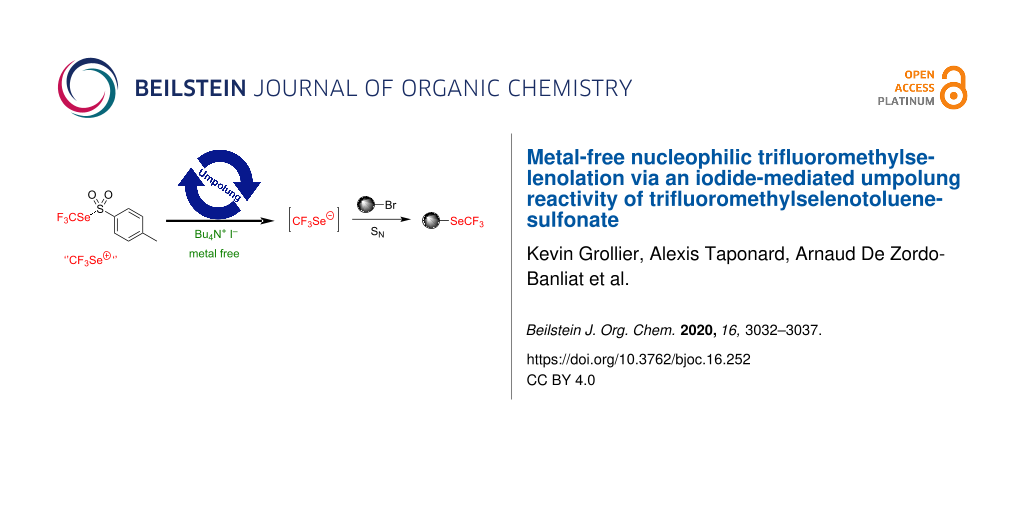
Abstract
We report herein a practical method to generate CF3Se− (and RFSe−) anions from shelf-stable reagents under iodide activation. Metal-free nucleophilic trifluoromethylselenolations have been then performed with this in situ-generated anion. Perfluoroalkylselenolations have also been described.

Graphical Abstract
Introduction
Because of the peculiar properties of the fluorine atom, fluorinated compounds gained a growing interest over the last decades and found applications in a large panel of fields from materials to life sciences [1-15]. Fluorinated motifs bring to molecules specific and often unique electronic and physicochemical characteristics. In order to design new substrates with targeted properties, a modulation of the properties of the introduced substituents became fundamental. In this context, the development of innovative fluorinated groups recently emerged, in particular by combining heteroatoms, such as chalcogens, and fluorinated moieties [16].
Despite, the negative reputation of selenium due to its toxicity at high doses, it is an essential trace element for human physiology and biochemistry [17-20]. Furthermore, selenolated compounds found valuable applications in materials [21-23], life sciences [19,20,24-29], and drug design [30-33]. Consequently, the merging of fluorinated moieties, such as CF3 with selenium could constitute an interesting motif in the design of new molecules, in particular in medicinal chemistry or agrochemistry. Even if, to date, there are no CF3Se-containing pharmaceuticals registered [15], a recent work has demonstrated the promising development of trifluoromethylselenolated nonsteroidal anti-inflammatory drugs as potential anticancer drugs [34].
Over the last years, trifluoromethylselenolation reactions have gained a rising infatuation but, despite this recent interest, methods to introduce the CF3Se group into organic substrates remain limited [35,36].
One of the simplest ways to achieve trifluoromethylselenolated compounds is the direct nucleophilic substitution of suitable leaving groups to form the CF3Se–C(sp3) bond. This chemistry is the prerogative of the CF3Se− anion (Scheme 1) [37-40]. However, the formation of this selenium species requires the tedious use of red elemental selenium [41] and also suffers from stability issues. To circumvent these drawbacks, a copper complex has been developed, but in this case the use of a stoichiometric amount of the metal is required [37-39].
![[1860-5397-16-252-i1]](/bjoc/content/inline/1860-5397-16-252-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: State of the art concerning the direct nucleophilic trifluoromethylselenolation.
Scheme 1: State of the art concerning the direct nucleophilic trifluoromethylselenolation.
Only a few years ago, trifluoromethylselenotoluenesulfonate (1a) has been developed as an efficient reagent to perform electrophilic or radical trifluoromethylselenolations [42-47]. Very recently, we demonstrated that under reductive conditions, such compounds succeeded to perform nucleophilic substitutions [48]. In this reaction, the CF3Se− anion was in situ generated by reduction through a double electron transfer of 1a with TDAE (tetrakis(dimethylamino)ethylene). Even though this umpolung strategy is efficient, the use of the sensitive TDAE, a strong reducing agent, could constitute a drawback for some applications. Consequently, we decided to develop a new umpolung method in non-reductive conditions.
Results and Discussion
A few years ago, we have demonstrated that trifluoromethanesulfenamides, electrophilic trifluoromethylthiolation reagents, could also perform nucleophilic trifluoromethylthiolations through the transient formation of a CF3SI species which presented an inverted polarity [49,50]. Based on a similar approach, we hypothesized that the CF3SeI species could also possess the CF3Seδ−–Iδ+ inverted polarity. Thus, based on the previously developed conditions, reagent 1a was reacted with benzyl bromide (2a) in the presence of tetrabutylammonium iodide (TBAI) in acetone at 40 °C (Table 1, entry 1).
Table 1: Reaction between 1a and 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-252-i4.svg?max-width=637&scale=1.0)
|
|||||||
| entry |
1a
(equiv) |
TBAI
(equiv) |
2a
(equiv) |
solvent |
T
(°C) |
t
(h) |
3a
(%)a |
| 1 | 1 | 2 | 1 | acetone | 40 | 15 | 52 |
| 2 | 1 | 2 | 1 | CH3CN | 40 | 15 | 54 |
| 3 | 1 | 2 | 1 | THF | 40 | 15 | 61 |
| 4 | 1 | 2 | 1 | THF | 40 | 4 | 62 |
| 5 | 1 | 2 | 1 | THF | 50 | 4 | 59 |
| 6 | 1 | 2 | 1 | THF | 60 | 4 | 54 |
| 7 | 1 | 2 | 1 | THF | 25 | 4 | 47 |
| 8 | 1.5 | 3 | 1 | THF | 40 | 4 | 55 |
| 9 | 1 | 2 | 2 | THF | 40 | 4 | 89 |
aYields determined by 19F NMR spectroscopy with PhOCF3 as an internal standard.
The observed result was moderate (Table 1, entry 1). Other solvents, which led also to satisfactory yields in the “sulfur series” were then tested. Acetonitrile did not improve the yield, however, a better one was obtained in THF (Table 1, entries 2 and 3). Interestingly, a shorter reaction time (4 h instead of 15 h) provided similar results (Table 1, entries 3 and 4). At higher temperatures, the results were not improved and even a slight decrease of the yield was observed, possibly due to an increased degradation of CF3SeI or CF3Se− (Table 1, entries 5 and 6). A lower temperature also decreased the yield (Table 1, entry 7). With an excess of reagents 1a and TBAI, leading to an excess of the CF3Se− species, no significant improvement was observed (Table 1, entry 8). On contrary, the use of 2 equivalents of the electrophile 2a, to ameliorate the CF3Se− anion trapping, had a significant effect and gave a very good yield of the product (Table 1, entry 9). With the optimal conditions in hand, the reaction was exemplified with various other electrophiles (Scheme 2).
![[1860-5397-16-252-i2]](/bjoc/content/inline/1860-5397-16-252-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The nucleophilic fluoroalkylselenolation of alkyl bromides. Yields were determined by 19F NMR spectroscopy with PhOCF3 as an internal standard and yields of isolated products are shown in parentheses. aWith 1 equiv of electrophile 2. bStarting from propargyl chloride.
Scheme 2: The nucleophilic fluoroalkylselenolation of alkyl bromides. Yields were determined by 19F NMR spect...
The reaction gave generally good results with reactive electrophiles such as benzylic, allylic or propargylic ones (3a–k). Noteworthy, in the reaction with 2-(bromomethyl)pyridine (2g) only 1 equivalent was required, maybe due to a higher reactivity. Furthermore, the reaction seems to be very sensitive to steric hindrance as illustrated by the low yield obtained for 3b. In contrast, in the aliphatic series, only low yields were observed (3m,n) except for the activated α-bromo acetophenone (3l). This led us to suppose that CF3Se− might be a poor nucleophile, which is confirmed by the medium yield observed with the propargylic substrate 3k, where the chloride starting material was used instead of the bromide as for the other compounds. Noteworthy, because of the volatility of the obtained products, the isolated yields were sometimes significantly below the NMR yields.
Higher fluorinated homologs of 1a were also synthesized. Consequently, an extension of this method was considered with pentafluoroethylated and tridecafluorohexylated reagents 1b and 1c. Good yields were obtained, in particular for 5a which constitutes, to the best of our knowledge, the first example of a direct nucleophilic tridecafluorohexylselenolation.
From a mechanistic point of view, a pathway inspired by the reaction described with trifluoromethanesulfenamides was postulated (Scheme 3) [49].
![[1860-5397-16-252-i3]](/bjoc/content/inline/1860-5397-16-252-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The first equivalent of iodide (from TBAI) reacts with the reagent 1a to produce the transient species CF3SeI with an inverted polarity on the selenium atom. This compound then undergoes the attack of the second equivalent of iodide to generate the CF3Se− anion with releasing of I2. Finally, the nucleophilic CF3Se− can substitute the leaving group onto the electrophilic substrate 2. However, because of the release of I2, as side reaction the oxidation of the CF3Se− anion can be also envisaged. This was confirmed by the formation of 25–30% of CF3SeSeCF3 when 1 equivalent of 2a was used (Table 1, entries 3 and 4). Consequently, the nucleophilic substitution is in competition with this relatively fast oxidation. By adding an excess of the electrophile 2, the substitution is favored detrimentally to the oxidation. Nevertheless, with weaker or hindered electrophiles, the oxidation reaction is favored compared to the slower substitution. This is adequate with the observed results. Noteworthy, the supposed formation of I2 was strengthened by the appearance of a red-brown color of the reaction media which faded after the addition of sodium thiosulfate. As demonstrated in the sulfur series [49], the in situ formation of an alkyl iodide from 2, through a Finkelstein reaction, can be also envisaged. This would not impact the reaction pathway since the released bromide can also activate the CF3SeI species to provide the expected CF3Se− anion.
Conclusion
To conclude, trifluoromethylselenotoluenesulfonate confirmed to be a versatile reagent able to perform electrophilic, radical or nucleophilic reactions depending on the conditions. The iodide-mediated, metal-free method is complementary to the previous one using TDAE. Thus, the umpolung reactivity of trifluoromethylselenotoluenesulfonate can be performed under reductive or oxidative conditions. Furthermore, this method was extended to higher fluorinated homologs allowing the first nucleophilic tridecafluorohexylselenolation.
Experimental
Typical procedure: In a 10 mL flame-dried flask tube equipped with a magnetic stirring bar was added 1a–c (0.2 mmol, 1 equiv) followed by 0.4 mL of dry THF. Then, compound 2a–n (0.4 mmol, 2 equiv) was added followed by TBAI (0.4 mmol, 2 equiv). The tube is then sealed and the reaction mixture stirred at 40 °C for 4 h. The conversion was checked by 19F NMR spectroscopy with PhOCF3 as internal standard. After completion, the reaction mixture was partitioned between Et2O or pentane and water. The aqueous layer was extracted with Et2O and pentane and the combined organic layers were dried over MgSO4, filtered and concentrated to dryness. The crude residue was purified by chromatography to afford the desired products 3, 4, or 5.
Supporting Information
| Supporting Information File 1: Additional experimental and analytical data. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/s0022-1139(01)00375-x
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788
Return to citation in text: [1] -
Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840
Return to citation in text: [1] -
Han, J.; Remete, A. M.; Dobson, L. S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V. A.; O’Hagan, D. J. Fluorine Chem. 2020, 239, 109639. doi:10.1016/j.jfluchem.2020.109639
Return to citation in text: [1] -
Cheng, M.; Guo, C.; Gross, M. L. Angew. Chem., Int. Ed. 2020, 59, 5880–5889. doi:10.1002/anie.201907662
Return to citation in text: [1] -
Johnson, B. M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N. A. J. Med. Chem. 2020, 63, 6315–6386. doi:10.1021/acs.jmedchem.9b01877
Return to citation in text: [1] -
Jeschke, P. Pest Manage. Sci. 2010, 66, 10–27. doi:10.1002/ps.1829
Return to citation in text: [1] -
Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467
Return to citation in text: [1] -
Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014
Return to citation in text: [1] -
Pagliaro, M.; Ciriminna, R. J. Mater. Chem. 2005, 15, 4981–4991. doi:10.1039/b507583c
Return to citation in text: [1] -
Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f
Return to citation in text: [1] -
Chopra, D.; Row, T. N. G. CrystEngComm 2011, 13, 2175–2186. doi:10.1039/c0ce00538j
Return to citation in text: [1] -
Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830
Return to citation in text: [1] [2] -
Cahard, D.; Ma, J.-A., Eds. Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Wiley-VCH: Weinheim, Germany, 2020. doi:10.1002/9783527824342
Return to citation in text: [1] -
EFSA Panel on Dietetic Products, Nutrition and Allergies. EFSA J. 2014, 12, 3846. doi:10.2903/j.efsa.2014.3846
Return to citation in text: [1] -
European Food Safety Authority (EFSA). EFSA Supporting Publ. 2017, 14, e15121E. doi:10.2903/sp.efsa.2017.e15121
Return to citation in text: [1] -
Rayman, M. P. Lancet 2000, 356, 233–241. doi:10.1016/s0140-6736(00)02490-9
Return to citation in text: [1] [2] -
Mousa, R.; Notis Dardashti, R.; Metanis, N. Angew. Chem., Int. Ed. 2017, 56, 15818–15827. doi:10.1002/anie.201706876
Return to citation in text: [1] [2] -
Romashov, L. V.; Ananikov, V. P. Chem. – Eur. J. 2013, 19, 17640–17660. doi:10.1002/chem.201302115
Return to citation in text: [1] -
Guo, W.; Fu, Y. Chem. – Eur. J. 2020, 26, 13322–13331. doi:10.1002/chem.202000878
Return to citation in text: [1] -
Li, Q.; Zhang, Y.; Chen, Z.; Pan, X.; Zhang, Z.; Zhu, J.; Zhu, X. Org. Chem. Front. 2020, 7, 2815–2841. doi:10.1039/d0qo00640h
Return to citation in text: [1] -
Mugesh, G.; du Mont, W.-W.; Sies, H. Chem. Rev. 2001, 101, 2125–2180. doi:10.1021/cr000426w
Return to citation in text: [1] -
Rocha, J. B. T.; Piccoli, B. C.; Oliveira, C. S. ARKIVOC 2017, No. ii, 457–491. doi:10.24820/ark.5550190.p009.784
Return to citation in text: [1] -
Short, S. P.; Pilat, J. M.; Williams, C. S. Free Radical Biol. Med. 2018, 127, 26–35. doi:10.1016/j.freeradbiomed.2018.05.066
Return to citation in text: [1] -
Solovyev, N.; Drobyshev, E.; Bjørklund, G.; Dubrovskii, Y.; Lysiuk, R.; Rayman, M. P. Free Radical Biol. Med. 2018, 127, 124–133. doi:10.1016/j.freeradbiomed.2018.02.030
Return to citation in text: [1] -
Antoniadou, I.; Kouskou, M.; Arsiwala, T.; Singh, N.; Vasudevan, S. R.; Fowler, T.; Cadirci, E.; Churchill, G. C.; Sharp, T. Br. J. Pharmacol. 2018, 175, 2599–2610. doi:10.1111/bph.14179
Return to citation in text: [1] -
Jain, V. K.; Priyadarsini, K. I., Eds. Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Royal Society of Chemistry: Cambridge. U.K., 2018. doi:10.1039/9781788011907
Return to citation in text: [1] -
Singh, N.; Halliday, A. C.; Thomas, J. M.; Kuznetsova, O. V.; Baldwin, R.; Woon, E. C. Y.; Aley, P. K.; Antoniadou, I.; Sharp, T.; Vasudevan, S. R.; Churchill, G. C. Nat. Commun. 2013, 4, 1332. doi:10.1038/ncomms2320
Return to citation in text: [1] -
Thangamani, S.; Younis, W.; Seleem, M. N. Sci. Rep. 2015, 5, 11596. doi:10.1038/srep11596
Return to citation in text: [1] -
Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A. P. Free Radical Biol. Med. 2018, 127, 80–97. doi:10.1016/j.freeradbiomed.2018.05.001
Return to citation in text: [1] -
Alcolea, V.; Pérez-Silanes, S. Eur. J. Med. Chem. 2020, 206, 112673. doi:10.1016/j.ejmech.2020.112673
Return to citation in text: [1] -
He, X.; Zhong, M.; Li, S.; Li, X.; Li, Y.; Li, Z.; Gao, Y.; Ding, F.; Wen, D.; Lei, Y.; Zhang, Y. Eur. J. Med. Chem. 2020, 208, 112864. doi:10.1016/j.ejmech.2020.112864
Return to citation in text: [1] -
Tlili, A.; Ismalaj, E.; Glenadel, Q.; Ghiazza, C.; Billard, T. Chem. – Eur. J. 2018, 24, 3659–3670. doi:10.1002/chem.201704637
Return to citation in text: [1] -
Billard, T.; Toulgoat, F. When Fluorine Meets Selenium. In Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Cahard, D.; Ma, J.-A., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp 691–721. doi:10.1002/9783527824342.ch23
Return to citation in text: [1] -
Chen, C.; Ouyang, L.; Lin, Q.; Liu, Y.; Hou, C.; Yuan, Y.; Weng, Z. Chem. – Eur. J. 2014, 20, 657–661. doi:10.1002/chem.201303934
Return to citation in text: [1] [2] -
Rong, M.; Huang, R.; You, Y.; Weng, Z. Tetrahedron 2014, 70, 8872–8878. doi:10.1016/j.tet.2014.09.091
Return to citation in text: [1] [2] -
Yang, Y.; Lin, X.; Zheng, Z.; Lin, G.; Zhang, Y.; You, Y.; Weng, Z. J. Fluorine Chem. 2017, 204, 1–5. doi:10.1016/j.jfluchem.2017.10.001
Return to citation in text: [1] [2] -
Dong, T.; He, J.; Li, Z.-H.; Zhang, C.-P. ACS Sustainable Chem. Eng. 2018, 6, 1327–1335. doi:10.1021/acssuschemeng.7b03673
Return to citation in text: [1] -
Tyrra, W.; Naumann, D.; Yagupolskii, Y. L. J. Fluorine Chem. 2003, 123, 183–187. doi:10.1016/s0022-1139(03)00118-0
Return to citation in text: [1] -
Glenadel, Q.; Ghiazza, C.; Tlili, A.; Billard, T. Adv. Synth. Catal. 2017, 359, 3414–3420. doi:10.1002/adsc.201700904
Return to citation in text: [1] -
Ghiazza, C.; Tlili, A.; Billard, T. Beilstein J. Org. Chem. 2017, 13, 2626–2630. doi:10.3762/bjoc.13.260
Return to citation in text: [1] -
Ghiazza, C.; Debrauwer, V.; Billard, T.; Tlili, A. Chem. – Eur. J. 2018, 24, 97–100. doi:10.1002/chem.201705231
Return to citation in text: [1] -
Ghiazza, C.; Khrouz, L.; Monnereau, C.; Billard, T.; Tlili, A. Chem. Commun. 2018, 54, 9909–9912. doi:10.1039/c8cc05256e
Return to citation in text: [1] -
Ghiazza, C.; Debrauwer, V.; Monnereau, C.; Khrouz, L.; Médebielle, M.; Billard, T.; Tlili, A. Angew. Chem., Int. Ed. 2018, 57, 11781–11785. doi:10.1002/anie.201806165
Angew. Chem. 2018, 130, 11955–11959. doi:10.1002/ange.201806165
Return to citation in text: [1] -
Ghiazza, C.; Monnereau, C.; Khrouz, L.; Médebielle, M.; Billard, T.; Tlili, A. Synlett 2019, 30, 777–782. doi:10.1055/s-0037-1610347
Return to citation in text: [1] -
Ghiazza, C.; Kataria, A.; Tlili, A.; Toulgoat, F.; Billard, T. Asian J. Org. Chem. 2019, 8, 675–678. doi:10.1002/ajoc.201900027
Return to citation in text: [1] -
Glenadel, Q.; Bordy, M.; Alazet, S.; Tlili, A.; Billard, T. Asian J. Org. Chem. 2016, 5, 428–433. doi:10.1002/ajoc.201600003
Return to citation in text: [1] [2] [3] -
Glenadel, Q.; Tlili, A.; Billard, T. Eur. J. Org. Chem. 2016, 1955–1957. doi:10.1002/ejoc.201600197
Return to citation in text: [1]
| 49. | Glenadel, Q.; Bordy, M.; Alazet, S.; Tlili, A.; Billard, T. Asian J. Org. Chem. 2016, 5, 428–433. doi:10.1002/ajoc.201600003 |
| 1. | Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/s0022-1139(01)00375-x |
| 2. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351 |
| 3. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 4. | Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788 |
| 5. | Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840 |
| 6. | Han, J.; Remete, A. M.; Dobson, L. S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V. A.; O’Hagan, D. J. Fluorine Chem. 2020, 239, 109639. doi:10.1016/j.jfluchem.2020.109639 |
| 7. | Cheng, M.; Guo, C.; Gross, M. L. Angew. Chem., Int. Ed. 2020, 59, 5880–5889. doi:10.1002/anie.201907662 |
| 8. | Johnson, B. M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N. A. J. Med. Chem. 2020, 63, 6315–6386. doi:10.1021/acs.jmedchem.9b01877 |
| 9. | Jeschke, P. Pest Manage. Sci. 2010, 66, 10–27. doi:10.1002/ps.1829 |
| 10. | Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467 |
| 11. | Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014 |
| 12. | Pagliaro, M.; Ciriminna, R. J. Mater. Chem. 2005, 15, 4981–4991. doi:10.1039/b507583c |
| 13. | Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f |
| 14. | Chopra, D.; Row, T. N. G. CrystEngComm 2011, 13, 2175–2186. doi:10.1039/c0ce00538j |
| 15. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 19. | Rayman, M. P. Lancet 2000, 356, 233–241. doi:10.1016/s0140-6736(00)02490-9 |
| 20. | Mousa, R.; Notis Dardashti, R.; Metanis, N. Angew. Chem., Int. Ed. 2017, 56, 15818–15827. doi:10.1002/anie.201706876 |
| 24. | Mugesh, G.; du Mont, W.-W.; Sies, H. Chem. Rev. 2001, 101, 2125–2180. doi:10.1021/cr000426w |
| 25. | Rocha, J. B. T.; Piccoli, B. C.; Oliveira, C. S. ARKIVOC 2017, No. ii, 457–491. doi:10.24820/ark.5550190.p009.784 |
| 26. | Short, S. P.; Pilat, J. M.; Williams, C. S. Free Radical Biol. Med. 2018, 127, 26–35. doi:10.1016/j.freeradbiomed.2018.05.066 |
| 27. | Solovyev, N.; Drobyshev, E.; Bjørklund, G.; Dubrovskii, Y.; Lysiuk, R.; Rayman, M. P. Free Radical Biol. Med. 2018, 127, 124–133. doi:10.1016/j.freeradbiomed.2018.02.030 |
| 28. | Antoniadou, I.; Kouskou, M.; Arsiwala, T.; Singh, N.; Vasudevan, S. R.; Fowler, T.; Cadirci, E.; Churchill, G. C.; Sharp, T. Br. J. Pharmacol. 2018, 175, 2599–2610. doi:10.1111/bph.14179 |
| 29. | Jain, V. K.; Priyadarsini, K. I., Eds. Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Royal Society of Chemistry: Cambridge. U.K., 2018. doi:10.1039/9781788011907 |
| 49. | Glenadel, Q.; Bordy, M.; Alazet, S.; Tlili, A.; Billard, T. Asian J. Org. Chem. 2016, 5, 428–433. doi:10.1002/ajoc.201600003 |
| 50. | Glenadel, Q.; Tlili, A.; Billard, T. Eur. J. Org. Chem. 2016, 1955–1957. doi:10.1002/ejoc.201600197 |
| 21. | Romashov, L. V.; Ananikov, V. P. Chem. – Eur. J. 2013, 19, 17640–17660. doi:10.1002/chem.201302115 |
| 22. | Guo, W.; Fu, Y. Chem. – Eur. J. 2020, 26, 13322–13331. doi:10.1002/chem.202000878 |
| 23. | Li, Q.; Zhang, Y.; Chen, Z.; Pan, X.; Zhang, Z.; Zhu, J.; Zhu, X. Org. Chem. Front. 2020, 7, 2815–2841. doi:10.1039/d0qo00640h |
| 49. | Glenadel, Q.; Bordy, M.; Alazet, S.; Tlili, A.; Billard, T. Asian J. Org. Chem. 2016, 5, 428–433. doi:10.1002/ajoc.201600003 |
| 17. | EFSA Panel on Dietetic Products, Nutrition and Allergies. EFSA J. 2014, 12, 3846. doi:10.2903/j.efsa.2014.3846 |
| 18. | European Food Safety Authority (EFSA). EFSA Supporting Publ. 2017, 14, e15121E. doi:10.2903/sp.efsa.2017.e15121 |
| 19. | Rayman, M. P. Lancet 2000, 356, 233–241. doi:10.1016/s0140-6736(00)02490-9 |
| 20. | Mousa, R.; Notis Dardashti, R.; Metanis, N. Angew. Chem., Int. Ed. 2017, 56, 15818–15827. doi:10.1002/anie.201706876 |
| 42. | Glenadel, Q.; Ghiazza, C.; Tlili, A.; Billard, T. Adv. Synth. Catal. 2017, 359, 3414–3420. doi:10.1002/adsc.201700904 |
| 43. | Ghiazza, C.; Tlili, A.; Billard, T. Beilstein J. Org. Chem. 2017, 13, 2626–2630. doi:10.3762/bjoc.13.260 |
| 44. | Ghiazza, C.; Debrauwer, V.; Billard, T.; Tlili, A. Chem. – Eur. J. 2018, 24, 97–100. doi:10.1002/chem.201705231 |
| 45. | Ghiazza, C.; Khrouz, L.; Monnereau, C.; Billard, T.; Tlili, A. Chem. Commun. 2018, 54, 9909–9912. doi:10.1039/c8cc05256e |
| 46. |
Ghiazza, C.; Debrauwer, V.; Monnereau, C.; Khrouz, L.; Médebielle, M.; Billard, T.; Tlili, A. Angew. Chem., Int. Ed. 2018, 57, 11781–11785. doi:10.1002/anie.201806165
Angew. Chem. 2018, 130, 11955–11959. doi:10.1002/ange.201806165 |
| 47. | Ghiazza, C.; Monnereau, C.; Khrouz, L.; Médebielle, M.; Billard, T.; Tlili, A. Synlett 2019, 30, 777–782. doi:10.1055/s-0037-1610347 |
| 16. | Cahard, D.; Ma, J.-A., Eds. Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Wiley-VCH: Weinheim, Germany, 2020. doi:10.1002/9783527824342 |
| 48. | Ghiazza, C.; Kataria, A.; Tlili, A.; Toulgoat, F.; Billard, T. Asian J. Org. Chem. 2019, 8, 675–678. doi:10.1002/ajoc.201900027 |
| 35. | Tlili, A.; Ismalaj, E.; Glenadel, Q.; Ghiazza, C.; Billard, T. Chem. – Eur. J. 2018, 24, 3659–3670. doi:10.1002/chem.201704637 |
| 36. | Billard, T.; Toulgoat, F. When Fluorine Meets Selenium. In Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Cahard, D.; Ma, J.-A., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp 691–721. doi:10.1002/9783527824342.ch23 |
| 41. | Tyrra, W.; Naumann, D.; Yagupolskii, Y. L. J. Fluorine Chem. 2003, 123, 183–187. doi:10.1016/s0022-1139(03)00118-0 |
| 34. | He, X.; Zhong, M.; Li, S.; Li, X.; Li, Y.; Li, Z.; Gao, Y.; Ding, F.; Wen, D.; Lei, Y.; Zhang, Y. Eur. J. Med. Chem. 2020, 208, 112864. doi:10.1016/j.ejmech.2020.112864 |
| 37. | Chen, C.; Ouyang, L.; Lin, Q.; Liu, Y.; Hou, C.; Yuan, Y.; Weng, Z. Chem. – Eur. J. 2014, 20, 657–661. doi:10.1002/chem.201303934 |
| 38. | Rong, M.; Huang, R.; You, Y.; Weng, Z. Tetrahedron 2014, 70, 8872–8878. doi:10.1016/j.tet.2014.09.091 |
| 39. | Yang, Y.; Lin, X.; Zheng, Z.; Lin, G.; Zhang, Y.; You, Y.; Weng, Z. J. Fluorine Chem. 2017, 204, 1–5. doi:10.1016/j.jfluchem.2017.10.001 |
| 15. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 30. | Singh, N.; Halliday, A. C.; Thomas, J. M.; Kuznetsova, O. V.; Baldwin, R.; Woon, E. C. Y.; Aley, P. K.; Antoniadou, I.; Sharp, T.; Vasudevan, S. R.; Churchill, G. C. Nat. Commun. 2013, 4, 1332. doi:10.1038/ncomms2320 |
| 31. | Thangamani, S.; Younis, W.; Seleem, M. N. Sci. Rep. 2015, 5, 11596. doi:10.1038/srep11596 |
| 32. | Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A. P. Free Radical Biol. Med. 2018, 127, 80–97. doi:10.1016/j.freeradbiomed.2018.05.001 |
| 33. | Alcolea, V.; Pérez-Silanes, S. Eur. J. Med. Chem. 2020, 206, 112673. doi:10.1016/j.ejmech.2020.112673 |
| 37. | Chen, C.; Ouyang, L.; Lin, Q.; Liu, Y.; Hou, C.; Yuan, Y.; Weng, Z. Chem. – Eur. J. 2014, 20, 657–661. doi:10.1002/chem.201303934 |
| 38. | Rong, M.; Huang, R.; You, Y.; Weng, Z. Tetrahedron 2014, 70, 8872–8878. doi:10.1016/j.tet.2014.09.091 |
| 39. | Yang, Y.; Lin, X.; Zheng, Z.; Lin, G.; Zhang, Y.; You, Y.; Weng, Z. J. Fluorine Chem. 2017, 204, 1–5. doi:10.1016/j.jfluchem.2017.10.001 |
| 40. | Dong, T.; He, J.; Li, Z.-H.; Zhang, C.-P. ACS Sustainable Chem. Eng. 2018, 6, 1327–1335. doi:10.1021/acssuschemeng.7b03673 |
© 2020 Grollier et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)