chem. 2005, 17, 411–435. doi:10.1080/10610270500127089 |
| 31. | Van Rossom, W.; Caers, J.; Robeyns, K.; Van Meervelt, L.; Maes, W.; Dehaen, W. J. Org. Chem. 2012, 77, 2791–2797. doi:10.1021/jo300004p |
| 32. | Rudzevich, Y.; Cao, Y.; Rudzevich, V.; Böhmer, V. Chem. – Eur. J. 2008, 14, 3346–3354. doi:10.1002/chem.200701694 |
| 33. | Vysotsky, M. O.; Bolte, M.; Thondorf, I.; Böhmer, V. Chem. – Eur. J. 2003, 9, 3375–3382. doi:10.1002/chem.200304912 |
| 34. | Pelizzi, N.; Casnati, A.; Friggeri, A.; Ungaro, R. J. Chem. Soc., Perkin Trans. 2 1998, 1307–1312. doi:10.1039/a801762j |
| 35. | Staffilani, M.; Hancock, K. S. B.; Steed, J. W.; Holman, K. T.; Atwood, J. L.; Juneja, R. K.; Burkhalter, R. S. J. Am. Chem. Soc. 1997, 119, 6324–6335. doi:10.1021/ja9702172 |
| 36. | Scheerder, J.; Vreekamp, R. H.; Engbersen, J. F. J.; Verboom, W.; van Duynhoven, J. P. M.; Reinhoudt, D. N. J. Org. Chem. 1996, 61, 3476–3481. doi:10.1021/jo9600262 |
| 37. | Rincón, A. M.; Prados, P.; de Mendoza, J. J. Am. Chem. Soc. 2001, 123, 3493–3498. doi:10.1021/ja0036054 |
| 38. | Cho, Y. L.; Rudkevich, D. M.; Rebek, J., Jr. J. Am. Chem. Soc. 2000, 122, 9868–9869. doi:10.1021/ja002345n |
| 39. | Brody, M. S.; Schalley, C. A.; Rudkevich, D. M.; Rebek, J., Jr. Angew. Chem., Int. Ed. 1999, 38, 1640–1644. doi:10.1002/(sici)1521-3773(19990601)38:11<1640::aid-anie1640>3.0.co;2-y |
| 44. | Mačková, M.; Mikšátko, J.; Budka, J.; Eigner, V.; Cuřínová, P.; Lhoták, P. New J. Chem. 2015, 39, 1382–1389. doi:10.1039/c4nj01956c |
| 25. | Vicens, J.; Harrowfield, J.; Backlouti, L., Eds. Calixarenes in the Nanoworld; Springer: Dordrecht, Netherlands, 2007. doi:10.1007/978-1-4020-5022-4 |
| 26. | Mandolini, L.; Ungaro, R., Eds. Calixarenes in Action; Imperial College Press: London, U.K., 2000. doi:10.1142/p168 |
| 44. | Mačková, M.; Mikšátko, J.; Budka, J.; Eigner, V.; Cuřínová, P.; Lhoták, P. New J. Chem. 2015, 39, 1382–1389. doi:10.1039/c4nj01956c |
| 45. | Botha, F.; Budka, J.; Eigner, V.; Hudeček, O.; Vrzal, L.; Císařová, I.; Lhoták, P. Tetrahedron 2014, 70, 477–483. doi:10.1016/j.tet.2013.11.030 |
| 22. | Neri, P.; Sessler, J. L.; Wang, M.-X., Eds. Calixarenes and Beyond; Springer International Publishing: Basel, Switzerland, 2016. doi:10.1007/978-3-319-31867-7 |
| 23. | Gutsche, C. D. Calixarenes: An Introduction; RSC Publishing: Cambridge, U.K., 2008. doi:10.1039/9781847558190 |
| 24. | Asfari, Z.; Böhmer, V.; Harrowfield, J.; Vicens, J.; Saadioui, M., Eds. Calixarenes 2001; Kluwer Academic Publishers: Dordrecht, Netherlands, 2002. doi:10.1007/0-306-47522-7 |
| 42. | Stibor, I.; Budka, J.; Michlová, V.; Tkadlecová, M.; Pojarová, M.; Cuřínová, P.; Lhoták, P. New J. Chem. 2008, 32, 1597–1607. doi:10.1039/b802871k |
| 43. | Cuřínová, P.; Stibor, I.; Budka, J.; Sýkora, J.; Lang, K.; Lhoták, P. New J. Chem. 2009, 33, 612–619. doi:10.1039/b816790g |
| 42. | Stibor, I.; Budka, J.; Michlová, V.; Tkadlecová, M.; Pojarová, M.; Cuřínová, P.; Lhoták, P. New J. Chem. 2008, 32, 1597–1607. doi:10.1039/b802871k |
© 2020 Horáčková et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)