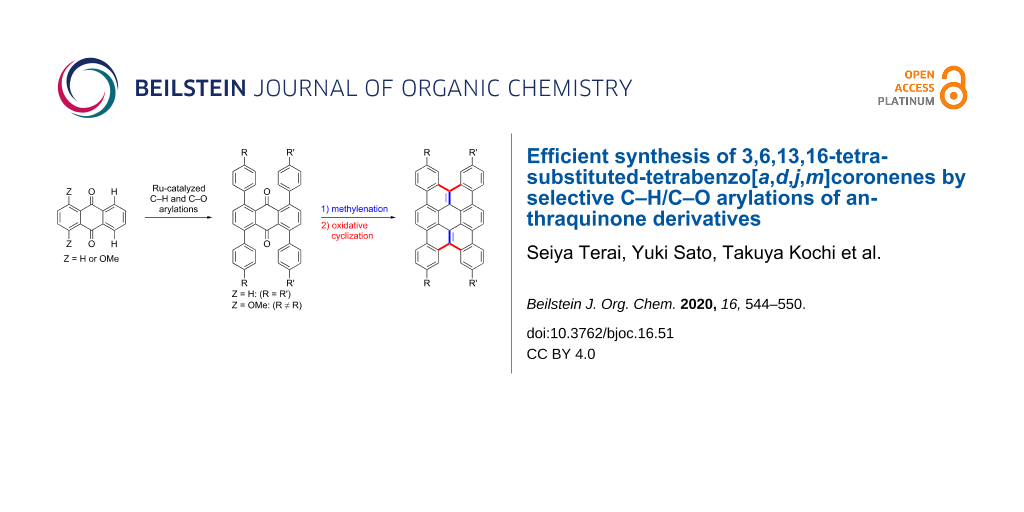
Abstract
An efficient synthesis of tetrabenzo[a,d,j,m]coronene derivatives having alkyl and alkoxy substituents at the 3, 6, 13, and 16-positions was achieved based on the ruthenium-catalyzed coupling reactions of anthraquinone derivatives with arylboronates via C–H and C–O bond cleavage. The reaction sequence involving the arylation, carbonyl methylenation, and oxidative cyclization effectively provided various tetrabenzo[a,d,j,m]coronenes in short steps from readily available starting materials. Tetrabenzo[a,d,j,m]coronenes possessing two different types of substituents were obtained selectively by sequential chemoselective C–O arylation and C–H arylation. The 1H NMR spectra of the tetrabenzo[a,d,j,m]coronene product indicated its self-assembling behavior in CDCl3.

Graphical Abstract
Introduction
Polycyclic aromatic hydrocarbons (PAHs) and their derivatives have attracted much attention from researchers due to their high potential as organic optoelectronic materials [1-3] and great efforts have been devoted to the development of their efficient synthetic methods [4]. One of the important steps in most of the PAH syntheses is the construction of carbon–carbon bonds between aryl groups to form biaryl frameworks. Traditionally, transition-metal-catalyzed cross-coupling reactions of aryl halides or pseudohalides with arylmetal reagents have been employed for the connection of two aryl units [5-9]. However, in search of more efficient synthetic routes, C–H arylation reactions have been increasingly utilized in recent years for the rapid expansion of the aromatic frameworks [10-15].
We have been working on the efficient syntheses of PAH derivatives based on the ruthenium-catalyzed C–H/C–O arylation reactions developed in our group [16-33]. In the presence of ruthenium carbonyl phosphine complexes, the reaction of aromatic ketones possessing C–H bonds or C–OR (R = alkyl) bonds with arylboronates provided C–H or C–O arylation products [21-33]. Anthraquinone was chosen as a convenient template for the PAH syntheses, because various anthraquinone derivatives possessing zero to four oxygen substituents at the ortho-positions are readily available and the regioselective arylation at the positions of either C–H or C–O bonds provided a variety of multiarylated anthraquinone derivatives [16-20]. Using this method, we have synthesized various π-extended aromatic compounds such as multiarylated acenes [16,18,20], dibenzo[h,rst]pentaphenes and dibenzo[fg,qr]pentacenes [19].
In the course of our reaction development, it was found that an introduction of two different aryl groups at the ortho-positions can be achieved by chemoselective C–O arylation of aromatic ketones possessing both C–H and C–O bonds and subsequent C–H arylation. We envisioned that the application of this strategy to the anthraquinone template would provide its derivatives possessing two different types of aryl groups in a designed manner and lead to the preparation of tetrabenzo[a,d,j,m]coronenes having two different types of substituents (Scheme 1). While 1,4,5,8-tetraarylanthraquinones prepared by the ruthenium-catalyzed C–H arylation of anthraquinone with a para-substituted arylboronate may be converted to 3,6,13,16-tetrasubstituted tetrabenzo[a,d,j,m]coronenes via carbonyl methylenation and oxidative cyclization, the two-step sequential C–O and C–H arylation of 1,4-dimethoxyanthraquinone followed by subsequent methylenation/cyclization would provide tetrabenzo[a,d,j,m]coronenes having two different types of substituents.
![[1860-5397-16-51-i1]](/bjoc/content/inline/1860-5397-16-51-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Projected synthetic routes for 3,6,13,16-tetrasubstituted tetrabenzo[a,d,j,m]coronenes.
Scheme 1: Projected synthetic routes for 3,6,13,16-tetrasubstituted tetrabenzo[a,d,j,m]coronenes.
Coronene derivatives such as hexabenzocoronenes and ovalenes have been extensively studied because of their optoelectronic and self-assembling properties [34-44]. Various synthetic methods have been developed for coronene derivatives in general [34-37], however, only a few strategies have been reported for the syntheses of tetrabenzo[a,d,j,m]coronenes [43,44]. Wu and co-workers reported the synthesis of 2,7,12,17-tetrasubstituted tetrabenzo[a,d,j,m]coronenes by using the Barton–Kellogg reaction, followed by dehydrogenative photocyclization, and FeCl3-mediated oxidative cyclization (Scheme 2a) [43]. Tao, Chao, and co-workers succeeded in the preparation of similar tetrasubstituted tetrabenzocoronenes by a Corey–Fuchs reaction, followed by a Suzuki–Miyaura cross-coupling, and a two-step dehydrogenative cyclization using DDQ and FeCl3 (Scheme 2b) [44]. However, 3,6,13,16-tetrasubstituted tetrabenzo[a,d,j,m]coronenes have not been reported using these reported methods.
![[1860-5397-16-51-i2]](/bjoc/content/inline/1860-5397-16-51-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reported syntheses of 2,7,12,17-tetrasubstituted tetrabenzo[a,d,j,m]coronenes.
Scheme 2: Reported syntheses of 2,7,12,17-tetrasubstituted tetrabenzo[a,d,j,m]coronenes.
Here, we describe a convenient method for the synthesis of 3,6,13,16-tetrasubstituted tetrabenzo[a,d,j,m]coronenes based on a ruthenium-catalyzed C–O/C–H multiarylation of anthraquinone and its derivatives.
Results and Discussion
Our efforts toward the synthesis of tetrabenzo[a,d,j,m]coronenes started with the preparation of tetraarylanthraquinones. Previously, we reported the synthesis of anthraquinone derivatives possessing four aryl groups at the 1, 4, 5, and 8-positions through a RuH2(CO)(PPh3)3-catalyzed C–H arylation of anthraquinone (1) with arylboronates 2 [16]. Using this method, we examined the synthesis of a tetraarylanthraquinone possessing four hexyloxy groups. When the reaction of 1 with 4-hexyloxyphenylboronate 2a was carried out in the presence of 20 mol % of RuH2(CO)(PPh3)3 (3) in refluxing pinacolone, the corresponding tetraarylation product 4aa was obtained in 57% yield (Scheme 3). Tetrakis(4-hexylphenyl)anthraquinone (4bb), the synthesis of which has been described in the above-mentioned publication [16], was also prepared using 4-hexylphenylboronate (2b) accordingly and used for further transformations. The reaction of 1 with 2b was also attempted using 10 mol % of 3, however, it only gave mono-, di- and triarylation products, and the tetraarylation product 4bb was not detected.
![[1860-5397-16-51-i3]](/bjoc/content/inline/1860-5397-16-51-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: C–H tetraarylation of anthraquinone (1).
Scheme 3: C–H tetraarylation of anthraquinone (1).
Tetraarylanthraquinones containing two different aryl groups were also prepared via the above-mentioned chemoselective C–O arylation and subsequent C–H arylation. The chemoselective C–O diarylation of 1,4-dimethoxyanthraquinone to form 1,4-diarylanthraquinones has been previously reported by our group [19], and 1,4-bis(4-hexyloxyphenyl)anthraquinone (5a) and 1,4-bis(4-hexylphenyl)anthraquinone (5b) were prepared accordingly. When the reaction of 5a with 4-methylphenylboronate 2c was conducted in the presence of 40 mol % of 3 in refluxing pinacolone, the ortho-C–H diarylation occurred giving the 1,4-bis(4-hexyloxyphenyl)-5,8-bis(4-methylphenyl)anthraquinone (4ac) in 91% yield. The C–H diarylation of 5b with 2a also proceeded smoothly to provide the corresponding tetraarylanthraquinone 4ba in 94% yield (Scheme 4).
![[1860-5397-16-51-i4]](/bjoc/content/inline/1860-5397-16-51-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: C–H diarylation of 1,4-diarylanthraquinones 5.
Scheme 4: C–H diarylation of 1,4-diarylanthraquinones 5.
Next, the conversion of tetraarylanthraquinones 4 to tetrabenzo[a,d,j,m]coronenes was investigated. After examining various carbonyl methylenation methods, we found that the dimethylenation products 6 can be obtained in high yields through methylation of the carbonyl groups, followed by dehydration. Thus, the reaction of 4aa with methyllithium and subsequent treatment of the crude diol with NaH2PO2·H2O and NaI in refluxing acetic acid gave the corresponding dimethylenation product 6aa in 96% yield (Table 1, entry 1). The oxidative cyclization of 6aa by treatment with 12 equiv of FeCl3 at 35 °C for 0.5 h provided the desired tetrabenzo[a,d,j,m]corronene derivative 7aa in 37% yield. This method was also applicable to the conversion of tetrakis(4-hexylphenyl)anthraquinone 4bb as well as the tetraarylanthraquinones containing two different aryl groups, 4ac and 4ba, leading to the corresponding tetrabenzo[a,d,j,m]corronene derivatives, 7bb, 7ac, and 7ba, respectively (Table 1, entries 2–4).
Table 1: Synthesis of 3,6,13,16-tetrasubstituted tetrabenzo[a,d,j,m]coronenes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-51-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | R1 | R2 | yield of 6 (%)a | yield of 7 (%)b |
| 1 | n-hexO | n-hexO | 6aa: 96 | 7aa: 37 |
| 2 | n-hex | n-hex | 6bb: 85 | 7bb: 73 |
| 3 | n-hexO | Me | 6ac: 78 | 7ac: 48 |
| 4 | n-hex | n-hexO | 6ba: 85 | 7ba: 48 |
aReaction conditions: 1) 4 (0.2 mmol), MeLi (2 mmol), THF (10 mL), −78 °C to rt, 2 h; 2) NaH2PO2·H2O (2 mmol), NaI (2 mmol), AcOH (6 mL), reflux, 2 h; breaction conditions: 6 (0.02 mmol), FeCl3 (0.24 mmol), MeNO2 (1 mL), CH2Cl2 (5 mL), gentle N2 bubbling, 35 °C, 0.5 h.
Next, the effect of the substituents in compounds 7 on their optical properties was studied by UV–vis spectroscopy (Figure 1). The UV–vis spectra of 7aa, 7bb, and 7ba were measured in chloroform, and the normalized UV–vis spectra are shown in Figure 1. These compounds showed similar peak patterns between 300 and 500 nm, and the π–π* transitions (p-band) for 7aa, 7bb, and 7ba were observed at 426 nm [11]. The peaks corresponding to n–π* transitions (α-band) were observed at 456–469 nm and were red-shifted with growing intensity as the number of hexyloxy groups increases.
![[1860-5397-16-51-1]](/bjoc/content/figures/1860-5397-16-51-1.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 1: Normalized UV–vis absorption spectra of 7aa, 7bb, and 7ba.
Figure 1: Normalized UV–vis absorption spectra of 7aa, 7bb, and 7ba.
The self-assembling property of tetrabenzo[a,d,j,m]coronene 7aa was also examined (Figure 2), as Wu and co-workers have studied the self-assembling property of the tetrabenzo[a,d,j,m]coronene derivative possessing n-octyl groups at the 2, 7, 12, and 17-positions by 1H NMR spectroscopy and suggested its intermolecular π–π interaction in solution [10]. When the 1H NMR spectrum of 7aa was measured at 2 × 10−3 M at rt, a considerable broadening of the signals in the aromatic region was observed. By increasing the temperature from rt to 60 °C, all signals in the aromatic region became sharper and low-field shifted. The dilution of the solution to 5 × 10−4 M at rt also led to a downfield shift and sharpening of the signals. These observations suggest that 7aa assembles by intermolecular π–π interaction in CDCl3 as was seen by Wu and co-workers for their compound possessing substituents at different positions.
![[1860-5397-16-51-2]](/bjoc/content/figures/1860-5397-16-51-2.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 2: Effects of the concentration and the temperature on the 1H NMR spectra of 7aa.
Figure 2: Effects of the concentration and the temperature on the 1H NMR spectra of 7aa.
Conclusion
The synthesis of a new class of tetrabenzo[a,d,j,m]coronene derivatives having alkyl and alkoxy-substituents at the 3, 6, 13, and 16-positions was achieved based on a ruthenium-catalyzed coupling reaction of anthraquinone derivatives with arylboronates via a C–H or/and C–O bond cleavage. The reaction sequence involving the arylation, carbonyl methylenation, and oxidative cyclization effectively provided various 3,6,13,16-tetrasubstituted tetrabenzo[a,d,j,m]coronenes in short steps from readily available starting materials. The tetrabenzo[a,d,j,m]coronenes possessing two different types of substituents were obtained selectively by sequential chemoselective C–O and C–H arylations. The strategy developed in this study should be useful for the synthesis of PAH derivatives with multiple substituents having different properties.
Supporting Information
| Supporting Information File 1: General experimental procedures, characterization data and NMR spectra of new compounds. | ||
| Format: PDF | Size: 1.8 MB | Download |
Funding
This work was supported, in part, by a Grant-in-Aid for Scientific Research on Innovative Areas 2707 “Middle molecule strategy” from the Ministry of Education, Culture, Sports, Science and Technology, Japan and by the Research Program of “Five-star Alliance” in “NJRC Mater. & Dev.”. T.K. is grateful for financial support provided by JSPS KAKENHI Grant Number JP18H04271 (Precisely Designed Catalysts with Customized Scaffolding).
References
-
Grimsdale, A. C.; Leok Chan, K.; Martin, R. E.; Jokisz, P. G.; Holmes, A. B. Chem. Rev. 2009, 109, 897–1091. doi:10.1021/cr000013v
Return to citation in text: [1] -
Feng, F.; Liu, L.; Wang, S. Nat. Protoc. 2010, 5, 1255–1264. doi:10.1038/nprot.2010.79
Return to citation in text: [1] -
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Narita, A.; Wang, X.-Y.; Feng, X.; Müllen, K. Chem. Soc. Rev. 2015, 44, 6616–6643. doi:10.1039/c5cs00183h
Return to citation in text: [1] -
Miyaura, N., Ed. Cross-Coupling Reactions: A Practical Guide; Top. Curr. Chem., Vol. 2019; 2002. doi:10.1007/3-540-45313-x
Return to citation in text: [1] -
Martin, R.; Buchwald, S. L. Acc. Chem. Res. 2008, 41, 1461–1473. doi:10.1021/ar800036s
Return to citation in text: [1] -
Nishihara, Y., Ed. Applied Cross-Coupling Reactions; Springer: Heidelberg, Germany, 2013. doi:10.1007/978-3-642-32368-3
Return to citation in text: [1] -
de Meijere, A.; Bräse, S.; Oestreich, M., Eds. Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527655588
Return to citation in text: [1] -
Tobisu, M.; Chatani, N. Acc. Chem. Res. 2015, 48, 1717–1726. doi:10.1021/acs.accounts.5b00051
Return to citation in text: [1] -
Hussain, I.; Singh, T. Adv. Synth. Catal. 2014, 356, 1661–1696. doi:10.1002/adsc.201400178
Return to citation in text: [1] [2] -
Kuninobu, Y.; Sueki, S. Synthesis 2015, 47, 3823–3845. doi:10.1055/s-0035-1560346
Return to citation in text: [1] [2] -
Segawa, Y.; Maekawa, T.; Itami, K. Angew. Chem., Int. Ed. 2015, 54, 66–81. doi:10.1002/anie.201403729
Return to citation in text: [1] -
Sharma, U.; Modak, A.; Maity, S.; Maji, A.; Maiti, D. Direct arylation via C–H activation. In New Trends in Cross-Coupling: Theory and Applications; Colacot, T. J., Ed.; RSC Publishing: Cambridge, UK, 2015; pp 551–609. doi:10.1039/9781782620259-00551
Return to citation in text: [1] -
Zha, G.-F.; Qin, H.-L.; Kantchev, E. A. B. RSC Adv. 2016, 6, 30875–30885. doi:10.1039/c6ra02742c
Return to citation in text: [1] -
Hagui, W.; Doucet, H.; Soulé, J.-F. Chem 2019, 5, 2006–2078. doi:10.1016/j.chempr.2019.06.005
Return to citation in text: [1] -
Kitazawa, K.; Kochi, T.; Sato, M.; Kakiuchi, F. Org. Lett. 2009, 11, 1951–1954. doi:10.1021/ol900393x
Return to citation in text: [1] [2] [3] [4] [5] -
Kitazawa, K.; Kochi, T.; Nitani, M.; Ie, Y.; Aso, Y.; Kakiuchi, F. Chem. Lett. 2011, 40, 300–302. doi:10.1246/cl.2011.300
Return to citation in text: [1] [2] -
Matsumura, D.; Kitazawa, K.; Terai, S.; Kochi, T.; Ie, Y.; Nitani, M.; Aso, Y.; Kakiuchi, F. Org. Lett. 2012, 14, 3882–3885. doi:10.1021/ol301608m
Return to citation in text: [1] [2] [3] -
Suzuki, Y.; Yamada, K.; Watanabe, K.; Kochi, T.; Ie, Y.; Aso, Y.; Kakiuchi, F. Org. Lett. 2017, 19, 3791–3794. doi:10.1021/acs.orglett.7b01666
Return to citation in text: [1] [2] [3] [4] -
Izumoto, A.; Kondo, H.; Kochi, T.; Kakiuchi, F. Synlett 2017, 28, 2609–2613. doi:10.1055/s-0036-1590937
Return to citation in text: [1] [2] [3] -
Kakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2003, 125, 1698–1699. doi:10.1021/ja029273f
Return to citation in text: [1] [2] -
Kakiuchi, F.; Matsuura, Y.; Kan, S.; Chatani, N. J. Am. Chem. Soc. 2005, 127, 5936–5945. doi:10.1021/ja043334n
Return to citation in text: [1] [2] -
Kitazawa, K.; Kotani, M.; Kochi, T.; Langeloth, M.; Kakiuchi, F. J. Organomet. Chem. 2010, 695, 1163–1167. doi:10.1016/j.jorganchem.2010.01.022
Return to citation in text: [1] [2] -
Hiroshima, S.; Matsumura, D.; Kochi, T.; Kakiuchi, F. Org. Lett. 2010, 12, 5318–5321. doi:10.1021/ol102325f
Return to citation in text: [1] [2] -
Ogiwara, Y.; Miyake, M.; Kochi, T.; Kakiuchi, F. Organometallics 2017, 36, 159–164. doi:10.1021/acs.organomet.6b00540
Return to citation in text: [1] [2] -
Koseki, Y.; Kitazawa, K.; Miyake, M.; Kochi, T.; Kakiuchi, F. J. Org. Chem. 2017, 82, 6503–6510. doi:10.1021/acs.joc.6b02623
Return to citation in text: [1] [2] -
Suzuki, I.; Kondo, H.; Kochi, T.; Kakiuchi, F. J. Org. Chem. 2019, 84, 12975–12982. doi:10.1021/acs.joc.9b01756
Return to citation in text: [1] [2] -
Kakiuchi, F.; Usui, M.; Ueno, S.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2004, 126, 2706–2707. doi:10.1021/ja0393170
Return to citation in text: [1] [2] -
Ueno, S.; Mizushima, E.; Chatani, N.; Kakiuchi, F. J. Am. Chem. Soc. 2006, 128, 16516–16517. doi:10.1021/ja067612p
Return to citation in text: [1] [2] -
Ogiwara, Y.; Kochi, T.; Kakiuchi, F. Org. Lett. 2011, 13, 3254–3257. doi:10.1021/ol2012007
Return to citation in text: [1] [2] -
Kondo, H.; Akiba, N.; Kochi, T.; Kakiuchi, F. Angew. Chem., Int. Ed. 2015, 54, 9293–9297. doi:10.1002/anie.201503641
Return to citation in text: [1] [2] -
Kondo, H.; Kochi, T.; Kakiuchi, F. Org. Lett. 2017, 19, 794–797. doi:10.1021/acs.orglett.6b03761
Return to citation in text: [1] [2] -
Kondo, H.; Kochi, T.; Kakiuchi, F. Chem. – Eur. J. 2020, 26, 1737–1741. doi:10.1002/chem.201905157
Return to citation in text: [1] [2] -
Feng, X.; Pisula, W.; Müllen, K. Pure Appl. Chem. 2009, 81, 2203–2224. doi:10.1351/pac-con-09-07-07
Return to citation in text: [1] [2] -
Hopf, H. Chem. Rec. 2014, 14, 979–1000. doi:10.1002/tcr.201402056
Return to citation in text: [1] [2] -
Zhang, Q.; Peng, H.; Zhang, G.; Lu, Q.; Chang, J.; Dong, Y.; Shi, X.; Wei, J. J. Am. Chem. Soc. 2014, 136, 5057–5064. doi:10.1021/ja413018f
Return to citation in text: [1] [2] -
Hirose, T.; Miyazaki, Y.; Watabe, M.; Akimoto, S.; Tachikawa, T.; Kodama, K.; Yasutake, M. Tetrahedron 2015, 71, 4714–4721. doi:10.1016/j.tet.2015.05.071
Return to citation in text: [1] [2] -
Witte, G.; Wöll, C. J. Mater. Res. 2004, 19, 1889–1916. doi:10.1557/jmr.2004.0251
Return to citation in text: [1] -
Wu, J.; Pisula, W.; Müllen, K. Chem. Rev. 2007, 107, 718–747. doi:10.1021/cr068010r
Return to citation in text: [1] -
Ren, S.; Yan, C.; Vak, D.; Jones, D. J.; Holmes, A. B.; Wong, W. W. H. Adv. Funct. Mater. 2012, 22, 2015–2026. doi:10.1002/adfm.201102141
Return to citation in text: [1] -
Wong, W. W. H.; Subbiah, J.; Puniredd, S. R.; Purushothaman, B.; Pisula, W.; Kirby, N.; Müllen, K.; Jones, D. J.; Holmes, A. B. J. Mater. Chem. 2012, 22, 21131–21137. doi:10.1039/c2jm34394k
Return to citation in text: [1] -
Dumslaff, T.; Yang, B.; Maghsoumi, A.; Velpula, G.; Mali, K. S.; Castiglioni, C.; De Feyter, S.; Tommasini, M.; Narita, A.; Feng, X.; Müllen, K. J. Am. Chem. Soc. 2016, 138, 4726–4729. doi:10.1021/jacs.6b01976
Return to citation in text: [1] -
Zhang, X.; Jiang, X.; Zhang, K.; Mao, L.; Luo, J.; Chi, C.; Chan, H. S. O.; Wu, J. J. Org. Chem. 2010, 75, 8069–8077. doi:10.1021/jo101701k
Return to citation in text: [1] [2] [3] -
Pola, S.; Kuo, C.-H.; Peng, W.-T.; Islam, M. M.; Chao, I.; Tao, Y.-T. Chem. Mater. 2012, 24, 2566–2571. doi:10.1021/cm301190c
Return to citation in text: [1] [2] [3]
| 10. | Hussain, I.; Singh, T. Adv. Synth. Catal. 2014, 356, 1661–1696. doi:10.1002/adsc.201400178 |
| 19. | Suzuki, Y.; Yamada, K.; Watanabe, K.; Kochi, T.; Ie, Y.; Aso, Y.; Kakiuchi, F. Org. Lett. 2017, 19, 3791–3794. doi:10.1021/acs.orglett.7b01666 |
| 11. | Kuninobu, Y.; Sueki, S. Synthesis 2015, 47, 3823–3845. doi:10.1055/s-0035-1560346 |
| 1. | Grimsdale, A. C.; Leok Chan, K.; Martin, R. E.; Jokisz, P. G.; Holmes, A. B. Chem. Rev. 2009, 109, 897–1091. doi:10.1021/cr000013v |
| 2. | Feng, F.; Liu, L.; Wang, S. Nat. Protoc. 2010, 5, 1255–1264. doi:10.1038/nprot.2010.79 |
| 3. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 16. | Kitazawa, K.; Kochi, T.; Sato, M.; Kakiuchi, F. Org. Lett. 2009, 11, 1951–1954. doi:10.1021/ol900393x |
| 17. | Kitazawa, K.; Kochi, T.; Nitani, M.; Ie, Y.; Aso, Y.; Kakiuchi, F. Chem. Lett. 2011, 40, 300–302. doi:10.1246/cl.2011.300 |
| 18. | Matsumura, D.; Kitazawa, K.; Terai, S.; Kochi, T.; Ie, Y.; Nitani, M.; Aso, Y.; Kakiuchi, F. Org. Lett. 2012, 14, 3882–3885. doi:10.1021/ol301608m |
| 19. | Suzuki, Y.; Yamada, K.; Watanabe, K.; Kochi, T.; Ie, Y.; Aso, Y.; Kakiuchi, F. Org. Lett. 2017, 19, 3791–3794. doi:10.1021/acs.orglett.7b01666 |
| 20. | Izumoto, A.; Kondo, H.; Kochi, T.; Kakiuchi, F. Synlett 2017, 28, 2609–2613. doi:10.1055/s-0036-1590937 |
| 21. | Kakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2003, 125, 1698–1699. doi:10.1021/ja029273f |
| 22. | Kakiuchi, F.; Matsuura, Y.; Kan, S.; Chatani, N. J. Am. Chem. Soc. 2005, 127, 5936–5945. doi:10.1021/ja043334n |
| 23. | Kitazawa, K.; Kotani, M.; Kochi, T.; Langeloth, M.; Kakiuchi, F. J. Organomet. Chem. 2010, 695, 1163–1167. doi:10.1016/j.jorganchem.2010.01.022 |
| 24. | Hiroshima, S.; Matsumura, D.; Kochi, T.; Kakiuchi, F. Org. Lett. 2010, 12, 5318–5321. doi:10.1021/ol102325f |
| 25. | Ogiwara, Y.; Miyake, M.; Kochi, T.; Kakiuchi, F. Organometallics 2017, 36, 159–164. doi:10.1021/acs.organomet.6b00540 |
| 26. | Koseki, Y.; Kitazawa, K.; Miyake, M.; Kochi, T.; Kakiuchi, F. J. Org. Chem. 2017, 82, 6503–6510. doi:10.1021/acs.joc.6b02623 |
| 27. | Suzuki, I.; Kondo, H.; Kochi, T.; Kakiuchi, F. J. Org. Chem. 2019, 84, 12975–12982. doi:10.1021/acs.joc.9b01756 |
| 28. | Kakiuchi, F.; Usui, M.; Ueno, S.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2004, 126, 2706–2707. doi:10.1021/ja0393170 |
| 29. | Ueno, S.; Mizushima, E.; Chatani, N.; Kakiuchi, F. J. Am. Chem. Soc. 2006, 128, 16516–16517. doi:10.1021/ja067612p |
| 30. | Ogiwara, Y.; Kochi, T.; Kakiuchi, F. Org. Lett. 2011, 13, 3254–3257. doi:10.1021/ol2012007 |
| 31. | Kondo, H.; Akiba, N.; Kochi, T.; Kakiuchi, F. Angew. Chem., Int. Ed. 2015, 54, 9293–9297. doi:10.1002/anie.201503641 |
| 32. | Kondo, H.; Kochi, T.; Kakiuchi, F. Org. Lett. 2017, 19, 794–797. doi:10.1021/acs.orglett.6b03761 |
| 33. | Kondo, H.; Kochi, T.; Kakiuchi, F. Chem. – Eur. J. 2020, 26, 1737–1741. doi:10.1002/chem.201905157 |
| 16. | Kitazawa, K.; Kochi, T.; Sato, M.; Kakiuchi, F. Org. Lett. 2009, 11, 1951–1954. doi:10.1021/ol900393x |
| 10. | Hussain, I.; Singh, T. Adv. Synth. Catal. 2014, 356, 1661–1696. doi:10.1002/adsc.201400178 |
| 11. | Kuninobu, Y.; Sueki, S. Synthesis 2015, 47, 3823–3845. doi:10.1055/s-0035-1560346 |
| 12. | Segawa, Y.; Maekawa, T.; Itami, K. Angew. Chem., Int. Ed. 2015, 54, 66–81. doi:10.1002/anie.201403729 |
| 13. | Sharma, U.; Modak, A.; Maity, S.; Maji, A.; Maiti, D. Direct arylation via C–H activation. In New Trends in Cross-Coupling: Theory and Applications; Colacot, T. J., Ed.; RSC Publishing: Cambridge, UK, 2015; pp 551–609. doi:10.1039/9781782620259-00551 |
| 14. | Zha, G.-F.; Qin, H.-L.; Kantchev, E. A. B. RSC Adv. 2016, 6, 30875–30885. doi:10.1039/c6ra02742c |
| 15. | Hagui, W.; Doucet, H.; Soulé, J.-F. Chem 2019, 5, 2006–2078. doi:10.1016/j.chempr.2019.06.005 |
| 16. | Kitazawa, K.; Kochi, T.; Sato, M.; Kakiuchi, F. Org. Lett. 2009, 11, 1951–1954. doi:10.1021/ol900393x |
| 5. | Miyaura, N., Ed. Cross-Coupling Reactions: A Practical Guide; Top. Curr. Chem., Vol. 2019; 2002. doi:10.1007/3-540-45313-x |
| 6. | Martin, R.; Buchwald, S. L. Acc. Chem. Res. 2008, 41, 1461–1473. doi:10.1021/ar800036s |
| 7. | Nishihara, Y., Ed. Applied Cross-Coupling Reactions; Springer: Heidelberg, Germany, 2013. doi:10.1007/978-3-642-32368-3 |
| 8. | de Meijere, A.; Bräse, S.; Oestreich, M., Eds. Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527655588 |
| 9. | Tobisu, M.; Chatani, N. Acc. Chem. Res. 2015, 48, 1717–1726. doi:10.1021/acs.accounts.5b00051 |
| 43. | Zhang, X.; Jiang, X.; Zhang, K.; Mao, L.; Luo, J.; Chi, C.; Chan, H. S. O.; Wu, J. J. Org. Chem. 2010, 75, 8069–8077. doi:10.1021/jo101701k |
| 4. | Narita, A.; Wang, X.-Y.; Feng, X.; Müllen, K. Chem. Soc. Rev. 2015, 44, 6616–6643. doi:10.1039/c5cs00183h |
| 44. | Pola, S.; Kuo, C.-H.; Peng, W.-T.; Islam, M. M.; Chao, I.; Tao, Y.-T. Chem. Mater. 2012, 24, 2566–2571. doi:10.1021/cm301190c |
| 19. | Suzuki, Y.; Yamada, K.; Watanabe, K.; Kochi, T.; Ie, Y.; Aso, Y.; Kakiuchi, F. Org. Lett. 2017, 19, 3791–3794. doi:10.1021/acs.orglett.7b01666 |
| 34. | Feng, X.; Pisula, W.; Müllen, K. Pure Appl. Chem. 2009, 81, 2203–2224. doi:10.1351/pac-con-09-07-07 |
| 35. | Hopf, H. Chem. Rec. 2014, 14, 979–1000. doi:10.1002/tcr.201402056 |
| 36. | Zhang, Q.; Peng, H.; Zhang, G.; Lu, Q.; Chang, J.; Dong, Y.; Shi, X.; Wei, J. J. Am. Chem. Soc. 2014, 136, 5057–5064. doi:10.1021/ja413018f |
| 37. | Hirose, T.; Miyazaki, Y.; Watabe, M.; Akimoto, S.; Tachikawa, T.; Kodama, K.; Yasutake, M. Tetrahedron 2015, 71, 4714–4721. doi:10.1016/j.tet.2015.05.071 |
| 16. | Kitazawa, K.; Kochi, T.; Sato, M.; Kakiuchi, F. Org. Lett. 2009, 11, 1951–1954. doi:10.1021/ol900393x |
| 18. | Matsumura, D.; Kitazawa, K.; Terai, S.; Kochi, T.; Ie, Y.; Nitani, M.; Aso, Y.; Kakiuchi, F. Org. Lett. 2012, 14, 3882–3885. doi:10.1021/ol301608m |
| 20. | Izumoto, A.; Kondo, H.; Kochi, T.; Kakiuchi, F. Synlett 2017, 28, 2609–2613. doi:10.1055/s-0036-1590937 |
| 43. | Zhang, X.; Jiang, X.; Zhang, K.; Mao, L.; Luo, J.; Chi, C.; Chan, H. S. O.; Wu, J. J. Org. Chem. 2010, 75, 8069–8077. doi:10.1021/jo101701k |
| 44. | Pola, S.; Kuo, C.-H.; Peng, W.-T.; Islam, M. M.; Chao, I.; Tao, Y.-T. Chem. Mater. 2012, 24, 2566–2571. doi:10.1021/cm301190c |
| 16. | Kitazawa, K.; Kochi, T.; Sato, M.; Kakiuchi, F. Org. Lett. 2009, 11, 1951–1954. doi:10.1021/ol900393x |
| 17. | Kitazawa, K.; Kochi, T.; Nitani, M.; Ie, Y.; Aso, Y.; Kakiuchi, F. Chem. Lett. 2011, 40, 300–302. doi:10.1246/cl.2011.300 |
| 18. | Matsumura, D.; Kitazawa, K.; Terai, S.; Kochi, T.; Ie, Y.; Nitani, M.; Aso, Y.; Kakiuchi, F. Org. Lett. 2012, 14, 3882–3885. doi:10.1021/ol301608m |
| 19. | Suzuki, Y.; Yamada, K.; Watanabe, K.; Kochi, T.; Ie, Y.; Aso, Y.; Kakiuchi, F. Org. Lett. 2017, 19, 3791–3794. doi:10.1021/acs.orglett.7b01666 |
| 20. | Izumoto, A.; Kondo, H.; Kochi, T.; Kakiuchi, F. Synlett 2017, 28, 2609–2613. doi:10.1055/s-0036-1590937 |
| 21. | Kakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2003, 125, 1698–1699. doi:10.1021/ja029273f |
| 22. | Kakiuchi, F.; Matsuura, Y.; Kan, S.; Chatani, N. J. Am. Chem. Soc. 2005, 127, 5936–5945. doi:10.1021/ja043334n |
| 23. | Kitazawa, K.; Kotani, M.; Kochi, T.; Langeloth, M.; Kakiuchi, F. J. Organomet. Chem. 2010, 695, 1163–1167. doi:10.1016/j.jorganchem.2010.01.022 |
| 24. | Hiroshima, S.; Matsumura, D.; Kochi, T.; Kakiuchi, F. Org. Lett. 2010, 12, 5318–5321. doi:10.1021/ol102325f |
| 25. | Ogiwara, Y.; Miyake, M.; Kochi, T.; Kakiuchi, F. Organometallics 2017, 36, 159–164. doi:10.1021/acs.organomet.6b00540 |
| 26. | Koseki, Y.; Kitazawa, K.; Miyake, M.; Kochi, T.; Kakiuchi, F. J. Org. Chem. 2017, 82, 6503–6510. doi:10.1021/acs.joc.6b02623 |
| 27. | Suzuki, I.; Kondo, H.; Kochi, T.; Kakiuchi, F. J. Org. Chem. 2019, 84, 12975–12982. doi:10.1021/acs.joc.9b01756 |
| 28. | Kakiuchi, F.; Usui, M.; Ueno, S.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2004, 126, 2706–2707. doi:10.1021/ja0393170 |
| 29. | Ueno, S.; Mizushima, E.; Chatani, N.; Kakiuchi, F. J. Am. Chem. Soc. 2006, 128, 16516–16517. doi:10.1021/ja067612p |
| 30. | Ogiwara, Y.; Kochi, T.; Kakiuchi, F. Org. Lett. 2011, 13, 3254–3257. doi:10.1021/ol2012007 |
| 31. | Kondo, H.; Akiba, N.; Kochi, T.; Kakiuchi, F. Angew. Chem., Int. Ed. 2015, 54, 9293–9297. doi:10.1002/anie.201503641 |
| 32. | Kondo, H.; Kochi, T.; Kakiuchi, F. Org. Lett. 2017, 19, 794–797. doi:10.1021/acs.orglett.6b03761 |
| 33. | Kondo, H.; Kochi, T.; Kakiuchi, F. Chem. – Eur. J. 2020, 26, 1737–1741. doi:10.1002/chem.201905157 |
| 34. | Feng, X.; Pisula, W.; Müllen, K. Pure Appl. Chem. 2009, 81, 2203–2224. doi:10.1351/pac-con-09-07-07 |
| 35. | Hopf, H. Chem. Rec. 2014, 14, 979–1000. doi:10.1002/tcr.201402056 |
| 36. | Zhang, Q.; Peng, H.; Zhang, G.; Lu, Q.; Chang, J.; Dong, Y.; Shi, X.; Wei, J. J. Am. Chem. Soc. 2014, 136, 5057–5064. doi:10.1021/ja413018f |
| 37. | Hirose, T.; Miyazaki, Y.; Watabe, M.; Akimoto, S.; Tachikawa, T.; Kodama, K.; Yasutake, M. Tetrahedron 2015, 71, 4714–4721. doi:10.1016/j.tet.2015.05.071 |
| 38. | Witte, G.; Wöll, C. J. Mater. Res. 2004, 19, 1889–1916. doi:10.1557/jmr.2004.0251 |
| 39. | Wu, J.; Pisula, W.; Müllen, K. Chem. Rev. 2007, 107, 718–747. doi:10.1021/cr068010r |
| 40. | Ren, S.; Yan, C.; Vak, D.; Jones, D. J.; Holmes, A. B.; Wong, W. W. H. Adv. Funct. Mater. 2012, 22, 2015–2026. doi:10.1002/adfm.201102141 |
| 41. | Wong, W. W. H.; Subbiah, J.; Puniredd, S. R.; Purushothaman, B.; Pisula, W.; Kirby, N.; Müllen, K.; Jones, D. J.; Holmes, A. B. J. Mater. Chem. 2012, 22, 21131–21137. doi:10.1039/c2jm34394k |
| 42. | Dumslaff, T.; Yang, B.; Maghsoumi, A.; Velpula, G.; Mali, K. S.; Castiglioni, C.; De Feyter, S.; Tommasini, M.; Narita, A.; Feng, X.; Müllen, K. J. Am. Chem. Soc. 2016, 138, 4726–4729. doi:10.1021/jacs.6b01976 |
| 43. | Zhang, X.; Jiang, X.; Zhang, K.; Mao, L.; Luo, J.; Chi, C.; Chan, H. S. O.; Wu, J. J. Org. Chem. 2010, 75, 8069–8077. doi:10.1021/jo101701k |
| 44. | Pola, S.; Kuo, C.-H.; Peng, W.-T.; Islam, M. M.; Chao, I.; Tao, Y.-T. Chem. Mater. 2012, 24, 2566–2571. doi:10.1021/cm301190c |
© 2020 Terai et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)