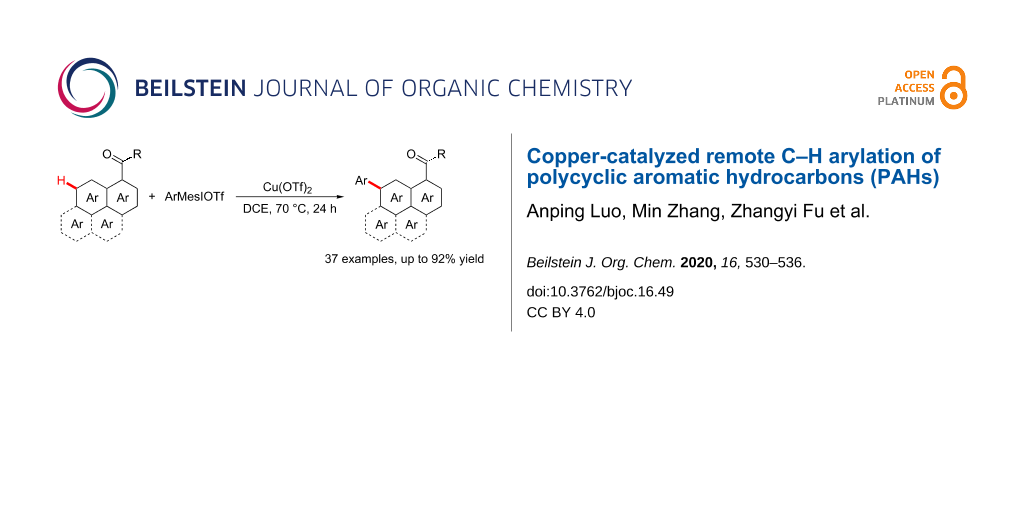
Abstract
The regioselective C–H arylation of substituted polycyclic aromatic hydrocarbons (PAHs) is a desired but challenging task. A copper-catalyzed C7–H arylation of 1-naphthamides has been developed by using aryliodonium salts as arylating reagents. This protocol does not need to use precious metal catalysts and tolerates wide variety of functional groups. Under standard conditions, the remote C–H arylation of other PAHs including phenanthrene-9-carboxamide, pyrene-1-carboxamide and fluoranthene-3-carboxamide has also accomplished, which provides an opportunity for the development of diverse organic optoelectronic materials.

Graphical Abstract
Introduction
Polycyclic aromatic hydrocarbons (PAHs) with rigid planar structure, such as naphthalene, phenanthrene, pyrene and their derivatives, can usually emit relatively strong fluorescence, and have been widely applied in many scientific areas including chemistry, biomedicine and materials science [1-6]. The arylation reaction of PAHs is an important strategy to further extend the π-conjugation length, which can effectively adjust the photophysical properties of molecules, thus having drawn much attention. Transition metal-catalyzed C–X/C–M cross-coupling reactions such as Suzuki and Stille couplings are the main approaches to achieve the arylation of PAHs [7-11]. However, the selective arylation of the C7-position of 1-naphthoic acid derivatives remains a challenging task due to the inaccessibility of the corresponding 7-halonaphthalene substrates [12].
Recently, transition metal-catalyzed C–H bond functionalization has emerged as a powerful tool to construct various biaryl skeletons [13-17]. The direct C7−H arylation of 1-naphthoic acid derivatives is undoubtedly a more effective route for the synthesis of 7-arylnaphthalene derivatives. Although the transition metal-catalyzed C2−H and C8−H arylations of 1-naphthoic acid derivatives have been widely reported, the studies on their C7−H arylation remain rare [18-25]. Our group has recently reported F+ reagent-promoted Pd-catalyzed C7–H arylation of 1‑naphthamides, but this method still suffers from a few disadvantages (Scheme 1) [26]. First, the precious metal palladium is employed as a catalyst. Moreover, stoichiometric F+ reagent is needed to oxidize Pd(II) species to more electrophilic high-valent cationic Pd(IV). In addition, this protocol is not compatible with other PAHs except naphthalene, such as phenanthrene, pyrene and fluoranthene, and cannot tolerate some special functional groups, such as alkenyl and alkynyl groups.
![[1860-5397-16-49-i1]](/bjoc/content/inline/1860-5397-16-49-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
As a component part of our ongoing research on direct C–H bond functionalization [20,27-29], we herein represent a copper-catalyzed remote C–H arylation of PAHs with aryliodonium salts as arylating reagents (Scheme 1). This protocol is compatible with different PAH substrates including 1-naphthamides, phenanthrene-9-carboxamide, pyrene-1-carboxamide and fluoranthene-3-carboxamide, which provides an opportunity for the development of diverse organic photoelectrical materials.
Results and Discussion
Our investigation commenced with the reaction between N-(tert-butyl)-1-naphthamide (1a) and mesityl(phenyl)iodonium triflate (2a, for detailed optimization, see Table S1, Supporting Information File 1). Initially, the reaction was performed in 1,2-dichloroethane (DCE, 1 mL) at 80 °C for 24 h in the presence of Cu(OTf)2 (10 mol %) as a catalyst. The direct C7–H arylation product N-(tert-butyl)-7-phenyl-1-naphthamide (3a) was obtained in 79% yield (Table 1, entry 1). Gratifyingly, when the reaction temperature was reduced to 70 ºC, 3a was obtained in 92% yield (Table 1, entry 2). The C7–H arylation could also occur with active copper powder as a catalyst (Table 1, entry 5). Other copper sources including CuO, CuCl and Cu(OAc)2 were also found to be effective catalysts in this reaction, albeit with slightly lower yields (Table 1, entries 6–8). The control experiment confirmed that this transformation did not occur in the absence of Cu catalyst (Table 1, entry 9). The screening of other solvents, such as dichloromethane (DCM), ortho-dichlorobenzene (ODCB), CHCl3 and PhCF3, indicated that DCE was still the best effective (Table 1, entries 10–13). Finally, the optimal reaction system was established, which composed of Cu(OTf)2 (10 mol %) in DCE (1.0 mL) at 70 °C under a nitrogen atmosphere for 24 hours.
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-49-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | [Cu] | T (°C) | Yield (%) |
| 1 | DCE | Cu(OTf)2 | 80 | 79 |
| 2 | DCE | Cu(OTf)2 | 70 | 92 |
| 3 | DCE | Cu(OTf)2 | 90 | 53 |
| 4 | DCE | Cu(OTf)2 | 60 | 41 |
| 5 | DCE | Cu | 70 | 54 |
| 6 | DCE | CuO | 70 | 81 |
| 7 | DCE | CuCl | 70 | 84 |
| 8 | DCE | Cu(OAc)2 | 70 | 80 |
| 9 | DCE | – | 70 | nd |
| 10 | DCM | Cu(OTf)2 | 70 | 38 |
| 11 | ODCB | Cu(OTf)2 | 70 | 77 |
| 12 | CHCl3 | Cu(OTf)2 | 70 | trace |
| 13 | PhCF3 | Cu(OTf)2 | 70 | trace |
aReaction conditions: 1a (0.2 mmol, 1.0 equiv), 2a (0.3 mmol, 1.5 equiv), [Cu] (10 mol %) and solvent (1 mL) under N2 for 24 h. Isolated yield. DCE = 1,2-dichloroethane. DCM = dichloromethane. ODCB = ortho-dichlorobenzene. nd: not detected.
With the optimal conditions in hand, we first examined the scope of aryliodonium salts. We were very pleased to find that a range of aryliodonium salts could be employed as arylating reagents, affording 7-arylated 1-naphthamides (3a–q) in moderate to excellent yields (Scheme 2). This protocol tolerated a wide variety of functional groups, including electron-donating methyl and methoxy groups, as well as electron-withdrawing ester, trifluoromethyl, fluoro, chloro, bromo, iodo and formyl groups. The arylating reactivity of aryliodonium salts with various substituents varied greatly due to the different electronic effects and steric hindrances. Arylating reagents with ortho-substituents led to slightly reduced yields (Scheme 2, 3f and 3i). Aryliodonium salts containing halogen substituents, especially bromo and iodo atoms, could afford the desired products in moderate to good yields (Scheme 2, 3j–n), making it possible to introduce useful functional groups into the products through the further transformation of corresponding aryl halides. 2-Naphthyliodonium salt could react smoothly with 1a to provide 3p in 66% yield (Scheme 2, 3p). Moreover, thiophen-2-yliodonium salt could be tolerated, albeit with a lower yield (Scheme 2, 3q).
![[1860-5397-16-49-i2]](/bjoc/content/inline/1860-5397-16-49-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of aryliodonium salts. Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol) in DCE (1 mL) at 70 °C under N2 for 24 h. Isolated yield. a60 °C. b80 °C. c50 °C.
Scheme 2: Scope of aryliodonium salts. Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol) in DCE (1 mL) at 70 °...
We next examined the scope of various naphthalene substrates (Scheme 3, 4a–l). The electronic effect of C4-substituents on N-(tert-butyl)-1-naphthamide was not obvious. The 4-substituted 1-naphthamide substrates, whether with electron-donating methyl and methoxy groups, or with electron-withdrawing phenyl, ester, fluoro and bromo groups, gave the corresponding products in good to excellent yields (Scheme 3, 4a–f). Substrates with C2-substituents also exhibited excellent reactivity, providing the desired products 4h and 4i in 85% and 80% yields, respectively (Scheme 3, 4h and 4i). Notably, 1-naphthamides with alkenyl (1l) and alkynyl (1m) groups were also suitable substrates for this direct C7−H arylation, affording 4k and 4l in good yields (Scheme 3, 4k and 4l). Furthermore, this Cu-catalyzed direct C−H arylation could tolerate other PAH substrates. The regioselective arylation of PAHs is challenging, and so far, there are no examples on the selective remote C–H arylation of phenanthrene-9-carboxamide, pyrene-1-carboxamide and fluoranthene-3-carboxamide. Gratifyingly, the remote C–H arylation of these PAH substrates occurred smoothly, giving the corresponding arylation products in moderate to good yields (Scheme 3, 4m–o).
![[1860-5397-16-49-i3]](/bjoc/content/inline/1860-5397-16-49-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of PAHs. Reaction conditions: 1 (0.2 mmol), 2a (0.3 mmol) in DCE (1 mL) at 70 °C under N2 for 24 h. Isolated yield. DCE = 1,2-dichloroethane.
Scheme 3: Scope of PAHs. Reaction conditions: 1 (0.2 mmol), 2a (0.3 mmol) in DCE (1 mL) at 70 °C under N2 for...
This catalytic system was also compatible with substrates bearing other directing groups except tert-butylaminocarbonyl (Scheme 3, 4p–s). When employing methylaminocarbonyl and cyclohexylaminocarbonyl as directing groups, the 7-arylation products of naphthalene rings were obtained in good yields (Scheme 3, 4p and 4q). A keto carbonyl group was also proved to be a suitable directing group, affording the corresponding arylation products in moderate yields (Scheme 3, 4r and 4s).
Considering that Cu(0), Cu(I) and Cu(II) all could catalyze this C−H arylation reaction and referring to previous research results [30-32], a Cu(I)/Cu(III) catalytic cycle was proposed (Scheme 4). First, Cu(I) is formed by the reduction or disproportionation of Cu(II). Then, aryliodonium salt oxidizes Cu(I) to highly electrophilic Cu(III)–aryl intermediate I. The coordination of the carbonyl oxygen to I gives intermediate II, which undergoes an aryl-transfer reaction via a Heck-like four-membered-ring transition state III to form the intermediate IV with Cu(III) and aryl group added at the C8- and C7-positions of the naphthalene ring, respectively. Finally, the breakdown of the C8–Cu bond delivers Cu(I), meantime, the OTf anion takes away the proton from the C7-position, affording the desired product 3 or 4.
![[1860-5397-16-49-i4]](/bjoc/content/inline/1860-5397-16-49-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Subsequently, the photophysical properties of the arylation products 4k, 4n and 4o were investigated (Figure 1). Their absorption bands cover from 300 nm to 400 nm, which corresponds to π−π* electron transition (Figure 1a and Table S2, Supporting Information File 1). The measurement of emission spectra demonstrates that 4k and 4n emit violet fluorescence with emission maxima at 395 nm and 390 nm, respectively, while 4o exhibits a sky-blue emission with an emission maximum at 477 nm (Figure 1b and Table S2, Supporting Information File 1).
![[1860-5397-16-49-1]](/bjoc/content/figures/1860-5397-16-49-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) UV-visible absorption spectra of 4k, 4n and 4o in toluene (1 × 10−5 mol/L). b) Emission spectra of 4k, 4n and 4o in toluene (1 × 10−5 mol/L).
Figure 1: a) UV-visible absorption spectra of 4k, 4n and 4o in toluene (1 × 10−5 mol/L). b) Emission spectra ...
Conclusion
In summary, we have developed a highly efficient strategy to accomplish the direct C7−H arylation of 1-naphthamides by the usage of Cu(II) as a catalyst and aryliodonium salts as arylating reagents, which features mild reaction conditions, excellent functional group tolerance, and moreover, does not need to use precious metal catalysts. This protocol is also compatible with other PAH substrates including phenanthrene-9-carboxamide, pyrene-1-carboxamide and fluoranthene-3-carboxamide, which provide an opportunity for the development of diverse organic photoelectrical materials.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, characterization data and copies of 1H and 13C NMR spectra of products. | ||
| Format: PDF | Size: 7.2 MB | Download |
References
-
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Sun, Z.; Ye, Q.; Chi, C.; Wu, J. Chem. Soc. Rev. 2012, 41, 7857–7889. doi:10.1039/c2cs35211g
Return to citation in text: [1] -
Figueira-Duarte, T. M.; Müllen, K. Chem. Rev. 2011, 111, 7260–7314. doi:10.1021/cr100428a
Return to citation in text: [1] -
Wu, J.; Pisula, W.; Müllen, K. Chem. Rev. 2007, 107, 718–747. doi:10.1021/cr068010r
Return to citation in text: [1] -
Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z
Return to citation in text: [1] -
Jiang, W.; Li, Y.; Wang, Z. Chem. Soc. Rev. 2013, 42, 6113–6127. doi:10.1039/c3cs60108k
Return to citation in text: [1] -
Stille, J. K. Angew. Chem., Int. Ed. Engl. 1986, 25, 508–524. doi:10.1002/anie.198605081
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Li, Y.; Gao, J.; Di Motta, S.; Negri, F.; Wang, Z. J. Am. Chem. Soc. 2010, 132, 4208–4213. doi:10.1021/ja100276x
Return to citation in text: [1] -
Zeng, W.; Gopalakrishna, T. Y.; Phan, H.; Tanaka, T.; Herng, T. S.; Ding, J.; Osuka, A.; Wu, J. J. Am. Chem. Soc. 2018, 140, 14054–14058. doi:10.1021/jacs.8b09075
Return to citation in text: [1] -
Hindenberg, P.; Busch, M.; Paul, A.; Bernhardt, M.; Gemessy, P.; Rominger, F.; Romero-Nieto, C. Angew. Chem., Int. Ed. 2018, 57, 15157–15161. doi:10.1002/anie.201809754
Return to citation in text: [1] -
Dewar, M. J. S.; Grisdale, P. J. J. Am. Chem. Soc. 1962, 84, 3541–3546. doi:10.1021/ja00877a024
Return to citation in text: [1] -
Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787–8863. doi:10.1021/acs.chemrev.6b00567
Return to citation in text: [1] -
Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174–238. doi:10.1021/cr0509760
Return to citation in text: [1] -
McGlacken, G. P.; Bateman, L. M. Chem. Soc. Rev. 2009, 38, 2447–2464. doi:10.1039/b805701j
Return to citation in text: [1] -
Ackermann, L.; Vicente, R.; Kapdi, A. R. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. doi:10.1002/anie.200902996
Return to citation in text: [1] -
Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074–1086. doi:10.1021/ar9000058
Return to citation in text: [1] -
Daugulis, O.; Zaitsev, V. G. Angew. Chem., Int. Ed. 2005, 44, 4046–4048. doi:10.1002/anie.200500589
Return to citation in text: [1] -
Biafora, A.; Krause, T.; Hackenberger, D.; Belitz, F.; Gooßen, L. J. Angew. Chem., Int. Ed. 2016, 55, 14752–14755. doi:10.1002/anie.201607270
Return to citation in text: [1] -
Shi, Y.; Zhang, L.; Lan, J.; Zhang, M.; Zhou, F.; Wei, W.; You, J. Angew. Chem., Int. Ed. 2018, 57, 9108–9112. doi:10.1002/anie.201804528
Return to citation in text: [1] [2] -
Tan, G.; You, Q.; Lan, J.; You, J. Angew. Chem., Int. Ed. 2018, 57, 6309–6313. doi:10.1002/anie.201802539
Return to citation in text: [1] -
Huang, L.; Weix, D. J. Org. Lett. 2016, 18, 5432–5435. doi:10.1021/acs.orglett.6b02862
Return to citation in text: [1] -
Yang, S.; Cheng, R.; Zhang, M.; Bin, Z.; You, J. ACS Catal. 2019, 9, 6188–6193. doi:10.1021/acscatal.9b01426
Return to citation in text: [1] -
Moon, S.; Nishii, Y.; Miura, M. Org. Lett. 2019, 21, 233–236. doi:10.1021/acs.orglett.8b03675
Return to citation in text: [1] -
Li, S.; Deng, G.-J.; Yin, F.; Li, C.-J.; Gong, H. Org. Chem. Front. 2017, 4, 417–420. doi:10.1039/c6qo00663a
Return to citation in text: [1] -
Zhang, M.; Luo, A.; Shi, Y.; Su, R.; Yang, Y.; You, J. ACS Catal. 2019, 9, 11802–11807. doi:10.1021/acscatal.9b04352
Return to citation in text: [1] -
Zhao, D.; Wang, W.; Yang, F.; Lan, J.; Yang, L.; Gao, G.; You, J. Angew. Chem., Int. Ed. 2009, 48, 3296–3300. doi:10.1002/anie.200900413
Return to citation in text: [1] -
Wu, J.; Cheng, Y.; Lan, J.; Wu, D.; Qian, S.; Yan, L.; He, Z.; Li, X.; Wang, K.; Zou, B.; You, J. J. Am. Chem. Soc. 2016, 138, 12803–12812. doi:10.1021/jacs.6b03890
Return to citation in text: [1] -
Zhang, L.; Wang, Y.; Shi, Y.; Wu, Y.; Lan, J.; Ma, W.; You, J. ACS Catal. 2019, 9, 5358–5364. doi:10.1021/acscatal.9b00925
Return to citation in text: [1] -
Phipps, R. J.; Gaunt, M. J. Science 2009, 323, 1593–1597. doi:10.1126/science.1169975
Return to citation in text: [1] -
Chen, B.; Hou, X.-L.; Li, Y.-X.; Wu, Y.-D. J. Am. Chem. Soc. 2011, 133, 7668–7671. doi:10.1021/ja201425e
Return to citation in text: [1] -
Yang, Y.; Li, R.; Zhao, Y.; Zhao, D.; Shi, Z. J. Am. Chem. Soc. 2016, 138, 8734–8737. doi:10.1021/jacs.6b05777
Return to citation in text: [1]
| 1. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 2. | Sun, Z.; Ye, Q.; Chi, C.; Wu, J. Chem. Soc. Rev. 2012, 41, 7857–7889. doi:10.1039/c2cs35211g |
| 3. | Figueira-Duarte, T. M.; Müllen, K. Chem. Rev. 2011, 111, 7260–7314. doi:10.1021/cr100428a |
| 4. | Wu, J.; Pisula, W.; Müllen, K. Chem. Rev. 2007, 107, 718–747. doi:10.1021/cr068010r |
| 5. | Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z |
| 6. | Jiang, W.; Li, Y.; Wang, Z. Chem. Soc. Rev. 2013, 42, 6113–6127. doi:10.1039/c3cs60108k |
| 18. | Daugulis, O.; Zaitsev, V. G. Angew. Chem., Int. Ed. 2005, 44, 4046–4048. doi:10.1002/anie.200500589 |
| 19. | Biafora, A.; Krause, T.; Hackenberger, D.; Belitz, F.; Gooßen, L. J. Angew. Chem., Int. Ed. 2016, 55, 14752–14755. doi:10.1002/anie.201607270 |
| 20. | Shi, Y.; Zhang, L.; Lan, J.; Zhang, M.; Zhou, F.; Wei, W.; You, J. Angew. Chem., Int. Ed. 2018, 57, 9108–9112. doi:10.1002/anie.201804528 |
| 21. | Tan, G.; You, Q.; Lan, J.; You, J. Angew. Chem., Int. Ed. 2018, 57, 6309–6313. doi:10.1002/anie.201802539 |
| 22. | Huang, L.; Weix, D. J. Org. Lett. 2016, 18, 5432–5435. doi:10.1021/acs.orglett.6b02862 |
| 23. | Yang, S.; Cheng, R.; Zhang, M.; Bin, Z.; You, J. ACS Catal. 2019, 9, 6188–6193. doi:10.1021/acscatal.9b01426 |
| 24. | Moon, S.; Nishii, Y.; Miura, M. Org. Lett. 2019, 21, 233–236. doi:10.1021/acs.orglett.8b03675 |
| 25. | Li, S.; Deng, G.-J.; Yin, F.; Li, C.-J.; Gong, H. Org. Chem. Front. 2017, 4, 417–420. doi:10.1039/c6qo00663a |
| 13. | Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787–8863. doi:10.1021/acs.chemrev.6b00567 |
| 14. | Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174–238. doi:10.1021/cr0509760 |
| 15. | McGlacken, G. P.; Bateman, L. M. Chem. Soc. Rev. 2009, 38, 2447–2464. doi:10.1039/b805701j |
| 16. | Ackermann, L.; Vicente, R.; Kapdi, A. R. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. doi:10.1002/anie.200902996 |
| 17. | Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074–1086. doi:10.1021/ar9000058 |
| 12. | Dewar, M. J. S.; Grisdale, P. J. J. Am. Chem. Soc. 1962, 84, 3541–3546. doi:10.1021/ja00877a024 |
| 7. | Stille, J. K. Angew. Chem., Int. Ed. Engl. 1986, 25, 508–524. doi:10.1002/anie.198605081 |
| 8. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 9. | Li, Y.; Gao, J.; Di Motta, S.; Negri, F.; Wang, Z. J. Am. Chem. Soc. 2010, 132, 4208–4213. doi:10.1021/ja100276x |
| 10. | Zeng, W.; Gopalakrishna, T. Y.; Phan, H.; Tanaka, T.; Herng, T. S.; Ding, J.; Osuka, A.; Wu, J. J. Am. Chem. Soc. 2018, 140, 14054–14058. doi:10.1021/jacs.8b09075 |
| 11. | Hindenberg, P.; Busch, M.; Paul, A.; Bernhardt, M.; Gemessy, P.; Rominger, F.; Romero-Nieto, C. Angew. Chem., Int. Ed. 2018, 57, 15157–15161. doi:10.1002/anie.201809754 |
| 30. | Phipps, R. J.; Gaunt, M. J. Science 2009, 323, 1593–1597. doi:10.1126/science.1169975 |
| 31. | Chen, B.; Hou, X.-L.; Li, Y.-X.; Wu, Y.-D. J. Am. Chem. Soc. 2011, 133, 7668–7671. doi:10.1021/ja201425e |
| 32. | Yang, Y.; Li, R.; Zhao, Y.; Zhao, D.; Shi, Z. J. Am. Chem. Soc. 2016, 138, 8734–8737. doi:10.1021/jacs.6b05777 |
| 20. | Shi, Y.; Zhang, L.; Lan, J.; Zhang, M.; Zhou, F.; Wei, W.; You, J. Angew. Chem., Int. Ed. 2018, 57, 9108–9112. doi:10.1002/anie.201804528 |
| 27. | Zhao, D.; Wang, W.; Yang, F.; Lan, J.; Yang, L.; Gao, G.; You, J. Angew. Chem., Int. Ed. 2009, 48, 3296–3300. doi:10.1002/anie.200900413 |
| 28. | Wu, J.; Cheng, Y.; Lan, J.; Wu, D.; Qian, S.; Yan, L.; He, Z.; Li, X.; Wang, K.; Zou, B.; You, J. J. Am. Chem. Soc. 2016, 138, 12803–12812. doi:10.1021/jacs.6b03890 |
| 29. | Zhang, L.; Wang, Y.; Shi, Y.; Wu, Y.; Lan, J.; Ma, W.; You, J. ACS Catal. 2019, 9, 5358–5364. doi:10.1021/acscatal.9b00925 |
| 26. | Zhang, M.; Luo, A.; Shi, Y.; Su, R.; Yang, Y.; You, J. ACS Catal. 2019, 9, 11802–11807. doi:10.1021/acscatal.9b04352 |
© 2020 Luo et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)