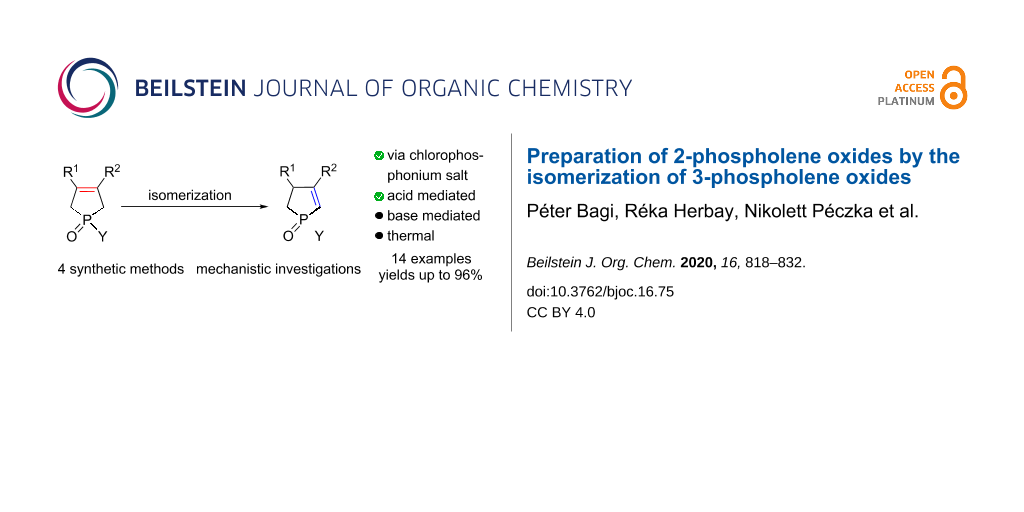
Abstract
A series of 1-substituted-3-methyl-2-phospholene oxides was prepared from the corresponding 3-phospholene oxides by double bond rearrangement. The 2-phospholene oxides could be obtained by heating the 3-phospholene oxides in methanesulfonic acid, or via the formation of cyclic chlorophosphonium salts. Whereas mixtures of the 2- and 3-phospholene oxides formed, when the isomerization of 3-phospholene oxides was attempted under thermal conditions, or in the presence of a base. The mechanisms of the various double bond migration pathways were elucidated by quantum chemical calculations.

Graphical Abstract
Introduction
P-Heterocyclic derivatives are valuable targets in synthetic organophosphorus chemistry [1-4]. A heterocyclic ring containing a P atom appears in many popular P(III)-ligands, such as BPE, DuPhos, TangPhos, DuanPhos, ZhangPhos [5-11], among which a number of species may be considered as privileged ligands for transition metal-catalyzed enantioselective transformations [12]. Among the P-heterocycles with various ring size, the five-membered derivatives received special attention due to their optimal bond angles and shapes [13,14]. Besides their application as ligands [13,15], the five-membered heterocycles may also be valuable organocatalysts in reactions involving a P(III)–P(V) redox cycle, such as in catalytic Wittig-, aza-Wittig-, Staudinger-, Appel- and other reactions [16-21]. The ring strain of the four- and five-membered derivatives makes them highly susceptible to deoxygenation, thus they are ideal organocatalysts in P(III)–P(V) redox-coupled transformations [18,22,23]. Polycyclic compounds incorporating a P-heterocyclic moiety are of special importance due to their optoelectronic properties and applications [24-26]. Moreover, a few biologically active 5-membered P-heterocyclic derivatives are also known, which showed promising antitumor [27-30], antifungal [31,32] or GABA receptor antagonist activity [33].
The most common synthesis of a given functionalized P-heterocyclic compound comprises the synthesis of the heterocyclic core, followed by its functionalization [27,29,34,35]. This latter step is of special importance for biologically active compounds, as specific functional groups are necessary to have a desired biological activity. Considering the functionalization of the five-membered P-heterocycles, the derivatives containing double bond(s) (i.e., phospholenes or phospholes) are of special importance, as a double bond may be functionalized by diverse reactions [36]. Considering the phospholenes, the position of the double bond (2- or 3-phospholenes) offers different possibilities for further transformations (Figure 1) [37-46]. Moreover, the presence of the double bond in the 2-phospholene derivatives is beneficial for having enhanced organocatalytic activity, in a few instances [47].
![[1860-5397-16-75-1]](/bjoc/content/figures/1860-5397-16-75-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples for catalytically or biologically active molecules containing five-membered P-heterocyclic rings. General strategies for the preparation and functionalization of 2- and 3-phospholene oxides.
Figure 1: Examples for catalytically or biologically active molecules containing five-membered P-heterocyclic...
The McCormack cycloaddition is a key reaction for the preparation of the five-membered P-heterocyclic scaffold, as both the 2- or 3-phospholene core can be prepared depending on the butadiene derivative and P-reagent used [36,48-50]. However, the long reaction times, as well as the moderate or low yields make this approach less appealing. More recent syntheses of the phospholene oxides involve either a ring-closing metathesis [35,51-54], or a double Arbuzov reaction of 1,4-dichlorobut-2-ene and a silylated P-reagent [34,35,55]. However, these syntheses generally afford the corresponding 3-phospholene oxides, and only a few examples are known to provide the corresponding 2-phospholene derivatives [56,57], which raises the necessity of complementary synthetic strategies for 2-phospholene oxides (Figure 1).
A convenient strategy may involve the isomerization of 3-phospholene oxides to the corresponding 2-phospholene oxides. A few isomerizations resulting 2-phospholene derivatives have already been observed and published in the literature [58-61]. However, either the isomerization remained incomplete [58,59], or it was an undesired side-reaction caused by the high temperature and the excess of the reagent [62-64]. Our recent synthetic efforts were directed to the novel preparation of cyclic chlorophospholenium chlorides, and the utilization of these reactive derivatives in silane-free deoxygenations or dynamic resolutions [65-67]. In these studies, the isomerization of the 1-chloro-3-methyl-1-phenyl-3-phospholenium chloride to the corresponding 2-phospholenium salt was also observed as a side-reaction upon prolonged reaction times. It was also shown that the corresponding 2-phospholene oxides are the thermodynamically more stable isomers.
This paper seeks simple synthetic methods for the preparation of 2-phospholene oxides via the isomerization of the corresponding 3-phospholene derivatives. The isomerization reactions were carried out under thermal conditions, in the presence of acids or bases or via the formation of chlorophosphonium salts. In our study, the 3-methyl-3-phospholene oxides were considered as model compounds, the double bond rearrangement of a 3-phospholene oxide and a 3,4-dimethyl-3-phospholene oxide was also investigated. Moreover, we wish to interpret the different isomerization mechanisms by quantumchemical calculations.
Results and Discussion
Preparation of 1-substituted-3-methyl-2-phospholene oxides (4) via chlorophospholenium chlorides (2 and 3)
The preparation of 2-phospholene oxides 4 was first attempted via the formation of chlorophospholenium salts 2 and 3. According to our procedure [65], the corresponding 1-substituted-3-methyl-3-phospholene oxide 1 was reacted with oxalyl chloride to give chloro-3-phospholenium salts 2 with complete conversion. In our experience, a synthetic protocol involving the use of oxalyl chloride in a slight excess, followed by the removal of the unreacted reagent under reduced pressure gave the corresponding chloro-3-methyl-3-phospholenium chlorides 2 in the most reproducible manner. The chloro-3-phospholenium salts 2 were then dissolved in chloroform, and were heated at 80 °C for 48 h. Upon this period, nearly complete isomerization of the chloro-3-phospholenium salts 2 to the corresponding chloro-2-phospholenium salts 3 occurred. The mechanism of this isomerization was investigated by quantumchemical calculations in our previous study [65]. However, the formation of the 2-phospholenium chlorides 3 was only postulated. Herein, two additional derivatives, the 4-methylphenyl- and ethyl-1-chloro-3-methyl-2-phospholenium chlorides 3c and 3h were isolated and characterized, which confirmed the key intermediate of the proposed mechanism. The chloro-2-phospholenium salts 3 were then hydrolyzed to give the corresponding 3-methyl-2-phospholene oxides 4 in an isomeric ratio above 97:3 and in yields of 71–96% (Table 1). If necessary, the traces of starting material 1 could be removed by simple column chromatography. Efficient chromatographic separation of 2- and 3-phospholene oxide isomers 4 and 1 could be accomplished in a mixture of acetone and 2-PrOH. The 2-phospholene oxides 4 obtained were characterized by spectroscopic methods (see Supporting Information File 1 for details).
Table 1: Preparation of 1-substituted-3-methyl-2-phospholene oxides 4 via chlorophospholenium chlorides (2 and 3).
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-75-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Y | Yield (%)a | Ratio of 4:1 (%)b |
| 1 | Ph (a) | 85 | 99:1 |
| 2 | 2-Me-C6H4 (b) | 84 | 97:3 |
| 3 | 4-Me-C6H4 (c) | 86 | 99:1 |
| 4 | 4-CF3-C6H4 (d) | 89 | 98:2 |
| 5 | 4-MeO-C6H4 (e) | 95 | 97:3 |
| 6 | 2,6-diMe-C6H3 (f) | 96 | 99:1 |
| 7 | 1-naphthyl (g) | 90 | 100:0 |
| 8 | Et (h) | 71 | 97:3 |
| 9 | n-Pr (i) | 75 | 99:1 |
| 10 | n-Bu (j) | 87 | 98:2 |
| 11 | iBu (k) | 87 | 99:1 |
| 12 | iPent (l) | 86 | 98:2 |
aIsolated yield of the mixture of 1 and 4; bdetermined by GC.
Preparation of 3-methyl-2-phospholene oxides 4 under acidic conditions
As the next step of this study, we wished to find another isomerization pathways leading to the corresponding 2-phospholene oxides 4, which does not require inert reaction conditions. Literature data indicated that such izomerizations may take place in the presence of acids [59,62,64]. 1-Phenyl-3-methyl-3-phospholene oxide (1a) was selected as the model compound, and a series of acids was tested. When phenylphospholene oxide 1a was heated in aqueous HCl, or in the presence of p-toluenesulfonic acid in a toluene solution significant isomerization could not be observed. When 1-phenyl-3-phospholene oxide (1a) was refluxed in trifluoroacetic acid, a mixture containing the corresponding 2- and 3-phospholene oxides (4a and 1a) in a ratio of 11:89 was obtained. However, when phospholene oxide 1a was heated in neat methanesulfonic acid at 160 °C for 24 h, 4a and 1a in a ratio of 97:3 were obtained, but the yield of the mixture was 57% due to decomposition caused by the vigorous reaction conditions. Based on this promising preliminary result with MeSO3H, the effect of both the reaction temperature and the reaction time was investigated. It was found that complete isomerization to the 2-phospholene oxide derivative 4a could not be reached with MeSO3H at 25 °C even after prolonged reaction times. Setting the reaction temperature to 50 °C and the reaction time to 60 h seemed to be the reasonable choice, as nearly the same isomeric composition (96:4) could be achieved as at 160 °C, but with significantly increased yield (up to 81%) (See Supporting Information File 1 for the optimization of the reaction conditions). Under these conditions, a series of 1-aryl- or 1-alkyl-3-methyl-3-phospholene oxides 1 was isomerized, and the products containing 2-phospholene oxides 4 in a range of 96–100% were obtained in yields of 81–96%. From the series of 3-phospholene oxides investigated, ethyl derivative 4h was somewhat the exception, as the yield of the crude product was only 55%, which contained 92% of desired 2-phospholene derivative 4h. This may be the consequence of a higher degree of decomposition occurring in case of this derivative (Table 2).
Table 2: Preparation of 1-substituted-3-methyl-2-phospholene oxides 4 in the presence of methanesulfonic acid.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-75-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Y | Yield (%)a | Ratio of 4:1 (%)b |
| 1 | Ph (a) | 81 | 96:4 |
| 2 | 2-Me-C6H4 (b) | 90 | 100:0 |
| 3 | 4-Me-C6H4 (c) | 94 | 99:1 |
| 4 | 4-CF3-C6H4 (d) | 88 | 98:2 |
| 5 | 4-MeO-C6H4 (e) | 87 | 97:3 |
| 6 | 2,6-diMe-C6H3 (f) | 94 | 99:1 |
| 7 | 1-naphthyl (g) | 96 | 98:2 |
| 8 | Et (h) | 55 | 92:8 |
| 9 | n-Pr (i) | 85 | 98:2 |
| 10 | n-Bu (j) | 95 | 99:1 |
| 11 | iBu (k) | 86 | 97:3 |
| 12 | iPent (l) | 84 | 98:2 |
aIsolated yield of the mixture of 1 and 4; bdetermined by GC.
Investigation of the isomerization of 3-phospholene oxides 1 to 2-phospholene oxides 4 under basic conditions
In the next stage, the base-mediated isomerization of 1-phenyl-3-methyl-3-phospholene oxide (1a) was also investigated. In the screening, several organic and inorganic bases, such as trimethylamine, N,N-diisopropylethylamine, pyridine, 4-dimethylaminopyridine, n-butyllithium, NaH and Cs2CO3 were tested. The initial experiments showed that practically no isomerization occurred when phenyl-3-phospholene oxide 1a was refluxed in toluene in the presence of organic bases (see Supporting Information File 1 for the results). Whereas the decomposition of the starting material was observed, when phenyl-3-phospholene oxide 1a was treated with n-butyllithium or NaH, as it was also observed by Stankevič et. al. [68] However, when phenyl-3-phospholene oxide was heated in the presence of 1 equiv of Cs2CO3 in toluene, the crude product obtained after the reaction contained the mixture of 1-phenyl-2-phospholene oxide 4a and the corresponding 3-phospholene oxide 1a in a 77:23 ratio. Based on this promising preliminary result, 1-ethyl-3-methyl-3-phospholene oxide (1h) was also included as a model compound for the alkyl derivatives. The isomerization of these two model compounds (1a and 1h) was tested in the presence of several inorganic bases, such as Na2CO3, K2CO3, Cs2CO3, NaOH and NaOEt in toluene, DMF or DMSO, with or without a phase transfer catalyst. The results indicated that the ratio of the 2- and 3-phospholene oxide isomers (4 and 1) did not exceed a ratio of ca. 80:20 and 70:30 in case of the phenyl- and the ethyl derivative, respectively, despite the base, solvent and reaction temperature used. The fact that practically the same results were obtained with a wide variety of bases suggested that the base-mediated isomerization leads to a thermodynamic equilibrium between phospholene oxides 1 and 4 catalyzed by the base (see Supporting Information File 1 for the results). In order to test this hypothesis, the two model compounds (1a and 1h) were refluxed with Cs2CO3 in DMF in the presence of TEBAC. The crude products obtained were reacted again two times under the same conditions. The results of these runs showed that the ratio of the corresponding 2- and 3-phospholene oxides (4a and 1a or 4h or 1h) remained practically unchanged throughout the reaction sequence (Table 3). These experiments suggested that the base-mediated isomerization leads to an equilibrium, which is in accordance with other reports in the literature [42]. Thus, this base-mediated isomerization was not studied further with other 3-phospholene oxide derivatives 1.
Table 3: Investigation of the isomerization of 1-phenyl- and 1-ethyl-3-methyl-3-phospholene oxides 1a or 1h to the corresponding 2-phospholene oxides 4 in the presence of Cs2CO3.
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-75-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Y | Run | Yield (%)a | Ratio of 4:1 (%)b |
| 1 | Ph (a) | I. | 77 | 84:16 |
| 2 | Ph (a) | II. | 76 | 80:20 |
| 3 | Ph (a) | III. | 67 | 81:19 |
| 4 | Et (h) | I. | 68 | 66:34 |
| 5 | Et (h) | II. | 78 | 67:33 |
| 6 | Et (h) | III. | 73 | 65:35 |
aIsolated yield of the mixture of 1 and 4; bdetermined by GC.
Isomerization of 3-phospholene oxides 1 to 2-phospholene oxides 4 under thermal conditions
The isomerization of 3-phospholene oxides 1 was also investigated under thermal conditions without using any reagent. In our initial attempts, 1-phenyl-3-methyl-3-phospholene oxide (1a) was refluxed in high boiling point solvents, such as toluene, DMF, or DMSO. However, the maximal content of phenyl-2-phospholene oxide 4a in the crude product was only 4%. In contrast, when neat 3-phospholene oxide 1a was heated to 200 °C without any solvent, the ratio of the 2- and 3-phospholene oxide 4a and 1a increased to 71:29 in the crude product after 24 h.
In order to study how the different substituents influence the rate of isomerization, a series of 1-substituted-3-methyl-3-phospholene oxides 1 was heated at 200 °C for 1 day in the absence of any solvent. The results indicated that the isomerization was not complete in any case even with prolonged reaction time, as the ratio of the 2- and 3-phospholene oxide isomers 4 and 1 was in the range of 43:57 and 60:40 for the alkyl derivatives (4h–l and 1h–l), and in between 63:37 and 85:15 for the arylphospholene oxides (4a–g and 1a–g). Due to the decomposition at the high temperature applied, yields of the crude products were moderate dropping in the range of 46–84% (Table 4). For the phenyl and the ethyl derivative (1a and 1h), the reaction time was extended to 5 days. However, this extension did not influence the ratio of the 2- and 3-phospholene oxide isomers (4 and 1) significantly (see Supporting Information File 1 for the results).
Table 4: Isomerization of 3-phospholene oxides 1 to 2-phospholene oxides 4 under thermal conditions.
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-75-i8.svg?max-width=637&scale=1.0)
|
|||
| Entry | Y | Yield (%)a | Ratio of 4:1 (%)b |
| 1 | Ph (a) | 58 | 71:29 |
| 2 | 2-Me-C6H4 (b) | 84 | 69:31 |
| 3 | 4-Me-C6H4 (c) | 83 | 63:37 |
| 4 | 4-CF3-C6H4 (d) | 81 | 72:28 |
| 5 | 4-MeO-C6H4 (e) | 82 | 73:27 |
| 6 | 2,6-diMe-C6H3 (f) | 75 | 85:15 |
| 7 | 1-naphthyl (g) | 69 | 73:27 |
| 8 | Et (h) | 50 | 55:45 |
| 9 | n-Pr (i) | 75 | 52:48 |
| 10 | n-Bu (j) | 68 | 49:51 |
| 11 | iBu (k) | 76 | 60:40 |
| 12 | iPent (l) | 46 | 43:57 |
aIsolated yield of the mixture of 1 and 4; bdetermined by GC.
Comparison of the isomerization of 1-phenyl-3-phospholene oxide (5), 1-phenyl-3-methyl-3-phospholene oxide (1a) and 1-phenyl-3,4-dimethyl-3-phospholene oxide (8) under the optimized conditions
As the last step of this study, it was investigated, how the methyl substituents on the P-heterocyclic core influence the outcome of the isomerization. Methyl group(s) in positon 3 and 4 represents a typical substitution pattern. Thus, the isomerization of 1-phenyl-3-phospholene oxide (5) and 1-phenyl-3,4-dimethyl-3-phospholene oxide (8) was investigated under the optimized conditions via the formation of chlorophospholenium salts (Scheme 1, route I), or using MeSO3H or Cs2CO3 (Scheme 1, routes II or III). These results were compared with those obtained for the 1-phenyl-3-methyl-3-phospholene oxide (1a). Scheme 1 shows that, considering all reaction pathways, the highest extent of isomerization was observed for 1-phenyl-3-phospholene oxide (5). Pure phenyl-2-phospholene oxide (7) could be prepared using MeSO3H, or via the formation of chlorophospholenium chloride (6), which indicates that these two routes may be applicable for the isomerization of other 1-substituted-3-phospholene oxides, as well. As it was expected, the lowest level of isomerization was observed in the presence of Cs2CO3, as the crude products contained the phenyl-2- and 3-phospholene oxides (7 and 5) in a ratio of 90:10.
![[1860-5397-16-75-i1]](/bjoc/content/inline/1860-5397-16-75-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Comparison of the isomerization of 1-phenyl-3-phospholene oxide (5), 1-phenyl-3-methyl-3-phospholene oxide (1a) and 1-phenyl-3,4-dimethyl-3-phospholene oxide (8). aIsolated yield of the mixture of 8–10, 1a–4a, 5–7, respectively. bDetermined by GC. cRatio of trans–cis isomers of 10: 44:56. dRatio of trans–cis isomers of 10: 45:55. eRatio of trans–cis isomers of 10: 40:60.
Scheme 1: Comparison of the isomerization of 1-phenyl-3-phospholene oxide (5), 1-phenyl-3-methyl-3-phospholen...
Considering the mono- and dimethyl-3-phospholene oxides 1a and 8, the ratio of the corresponding 2-phospholene oxides 4a or 10 was in the range of 84–96% for the 3-methyl-, and in the range of 34–86% for the 3,4-dimethyl derivative indicating that the additional methyl groups at position 3 or 4 hinder the migration of the double bond.
It is noteworthy, that in case of 1-phenyl-3,4-dimethyl-3-phospholene oxide (8), the isomerization involves the formation of an additional stereogenic center and the corresponding 2-phospholene oxide (10) was obtained as a mixture of cis- and trans-diastereomers. The ratio of these two diastereomers was in the range of 60:40 to 55:45. The cis- and trans-diastereomers (cis- and trans-10) were separated by column chromatography, and they were identified by nuclear Overhauser effect (NOE) NMR spectroscopy. In the ROESY spectrum of the cis-diastereomer (cis-10), the phenyl 1H resonances give NOE/ROE crosspeaks with the C(4)H and one of the C(5)H2 1H resonances. By contrast, in the spectrum of trans-diastereomer (trans-10), phenyl 1H resonances give NOE/ROE crosspeaks with C(4)-methyl and the other one of the C(5)H2 1H signals (see Supporting Information File 1 for details). Thus, based on the distinctive NOE patterns of phenyl resonances, the two diastereomers (cis- and trans-10) could be unambiguously identified.
Theoretical aspects of the isomerization of the 3-phospholene oxides 1 to 2-phospholene oxides 4
Thermodynamic description
The different isomerization pathways leading to the corresponding 2-phospholene oxides 4 were also investigated by quantum chemical calculations at MP2/6-31++G(d,p) and MP2/6-311++G(2d,2p) levels of theory, including the PCM solvent model with the parameters of THF. For these investigations, the 1-phenyl or 1-ethyl-3-methylphospholene oxides (1a or 1h) were considered as model compounds, but phospholene oxides incorporating substituted phenyl moiety (1c–f) were also calculated in a few instances. 1-Phenyl-3-phospholene oxide (5) and 1-phenyl-3,4-dimethyl-3-phospholene oxide (8) were also included in our calculations to interpret how the substituents on the P-heterocyclic ring affect the extent of the isomerization.
The conversion of 3-phospholene oxides 1 to the corresponding 2-phospholene derivatives 4 was first interpreted by means of thermodynamic data, and the olefinicity concept [69-72]. The calculations were performed at a high MP2/6-311G++(2d,2p) level of theory (Table 5). The reaction enthalpies (ΔH), as well as Gibbs free energies (ΔG) of the 1→4 transformation exhibited typically low values, and these reactions can be considered as nearly thermoneutral processes. Interestingly, the aromatic derivatives 1a or 1c–f prefer more the formation of the 2-phospholene oxide isomer (4a or 4c–f, Table 5, entries 3–7). In contrast, there is no benefit for the ethylphospholene oxides (1h or 4h) (Table 5, entry 8). The olefinicity concept and the olefinicity value (OL%) was developed to measure and describe quantitatively the degree of conjugation of an alkene group with the neighboring functional groups relative to ethylene (0%) and allyl anion (100%) as references on a linear scale. Large values mean a high degree of conjugations, while low values refer to a rather isolated double bond. The calculated olefinicity values (OL%) for 3- and 2-phospholene oxides 1 and 4 are within a narrow range, between 29–35%, and the change during the isomerization of 1→4 also confirms the thermodynamic data. When 3-phospholene oxide 1 exhibits a lower OL%, it is transformed in an exothermic fashion to the corresponding 2-phospholene oxide 4 having a higher OL% value. The phospholene oxides with aromatic substituents exhibit slightly higher OL% (ca. 4%) towards 2-phospholene oxide 4 than to the corresponding 3-phospholene isomer 1 (Table 5, entries 3–7). In contrast, the difference of the OL% values is almost negligible for the ethyl-3- or 2-phospholene oxides 1h or 4h (Table 5, entry 8). Both the thermodynamic data and the OL% values indicate that the additional methyl groups on the phospholene ring also influence the ratio of the corresponding 2- or 3-phospholene oxides 4 or 1. The calculations suggest that the 3-phospholene oxide isomer 8 is the most stable form in case of the phenyl-3,4-dimethylphospholene oxide, while the corresponding 2-phospholene oxides 4a or 7 are the preferred isomers for the unsubstituted or methyl-substituted phospholene oxide derivatives (Table 5, entries 1–3). According to the thermodynamic data without the consideration of the reaction mechanisms and transition states (Table 5), one may predict a well-balanced equilibrium between the 2- or 3-phospholene oxides 4 or 1 in a final state. The experimental results obtained under basic or thermal conditions support this equilibrium concept. In contrast, nearly complete isomerization was observed via the formation of chlorophospholenium chlorides or in the presence of methanesulfonic acid, which may indicate that the intermediates formed over the course of the reaction may perturb the thermodynamic equilibrium of the 2- or 3-phospholene oxides 4 or 1. It was shown in our previous study [65] that the relative stability shifts towards the 2-phospholene isomer, when chlorophospholenium salts are involved, and it is the underlying reason why the 2-phospholene oxides 4 can be prepared effectively by this reaction. Under strong acidic conditions, the P=O moiety tends to be protonated, which may be responsible for the complete isomerization in the presence of methanesulfonic acid. The thermodynamic data and the olefinicity values (OL%) calculated for the corresponding protonated derivatives 1+H+ and 4+H+ showed that indeed this protonation perturbs the original equilibrium significantly. The protonated 2-phospholene oxide 4+H+ became the most preferred isomer against protonated 3-phospholene oxide 1+H+ with the isomeric ratio exceeding 90:10 (Table 5, entries 12–16.). The aqueous work-up of the reaction mixture is an irreversible process, and it freezes the results of the acidic equilibrium of (1+H+)→(4+H+), providing the corresponding 2-phospholene-oxides 4 with good yields, despite the original enthalpy differences of 1 and 4. The calculations also showed that additional methyl groups on the P-heterocyclic ring influence the acid mediated isomerization (4 or 1, Table 5, entries 9–11.). The 2-phospholene oxides (4a or 7) are the preferred isomers for the unsubstituted and methyl-substituted phospholene core. In contrast, the calculations predicted a 73:27 ratio for the dimethyl-substituted 2- and 3-phoshopholene oxides (10 and 8) (Table 5, entry 10). These predictions showed strong correlation with the experimental results.
Table 5: Reaction enthalpies (ΔH in kJ mol−1) of the isomerization processes of 1→4 and (1+H+)→(4+H+) at MP2/6-311G++(2d,2p) level of theory. Their calculated olefinicity values (OL%) are also given in percentages at the same level of theory.
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-75-i9.svg?max-width=637&scale=1.0)
|
|||||||||
| R1 | R2 | Y | Entry |
ΔH
(1→4) |
ΔG
(1→4) |
1/4
ratio |
OL%(1)
OL%(4) |
ΔOL% | |
| H | H | Ph | 1 | −1.7 | 0.2 | 51:49 |
31.6%
29.8% |
−2.1% | |
| Me | Me | Ph | 2 | 9.5 | 6.1 | 83:17 |
31.0%
27.3% |
−3.7% | |
| a | H | Me | Ph | 3 | −4.7 | −2.5 | 35:65 |
31.9%
36.1% |
+4.2% |
| c | H | Me | 4-Me-C6H4 | 4 | −4.3 | −2.5 | 35:65 |
32.3%
36.3% |
+4.0% |
| f | H | Me | 4-OMe-C6H4 | 5 | −4.2 | −3.5 | 29:71 |
32.5%
36.3% |
+3.8% |
| e | H | Me | 4-CF3-C6H4 | 6 | −4.5 | −3.6 | 28:72 |
32.5%
36.7% |
+4.2% |
| g | H | Me | 2,6-diMe-C6H3 | 7 | −4.1 | −5.1 | 21:79 |
31.5%
35.2% |
+4.1% |
| h | H | Me | Et | 8 | +2.0 | −0.1 | 51:49 |
31.6%
29.8% |
−1.8% |
![[Graphic 6]](/bjoc/content/inline/1860-5397-16-75-i10.svg?max-width=637&scale=1.0)
|
|||||||||
| R1 | R2 | Y | Entry |
ΔH(1+H+
→4+H+) |
ΔG(1+H+
→4+H+) |
1+H+/ 4+H+ ratio |
OL%(1+H+)
OL%(4+H+) |
ΔOL% | |
| H | H | Ph | 9 | −11.3 | −11.1 | 6:94 |
54.1%
66.2% |
+12.1% | |
| Me | Me | Ph | 10 | −7.9 | −4.0 | 27:73 |
53.8%
67.1% |
+13.3% | |
| a | H | Me | Ph | 11 | −12.4 | −12.6 | 4:96 |
55.4%
69.1% |
+13.7% |
| c | H | Me | 4-Me-C6H4 | 12 | −10.6 | −10.9 | 6:94 |
55.7%
69.2% |
+13.5% |
| f | H | Me | 4-OMe-C6H4 | 13 | −10.1 | −10.3 | 7:93 |
55.0%
69.5% |
+14.5% |
| e | H | Me | 4-CF3-C6H4 | 14 | −13.7 | −13.6 | 3:97 |
55.1%
68.1% |
+13.0% |
| g | H | Me | 2,6-diMe-C6H3 | 15 | −9.3 | −9.9 | 7:93 |
54.4%
66.9% |
+12.5% |
| h | H | Me | Et | 16 | −8.8 | −9.2 | 9:91 |
52.8%
58.2% |
+5.4% |
Mechanistic investigations of the isomerization of 3-phospholene oxides 1 under thermal conditions
For the isomerization under thermal conditions, three mechanisms (mechanism A, B or C in Scheme 2) were outlined and examined by theoretical methods. Mechanism A involves a monomolecular rearrangement, involving zwitterionic structure 11 via high energy and a four-membered ring transition state [TS(1→11)]. This stressed structure has a rigid, entropically unbeneficial transition. The second transition state [TS(11→4)] represents a significantly lower energy gap with a more comfortable five-membered ring. As the thermal rearrangement was only feasible under solvent-free conditions, one may assume mechanism B, which involves the formation of phospholene oxide 1 dimers, and the second molecule only assists the rearrangement similarly to mechanism A. However, according to computations, in this arrangement the neighboring P=O group has only marginal beneficial effect on the activation enthalpies, compared to mechanism A. In the third possibility (mechanism C1), the proton from the C(2)H2 moiety can migrate to the P=O group in the second molecule, forming quasi-deprotonated (anionic) and protonated (cationic) molecules [TS(1→14)]. However, this arrangement is not stable, and a stabilized homodimer with a pentavalent P atom 14 is formed immediately. This quasi-autocatalytic process decreases significantly the enthalpy demand of the TS to a reliable level, and finally it gives the corresponding 2-phospholene oxide 4. More interestingly, mechanism C2 shows that the enthalpy level of the process decreases by 7–10 kJ mol−1 if a heterodimer (1–4) is formed instead of a homodimer (1–1). Thus, the 2-phospholene oxide 4 can enhance the rate of the isomerization by 6 times at temperature of the reaction (570 K).
![[1860-5397-16-75-i2]](/bjoc/content/inline/1860-5397-16-75-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Three possible reaction mechanisms considered in the theoretical studies for the isomerization of 3-phospholene oxides 1 under thermal conditions. *The calculated ΔG values for mechanism B and C were compensated by the dimerization entropy (ca. 128 J mol−1 K−1) due to the fact that the entropy obtained in vacuo calculations are not realistic in condensed phase.
Scheme 2: Three possible reaction mechanisms considered in the theoretical studies for the isomerization of 3...
In order to verify the computed reaction mechanism of the 3-phospholene oxide 1–2-phospholene oxide 4 rearrangement, standard kinetic experiments were run for the substituted aryl- or ethyl-3-methylphospholene oxides (1c–f or 1h) at a constant temperature. The reactions were carried out at 200 °C in neat, and the progression of the reactions was followed by gas chromatography for two days using biphenyl as internal standard. According to repeated experiments, the errors in measured relative concentrations were below 5%. The concentration curves undoubtedly refer to an equilibrium reaction, reaching a final concentration of 1 and 4 (Figure 2a). The results of this kinetic study indicated that the electron-donating groups on the phenyl ring accelerated, while the electron-withdrawing groups decelerated the isomerization in the first few hours.
![[1860-5397-16-75-2]](/bjoc/content/figures/1860-5397-16-75-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The full time experimental kinetic curves (a); The initial part of the kinetic curves of 1c–f and 1h (b); Initial and second fittings on the kinetic curve of 1c (c). For more kinetic curves, see Supporting Information File 1, Figure S1).
Figure 2: The full time experimental kinetic curves (a); The initial part of the kinetic curves of 1c–f and 1h...
All thermal isomerization processes lead to an equilibrium (Figure 2a). The mathematical analysis of the initial part of these kinetic curves resulted in a first order-like mechanism in contrast to other assumptions (e.g., second order) (Figure 2b). Interestingly, the later points started deviating from the initial linear fitting on the graph after the conversion of 3-phospholene oxides 1c–f or 1h reached the 15–20% (after 10 000 s). After 5 h (>20 000 s), the reaction rates of the first order approximation increased in all cases, and these values were doubled for phospholene oxides 1c, 1d and 1f (Figure 2c and Supporting Information File 1). This acceleration observed in the isomerization of the phospholene oxides is in accordance with the proposed mechanism (mechanism C2, Scheme 2), which predicted that the 2-phospholene oxides 4 can enhance the rate of the isomerization. Thus, the increasing amount of the corresponding 2-phospholene oxide 4 takes a more and more dominant role as the catalysis of the 3-phospholene oxide (1) 2-phospholene oxide (4) rearrangement, which explains the change in the reaction rate in the kinetic experiments.
Mechanistic studies of the isomerization of 3-phospholene oxides 1 under acidic conditions
The isomerization of 3-phospholene oxides 1 also proceeds in the presence of strong acids, such as methanesulfonic acid. On the one hand, the reaction rate increased in the presence of MeSO3H, on the other hand, the reaction mixture contained a much higher amount of the 2-phospholene oxide 4, as compared to the thermal- or base-catalyzed reactions. Scheme 3 shows the computed reaction mechanism of the acidic transformation, which starts from the complex formed from MeSO3H and the 3-phospholene oxide 16A, involving in a protonation equilibrium with 16B. In the next step, the acid migrates to position C(4), and the olefinic carbon atom can be easily protonated via a high enthalpy transition state [TS(17→18)], which is the rate determining step of this reaction sequence. We suppose that this rather high enthalpy is much lower due to the tunneling effect of the proton, whose energy gap is estimated to be reduced to about 130 kJ mol−1 in an optimal arrangement of the proton [73,74]. After the transition state, the mesylate anion attacks consecutively the position C(3), forming a neutral, relatively low-enthalpy intermediate 18. The departure of the mesylate anion from position C(3) attracts the proton from C(2) via a lower-enthalpy transition state [TS(17→18)] yielding the expected product, complexed by the acid 19A. Considering the equilibrium only between 16A–B and 19A–B, the product side is already shifted by −8.5 kJ mol−1 or −2.4 kJ mol−1, which makes the formation of 4a or 4h a beneficial process, meaning a shifted equilibrium towards the corresponding 2-phospholene oxide (4a or 4h). Comparing the calculations for the phenyl- and ethylphospholene derivatives, one can conclude that there is no significant difference in the reaction enthalpies for the aryl- and alkylphospholene oxide models.
![[1860-5397-16-75-i3]](/bjoc/content/inline/1860-5397-16-75-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Computed reaction mechanism of the 3-phospholene oxide (1) 2-phospholene oxide (4) isomerization under acidic conditions at MP2/6-311++G(2d,2p)//PCM(THF), including implicit solvent model.
Scheme 3: Computed reaction mechanism of the 3-phospholene oxide (1) 2-phospholene oxide (4) isomerization un...
Mechanistic investigations of the isomerization of 3-phospholene oxides 1 under basic conditions
The calculations showed that the reaction mechanism follows a simple sequence under basic conditions (Scheme 4). In the first step the base makes the reactant–base complex 20, and the consequent deprotonation occurs at position C(2), which is more favorable than the same process at position C(5) (by ca. 3–4 kJ mol−1). An anionic intermediate 21 is formed in this manner, and the related transition state represents a low enthalpy barrier (ca. 55 kJ mol−1), which is lowered by the tunneling effect of the proton. In the final step, the protonated base gives back the proton to the position C(4) also via a transition state with a low enthalpy barrier (ca. +20 kJ mol−1) yielding the corresponding 2-phospholene oxide 4 The calculations showed that the base does not affect the enthalpy difference between the 3- and 2-phospholene oxides 1 and 4 maintaining the thermodynamic equilibrium, it has only an accelerating catalytic effect on the reaction rate. Consequently, the expected ratio of the 2- and 3-phospholene oxides 4 and 1 is similar to the results obtained under thermal conditions. These theoretical findings are in accordance with the experimental results. Interestingly, the strength of the base applied may change the reaction rate significantly, as the reaction enthalpy of the intermediate 21 changes compared to 20. Pyridine is not able to take over the proton from the molecule (the structure 20 is not a minimum on the potential hypersurface (PES)). Triethylamine can initiate the deprotonation, but the reaction enthalpy towards 21 is very endothermic, making the reaction rate negligible, as observed in the experiment. In the case of KCO3− (instead of Cs2CO3), the deprotonation proceeds smoothly with almost a thermoneutral fashion, predicting a significant improvement in the reaction rate. According to the estimated enthalpy value of the TS for 20→21, one may expect a relatively fast process with Cs2CO3 even at moderate temperatures, however, the poor solubility of the inorganic salt demands a higher reaction temperature. Finally, the deprotonation by NaOEt is already a slightly exothermic process, suggesting a fast transformation.
![[1860-5397-16-75-i4]](/bjoc/content/inline/1860-5397-16-75-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Computed reaction mechanism of the 3-phospholene oxide (1) 2-phospholene oxide (4) isomerization under basic conditions computed at MP2/6-311G(2d,2p) level of theory, including implicit solvent model.
Scheme 4: Computed reaction mechanism of the 3-phospholene oxide (1) 2-phospholene oxide (4) isomerization un...
Conclusion
In this comprehensive study, the isomerization of 3-phospholene oxides 1 to the corresponding 2-phospholene oxides 4 was investigated. Complete isomerization took place either in the presence of methanesulfonic acid, or via the formation of chlorophospholenium salts (2 and 3), and a series of 1-substituted-3-methyl-2-phospholene oxides 4 were prepared. It was also shown that the isomerization may occur to some extent in the presence of inorganic bases, or at elevated temperatures. However, the isomerization remained incomplete under these conditions. It was found that methyl group(s) at positon 3 or 4 in the P-heterocyclic ring hinder the isomerization in all reaction pathways investigated. The mechanisms of acid- or base-mediated, as well as thermal isomerizations were elucidated by quantum chemical calculations, and the results justified why a given isomerization pathway leads to an equilibrium or proceeds until completion.
McCormack cycloaddition is still an established method for the synthesis of both 2- or 3-phospholene oxides. However, our paper outlined that the preparation of 2-phospholene oxide derivatives 4 is also feasible from 3-phospholene oxides 1 via the formation of chlorophosphonium salts or in the presence of MeSO3H. These isomerization reactions may be regarded as complementary procedures for the more recent preparation of the five-membered heterocyclic core, which generally affords the corresponding 3-phospholene oxide isomers 1. Our study also showed that high temperature or basic conditions should also be taken into consideration, when new or even existing reactions of 3-phospholene oxides 1 are carried out, in order to avoid the undesired isomerization, and thus diminished yields.
Experimental
General procedure for the preparation of 2-phospholene oxides 4, 7 or 10 via the formation of chlorophospholenium chlorides 3, 6 and 9: To the solution of 1.0 mmol of 3-phospholene oxide (1a: 0.19 g, 1b: 0.21 g, 1c: 0.21 g, 1d: 0.26 g, 1e: 0.22 g, 1f: 0.22 g, 1g: 0.24 g, 1h: 0.14 g, 1i: 0.16 g, 1j: 0.17 g, 1k: 0.17 g, 1l: 0.19 g, 5: 0.18 g, 8: 0.21 g) in 1.0 mL of dichloromethane, 0.094 mL (1.1 mmol) of oxalyl chloride was added dropwise at 0 °C in a sealed tube. The reaction mixture was stirred for 15 minutes, then the volatiles were evaporated to remove the solvent and the excess of the oxalyl chloride. The cyclic halophosphonium salt thus prepared was dissolved in 1.0 mL of chloroform, and the solution was stirred at 80 °C for 2 days. The reaction mixture was hydrolyzed with 1.0 mL of water. The phases were separated, and the aqueous phase was extracted with 5 × 1.0 mL of dichloromethane. The organic phases were combined, washed with 2.0 mL of saturated NaHCO3, dried (Na2SO4) and evaporated. The crude reaction mixture was filtered through a plug of silica with 3% methanol in dichloromethane to give the corresponding 2-phospholene oxide (4, 7 or 10). The results are summarized in Table 1 and Scheme 1.
General procedure for the preparation of 2-phospholene oxides (4, 7 or 10) using methanesulfonic acid: 1.0 mmol of the 3-phospholene oxide (1a: 0.19 g, 1b: 0.21 g, 1c: 0.21 g, 1d: 0.26 g, 1e: 0.22 g, 1f: 0.22 g, 1g: 0.24 g, 1h: 0.14 g, 1i: 0.16 g, 1j: 0.17 g, 1k: 0.17 g, 1l: 0.19 g, 5: 0.18 g, 8: 0.21 g) was dissolved in 1.0 mL of methanesulfonic acid, and the reaction mixture was heated at 50 °C for 60 h. The reaction mixture was then cooled to 0 °C, and 2.0 mL of water was added. Then, the reaction mixture was neutralized with saturated NaHCO3 at 0 °C. The solution was extracted with 5 × 3.0 mL dichloromethane. The organic phase dried (Na2SO4), evaporated to give 0.15 g (81%) of 1-phenyl-3-methyl-2-phospholene oxide (4a). The results are summarized in Table 2 and Scheme 1.
Supporting Information
The general methods and instruments, the preparation of the starting materials, a few experimental procedures, the details of the optimization, kinetic studies and computations, as well as the characterization and NMR spectra of the new compounds can be found in the Supporting Information.
| Supporting Information File 1: General methods, experimental and analytical data. | ||
| Format: PDF | Size: 4.4 MB | Download |
References
-
Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, NY, USA, 2000.
Return to citation in text: [1] -
Bansal, R. K., Ed. Phosphorous Heterocycles I; Springer: Berlin, Germany, 2009. doi:10.1007/978-3-642-00338-7
Return to citation in text: [1] -
Bansal, R. K., Ed. Phosphorus Heterocycles II; Springer: Berlin, Germany, 2010. doi:10.1007/978-3-642-12254-5
Return to citation in text: [1] -
Keglevich, G. Curr. Org. Chem. 2019, 23, 1342–1355. doi:10.2174/1385272823666190726093322
Return to citation in text: [1] -
Burk, M. J. Acc. Chem. Res. 2000, 33, 363–372. doi:10.1021/ar990085c
Return to citation in text: [1] -
Clark, T.; Landis, C. Tetrahedron: Asymmetry 2004, 15, 2123–2137. doi:10.1016/j.tetasy.2004.06.025
Return to citation in text: [1] -
Tang, W.; Zhang, X. Angew. Chem., Int. Ed. 2002, 41, 1612–1614. doi:10.1002/1521-3773(20020503)41:9<1612::aid-anie1612>3.0.co;2-h
Return to citation in text: [1] -
Liu, D.; Zhang, X. Eur. J. Org. Chem. 2005, 646–649. doi:10.1002/ejoc.200400690
Return to citation in text: [1] -
Zhang, X.; Huang, K.; Hou, G.; Cao, B.; Zhang, X. Angew. Chem., Int. Ed. 2010, 49, 6421–6424. doi:10.1002/anie.201002990
Return to citation in text: [1] -
Tang, W.; Qu, B.; Capacci, A. G.; Rodriguez, S.; Wei, X.; Haddad, N.; Narayanan, B.; Ma, S.; Grinberg, N.; Yee, N. K.; Krishnamurthy, D.; Senanayake, C. H. Org. Lett. 2010, 12, 176–179. doi:10.1021/ol9025815
Return to citation in text: [1] -
Li, G.; Wang, X.-j.; Zhang, Y.; Tan, Z.; DeCroos, P.; Lorenz, J. C.; Wei, X.; Grinberg, N.; Yee, N. K.; Senanayake, C. H. J. Org. Chem. 2017, 82, 5456–5460. doi:10.1021/acs.joc.7b00491
Return to citation in text: [1] -
Zhou, Q.-L., Ed. Privileged Chiral Ligands and Catalysts; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527635207
Return to citation in text: [1] -
Kollár, L.; Keglevich, G. Chem. Rev. 2010, 110, 4257–4302. doi:10.1021/cr900364c
Return to citation in text: [1] [2] -
Fourmy, K.; Nguyen, D. H.; Dechy-Cabaret, O.; Gouygou, M. Catal. Sci. Technol. 2015, 5, 4289–4323. doi:10.1039/c4cy01701c
Return to citation in text: [1] -
Bagi, P.; Kovács, T.; Szilvási, T.; Pongrácz, P.; Kollár, L.; Drahos, L.; Fogassy, E.; Keglevich, G. J. Organomet. Chem. 2014, 751, 306–313. doi:10.1016/j.jorganchem.2013.03.045
Return to citation in text: [1] -
Voituriez, A.; Saleh, N. Tetrahedron Lett. 2016, 57, 4443–4451. doi:10.1016/j.tetlet.2016.08.036
Return to citation in text: [1] -
Lao, Z.; Toy, P. H. Beilstein J. Org. Chem. 2016, 12, 2577–2587. doi:10.3762/bjoc.12.253
Return to citation in text: [1] -
Zhao, W.; Yan, P. K.; Radosevich, A. T. J. Am. Chem. Soc. 2015, 137, 616–619. doi:10.1021/ja511889y
Return to citation in text: [1] [2] -
Schoene, J.; Bel Abed, H.; Schmieder, P.; Christmann, M.; Nazaré, M. Chem. – Eur. J. 2018, 24, 9090–9100. doi:10.1002/chem.201800763
Return to citation in text: [1] -
Han, X.; Saleh, N.; Retailleau, P.; Voituriez, A. Org. Lett. 2018, 20, 4584–4588. doi:10.1021/acs.orglett.8b01870
Return to citation in text: [1] -
Cai, L.; Zhang, K.; Chen, S.; Lepage, R. J.; Houk, K. N.; Krenske, E. H.; Kwon, O. J. Am. Chem. Soc. 2019, 141, 9537–9542. doi:10.1021/jacs.9b04803
Return to citation in text: [1] -
Keglevich, G.; Fekete, M.; Chuluunbaatar, T.; Dobó, A.; Harmat, V.; Tőke, L. J. Chem. Soc., Perkin Trans. 1 2000, 4451–4455. doi:10.1039/b005380p
Return to citation in text: [1] -
O'Brien, C. J.; Tellez, J. L.; Nixon, Z. S.; Kang, L. J.; Carter, A. L.; Kunkel, S. R.; Przeworski, K. C.; Chass, G. A. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. doi:10.1002/anie.200902525
Return to citation in text: [1] -
Joly, D.; Bouit, P.-A.; Hissler, M. J. Mater. Chem. C 2016, 4, 3686–3698. doi:10.1039/c6tc00590j
Return to citation in text: [1] -
Duffy, M. P.; Delaunay, W.; Bouit, P.-A.; Hissler, M. Chem. Soc. Rev. 2016, 45, 5296–5310. doi:10.1039/c6cs00257a
Return to citation in text: [1] -
Regulska, E.; Romero-Nieto, C. Dalton Trans. 2018, 47, 10344–10359. doi:10.1039/c8dt01485j
Return to citation in text: [1] -
Yamada, M.; Yamashita, M.; Suyama, T.; Yamashita, J.; Asai, K.; Niimi, T.; Ozaki, N.; Fujie, M.; Maddali, K.; Nakamura, S.; Ohnishi, K. Bioorg. Med. Chem. Lett. 2010, 20, 5943–5946. doi:10.1016/j.bmcl.2010.01.061
Return to citation in text: [1] [2] -
Nakamura, S.; Yamashita, M.; Yokota, D.; Hirano, I.; Ono, T.; Fujie, M.; Shibata, K.; Niimi, T.; Suyama, T.; Maddali, K.; Asai, K.; Yamashita, J.; Iguchi, Y.; Ohnishi, K. Invest. New Drugs 2010, 28, 381–391. doi:10.1007/s10637-009-9255-3
Return to citation in text: [1] -
Yamaoka, M.; Yamashita, M.; Yamada, M.; Fujie, M.; Kiyofuji, K.; Ozaki, N.; Asai, K.; Niimi, T.; Suyama, T.; Yamashita, J.; Sawada, A.; Makita, R.; Sugiyama, M.; Toda, M.; Nakamura, S.; Ohnishi, K. Pure Appl. Chem. 2012, 84, 37–48. doi:10.1351/pac-con-11-10-09
Return to citation in text: [1] [2] -
Clarion, L.; Jacquard, C.; Sainte-Catherine, O.; Loiseau, S.; Filippini, D.; Hirlemann, M.-H.; Volle, J.-N.; Virieux, D.; Lecouvey, M.; Pirat, J.-L.; Bakalara, N. J. Med. Chem. 2012, 55, 2196–2211. doi:10.1021/jm201428a
Return to citation in text: [1] -
Yudelevich, V. I.; Belakhov, V. V.; Belyaeva, T. N.; Sergienko, L. V.; Komarov, E. V.; Ionin, B. I.; Mikhailova, M. A.; Araviiskii, R. A. Pharm. Chem. J. 1983, 17, 866–872. doi:10.1007/bf00765969
Return to citation in text: [1] -
Machetti, F.; Anichini, B.; Cicchi, S.; Brandi, A.; Wieczorek, W.; Pietrusiewicz, K. M.; Gehret, J.-C. J. Heterocycl. Chem. 1996, 33, 1091–1098. doi:10.1002/jhet.5570330416
Return to citation in text: [1] -
Gavande, N.; Yamamoto, I.; Salam, N. K.; Ai, T.-H.; Burden, P. M.; Johnston, G. A. R.; Hanrahan, J. R.; Chebib, M. ACS Med. Chem. Lett. 2011, 2, 11–16. doi:10.1021/ml1001344
Return to citation in text: [1] -
Páv, O.; Zborníková, E.; Buděšínský, M.; Rosenberg, I. Tetrahedron 2013, 69, 9120–9129. doi:10.1016/j.tet.2013.08.021
Return to citation in text: [1] [2] -
Páv, O.; Buděšínský, M.; Rosenberg, I. Tetrahedron 2017, 73, 5220–5228. doi:10.1016/j.tet.2017.07.020
Return to citation in text: [1] [2] [3] -
Quin, L. D. The Heterocyclic Chemistry of Phosphorus: Systems Based on the Phosphorus-Carbon Bond; Wiley-Interscience: New York, NY, USA, 1981.
Return to citation in text: [1] [2] -
Chan, T. H.; Wong, L. T. L. Can. J. Chem. 1971, 49, 530–531. doi:10.1139/v71-085
Return to citation in text: [1] -
Amin, M.; Holah, D. G.; Hughes, A. N.; Rukachaisirikul, T. J. Heterocycl. Chem. 1985, 22, 513–522. doi:10.1002/jhet.5570220262
Return to citation in text: [1] -
Pietrusiewicz, K. M.; Flis, A.; Ujj, V.; Körtvélyesi, T.; Drahos, L.; Pongrácz, P.; Kollár, L.; Keglevich, G. Heteroat. Chem. 2011, 22, 730–736. doi:10.1002/hc.20741
Return to citation in text: [1] -
Jablonkai, E.; Drahos, L.; Drzazga, Z.; Pietrusiewicz, K. M.; Keglevich, G. Heteroat. Chem. 2012, 23, 539–544. doi:10.1002/hc.21047
Return to citation in text: [1] -
Yoshimura, A.; Nguyen, K. C.; Rohde, G. T.; Postnikov, P. S.; Yusubov, M. S.; Zhdankin, V. V. J. Org. Chem. 2017, 82, 11742–11751. doi:10.1021/acs.joc.7b01462
Return to citation in text: [1] -
Lim, K. M.-H.; Hayashi, T. J. Am. Chem. Soc. 2017, 139, 8122–8125. doi:10.1021/jacs.7b04570
Return to citation in text: [1] [2] -
Keglevich, G.; Újszászy, K.; Szöllősy, Á.; Ludányi, K.; Tőke, L. J. Organomet. Chem. 1996, 516, 139–145. doi:10.1016/0022-328x(96)06134-7
Return to citation in text: [1] -
Desmazeau, P.; Legros, J.-Y.; Fiaud, J.-C. Tetrahedron Lett. 1998, 39, 6707–6710. doi:10.1016/s0040-4039(98)01401-4
Return to citation in text: [1] -
Pakulski, Z.; Kwiatosz, R.; Pietrusiewicz, K. M. Tetrahedron 2005, 61, 1481–1492. doi:10.1016/j.tet.2004.12.004
Return to citation in text: [1] -
de Azambuja, F.; Carmona, R. C.; Chorro, T. H. D.; Heerdt, G.; Correia, C. R. D. Chem. – Eur. J. 2016, 22, 11205–11209. doi:10.1002/chem.201602572
Return to citation in text: [1] -
Hamstra, D. F. J.; Lenstra, D. C.; Koenders, T. J.; Rutjes, F. P. J. T.; Mecinović, J. Org. Biomol. Chem. 2017, 15, 6426–6432. doi:10.1039/c7ob01510k
Return to citation in text: [1] -
McCormack, W. B. Org. Synth. 1963, 43, 73. doi:10.15227/orgsyn.043.0073
Return to citation in text: [1] -
Mann, F. G. The Heterocyclic Derivatives of Phosphorus, Arsenic, Antimony and Bismuth; Wiley-Interscience: London, 1970.
Return to citation in text: [1] -
Keglevich, G.; Szelke, H. Houben–Weyl Methods of Molecular Transformations; Georg Thieme Verlag: Stuttgart, Germany, 2007; pp 745–758.
Return to citation in text: [1] -
Trevitt, M.; Gouverneur, V. Tetrahedron Lett. 1999, 40, 7333–7336. doi:10.1016/s0040-4039(99)01534-8
Return to citation in text: [1] -
Bujard, M.; Gouverneur, V.; Mioskowski, C. J. Org. Chem. 1999, 64, 2119–2123. doi:10.1021/jo981795j
Return to citation in text: [1] -
McReynolds, M. D.; Dougherty, J. M.; Hanson, P. R. Chem. Rev. 2004, 104, 2239–2258. doi:10.1021/cr020109k
Return to citation in text: [1] -
Harvey, J. S.; Malcolmson, S. J.; Dunne, K. S.; Meek, S. J.; Thompson, A. L.; Schrock, R. R.; Hoveyda, A. H.; Gouverneur, V. Angew. Chem., Int. Ed. 2009, 48, 762–766. doi:10.1002/anie.200805066
Return to citation in text: [1] -
Montchamp, J.-L.; Tian, F.; Frost, J. W. J. Org. Chem. 1995, 60, 6076–6081. doi:10.1021/jo00124a018
Return to citation in text: [1] -
Wu, X.; O’Brien, P.; Ellwood, S.; Secci, F.; Kelly, B. Org. Lett. 2013, 15, 192–195. doi:10.1021/ol303253h
Return to citation in text: [1] -
Carr, D. J.; Kudavalli, J. S.; Dunne, K. S.; Müller-Bunz, H.; Gilheany, D. G. J. Org. Chem. 2013, 78, 10500–10505. doi:10.1021/jo401318g
Return to citation in text: [1] -
Hunger, K.; Hasserodt, U.; Korte, F. Tetrahedron 1964, 20, 1593–1604. doi:10.1016/s0040-4020(01)99156-2
Return to citation in text: [1] [2] -
Quin, L. D.; Gratz, J. P.; Barket, T. P. J. Org. Chem. 1968, 33, 1034–1041. doi:10.1021/jo01267a020
Return to citation in text: [1] [2] [3] -
Hintermann, L.; Schmitz, M. Adv. Synth. Catal. 2008, 350, 1469–1473. doi:10.1002/adsc.200800226
Return to citation in text: [1] -
Wu, Y.; Singh, R. P.; Deng, L. J. Am. Chem. Soc. 2011, 133, 12458–12461. doi:10.1021/ja205674x
Return to citation in text: [1] -
Kiss, N. Z.; Ludányi, K.; Drahos, L.; Keglevich, G. Synth. Commun. 2009, 39, 2392–2404. doi:10.1080/00397910802654880
Return to citation in text: [1] [2] -
Bálint, E.; Jablonkai, E.; Bálint, M.; Keglevich, G. Heteroat. Chem. 2010, 21, 211–214. doi:10.1002/hc.20596
Return to citation in text: [1] -
Keglevich, G.; Rádai, Z.; Harsági, N.; Szigetvári, Á.; Kiss, N. Z. Heteroat. Chem. 2017, 28, e21394. doi:10.1002/hc.21394
Return to citation in text: [1] [2] -
Herbay, R.; Bagi, P.; Mucsi, Z.; Mátravölgyi, B.; Drahos, L.; Fogassy, E.; Keglevich, G. Tetrahedron Lett. 2017, 58, 458–461. doi:10.1016/j.tetlet.2016.12.059
Return to citation in text: [1] [2] [3] [4] -
Bagi, P.; Herbay, R.; Ábrányi-Balogh, P.; Mátravölgyi, B.; Fogassy, E.; Keglevich, G. Tetrahedron 2018, 74, 5850–5857. doi:10.1016/j.tet.2018.07.058
Return to citation in text: [1] -
Herbay, R.; Péczka, N.; Györke, G.; Bagi, P.; Fogassy, E.; Keglevich, G. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 610–613. doi:10.1080/10426507.2018.1553043
Return to citation in text: [1] -
Stankevič, M.; Pisklak, J.; Włodarczyk, K. Tetrahedron 2016, 72, 810–824. doi:10.1016/j.tet.2015.12.043
Return to citation in text: [1] -
Mucsi, Z.; Chass, G. A.; Viskolcz, B.; Csizmadia, I. G. J. Phys. Chem. A 2009, 113, 7953–7962. doi:10.1021/jp805325a
Return to citation in text: [1] -
Novak, T.; Mucsi, Z.; Balázs, B.; Keresztély, L.; Blaskó, G.; Nyerges, M. Synlett 2010, 2411–2414. doi:10.1055/s-0030-1258036
Return to citation in text: [1] -
Mucsi, Z.; Porcs-Makkay, M.; Simig, G.; Csizmadia, I. G.; Volk, B. J. Org. Chem. 2012, 77, 7282–7290. doi:10.1021/jo300715d
Return to citation in text: [1] -
Kondacs, L. A.; Pilipecz, M. V.; Mucsi, Z.; Balázs, B.; Gáti, T.; Nyerges, M.; Dancsó, A.; Nemes, P. Eur. J. Org. Chem. 2015, 6872–6890. doi:10.1002/ejoc.201500905
Return to citation in text: [1] -
Caldin, E. F. Chem. Rev. 1969, 69, 135–156. doi:10.1021/cr60257a006
Return to citation in text: [1] -
Bell, R. P. Chem. Soc. Rev. 1974, 3, 513–544. doi:10.1039/cs9740300513
Return to citation in text: [1]
| 42. | Lim, K. M.-H.; Hayashi, T. J. Am. Chem. Soc. 2017, 139, 8122–8125. doi:10.1021/jacs.7b04570 |
| 69. | Mucsi, Z.; Chass, G. A.; Viskolcz, B.; Csizmadia, I. G. J. Phys. Chem. A 2009, 113, 7953–7962. doi:10.1021/jp805325a |
| 70. | Novak, T.; Mucsi, Z.; Balázs, B.; Keresztély, L.; Blaskó, G.; Nyerges, M. Synlett 2010, 2411–2414. doi:10.1055/s-0030-1258036 |
| 71. | Mucsi, Z.; Porcs-Makkay, M.; Simig, G.; Csizmadia, I. G.; Volk, B. J. Org. Chem. 2012, 77, 7282–7290. doi:10.1021/jo300715d |
| 72. | Kondacs, L. A.; Pilipecz, M. V.; Mucsi, Z.; Balázs, B.; Gáti, T.; Nyerges, M.; Dancsó, A.; Nemes, P. Eur. J. Org. Chem. 2015, 6872–6890. doi:10.1002/ejoc.201500905 |
| 65. | Herbay, R.; Bagi, P.; Mucsi, Z.; Mátravölgyi, B.; Drahos, L.; Fogassy, E.; Keglevich, G. Tetrahedron Lett. 2017, 58, 458–461. doi:10.1016/j.tetlet.2016.12.059 |
| 1. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, NY, USA, 2000. |
| 2. | Bansal, R. K., Ed. Phosphorous Heterocycles I; Springer: Berlin, Germany, 2009. doi:10.1007/978-3-642-00338-7 |
| 3. | Bansal, R. K., Ed. Phosphorus Heterocycles II; Springer: Berlin, Germany, 2010. doi:10.1007/978-3-642-12254-5 |
| 4. | Keglevich, G. Curr. Org. Chem. 2019, 23, 1342–1355. doi:10.2174/1385272823666190726093322 |
| 13. | Kollár, L.; Keglevich, G. Chem. Rev. 2010, 110, 4257–4302. doi:10.1021/cr900364c |
| 15. | Bagi, P.; Kovács, T.; Szilvási, T.; Pongrácz, P.; Kollár, L.; Drahos, L.; Fogassy, E.; Keglevich, G. J. Organomet. Chem. 2014, 751, 306–313. doi:10.1016/j.jorganchem.2013.03.045 |
| 47. | Hamstra, D. F. J.; Lenstra, D. C.; Koenders, T. J.; Rutjes, F. P. J. T.; Mecinović, J. Org. Biomol. Chem. 2017, 15, 6426–6432. doi:10.1039/c7ob01510k |
| 13. | Kollár, L.; Keglevich, G. Chem. Rev. 2010, 110, 4257–4302. doi:10.1021/cr900364c |
| 14. | Fourmy, K.; Nguyen, D. H.; Dechy-Cabaret, O.; Gouygou, M. Catal. Sci. Technol. 2015, 5, 4289–4323. doi:10.1039/c4cy01701c |
| 36. | Quin, L. D. The Heterocyclic Chemistry of Phosphorus: Systems Based on the Phosphorus-Carbon Bond; Wiley-Interscience: New York, NY, USA, 1981. |
| 48. | McCormack, W. B. Org. Synth. 1963, 43, 73. doi:10.15227/orgsyn.043.0073 |
| 49. | Mann, F. G. The Heterocyclic Derivatives of Phosphorus, Arsenic, Antimony and Bismuth; Wiley-Interscience: London, 1970. |
| 50. | Keglevich, G.; Szelke, H. Houben–Weyl Methods of Molecular Transformations; Georg Thieme Verlag: Stuttgart, Germany, 2007; pp 745–758. |
| 12. | Zhou, Q.-L., Ed. Privileged Chiral Ligands and Catalysts; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527635207 |
| 36. | Quin, L. D. The Heterocyclic Chemistry of Phosphorus: Systems Based on the Phosphorus-Carbon Bond; Wiley-Interscience: New York, NY, USA, 1981. |
| 5. | Burk, M. J. Acc. Chem. Res. 2000, 33, 363–372. doi:10.1021/ar990085c |
| 6. | Clark, T.; Landis, C. Tetrahedron: Asymmetry 2004, 15, 2123–2137. doi:10.1016/j.tetasy.2004.06.025 |
| 7. | Tang, W.; Zhang, X. Angew. Chem., Int. Ed. 2002, 41, 1612–1614. doi:10.1002/1521-3773(20020503)41:9<1612::aid-anie1612>3.0.co;2-h |
| 8. | Liu, D.; Zhang, X. Eur. J. Org. Chem. 2005, 646–649. doi:10.1002/ejoc.200400690 |
| 9. | Zhang, X.; Huang, K.; Hou, G.; Cao, B.; Zhang, X. Angew. Chem., Int. Ed. 2010, 49, 6421–6424. doi:10.1002/anie.201002990 |
| 10. | Tang, W.; Qu, B.; Capacci, A. G.; Rodriguez, S.; Wei, X.; Haddad, N.; Narayanan, B.; Ma, S.; Grinberg, N.; Yee, N. K.; Krishnamurthy, D.; Senanayake, C. H. Org. Lett. 2010, 12, 176–179. doi:10.1021/ol9025815 |
| 11. | Li, G.; Wang, X.-j.; Zhang, Y.; Tan, Z.; DeCroos, P.; Lorenz, J. C.; Wei, X.; Grinberg, N.; Yee, N. K.; Senanayake, C. H. J. Org. Chem. 2017, 82, 5456–5460. doi:10.1021/acs.joc.7b00491 |
| 37. | Chan, T. H.; Wong, L. T. L. Can. J. Chem. 1971, 49, 530–531. doi:10.1139/v71-085 |
| 38. | Amin, M.; Holah, D. G.; Hughes, A. N.; Rukachaisirikul, T. J. Heterocycl. Chem. 1985, 22, 513–522. doi:10.1002/jhet.5570220262 |
| 39. | Pietrusiewicz, K. M.; Flis, A.; Ujj, V.; Körtvélyesi, T.; Drahos, L.; Pongrácz, P.; Kollár, L.; Keglevich, G. Heteroat. Chem. 2011, 22, 730–736. doi:10.1002/hc.20741 |
| 40. | Jablonkai, E.; Drahos, L.; Drzazga, Z.; Pietrusiewicz, K. M.; Keglevich, G. Heteroat. Chem. 2012, 23, 539–544. doi:10.1002/hc.21047 |
| 41. | Yoshimura, A.; Nguyen, K. C.; Rohde, G. T.; Postnikov, P. S.; Yusubov, M. S.; Zhdankin, V. V. J. Org. Chem. 2017, 82, 11742–11751. doi:10.1021/acs.joc.7b01462 |
| 42. | Lim, K. M.-H.; Hayashi, T. J. Am. Chem. Soc. 2017, 139, 8122–8125. doi:10.1021/jacs.7b04570 |
| 43. | Keglevich, G.; Újszászy, K.; Szöllősy, Á.; Ludányi, K.; Tőke, L. J. Organomet. Chem. 1996, 516, 139–145. doi:10.1016/0022-328x(96)06134-7 |
| 44. | Desmazeau, P.; Legros, J.-Y.; Fiaud, J.-C. Tetrahedron Lett. 1998, 39, 6707–6710. doi:10.1016/s0040-4039(98)01401-4 |
| 45. | Pakulski, Z.; Kwiatosz, R.; Pietrusiewicz, K. M. Tetrahedron 2005, 61, 1481–1492. doi:10.1016/j.tet.2004.12.004 |
| 46. | de Azambuja, F.; Carmona, R. C.; Chorro, T. H. D.; Heerdt, G.; Correia, C. R. D. Chem. – Eur. J. 2016, 22, 11205–11209. doi:10.1002/chem.201602572 |
| 27. | Yamada, M.; Yamashita, M.; Suyama, T.; Yamashita, J.; Asai, K.; Niimi, T.; Ozaki, N.; Fujie, M.; Maddali, K.; Nakamura, S.; Ohnishi, K. Bioorg. Med. Chem. Lett. 2010, 20, 5943–5946. doi:10.1016/j.bmcl.2010.01.061 |
| 28. | Nakamura, S.; Yamashita, M.; Yokota, D.; Hirano, I.; Ono, T.; Fujie, M.; Shibata, K.; Niimi, T.; Suyama, T.; Maddali, K.; Asai, K.; Yamashita, J.; Iguchi, Y.; Ohnishi, K. Invest. New Drugs 2010, 28, 381–391. doi:10.1007/s10637-009-9255-3 |
| 29. | Yamaoka, M.; Yamashita, M.; Yamada, M.; Fujie, M.; Kiyofuji, K.; Ozaki, N.; Asai, K.; Niimi, T.; Suyama, T.; Yamashita, J.; Sawada, A.; Makita, R.; Sugiyama, M.; Toda, M.; Nakamura, S.; Ohnishi, K. Pure Appl. Chem. 2012, 84, 37–48. doi:10.1351/pac-con-11-10-09 |
| 30. | Clarion, L.; Jacquard, C.; Sainte-Catherine, O.; Loiseau, S.; Filippini, D.; Hirlemann, M.-H.; Volle, J.-N.; Virieux, D.; Lecouvey, M.; Pirat, J.-L.; Bakalara, N. J. Med. Chem. 2012, 55, 2196–2211. doi:10.1021/jm201428a |
| 33. | Gavande, N.; Yamamoto, I.; Salam, N. K.; Ai, T.-H.; Burden, P. M.; Johnston, G. A. R.; Hanrahan, J. R.; Chebib, M. ACS Med. Chem. Lett. 2011, 2, 11–16. doi:10.1021/ml1001344 |
| 24. | Joly, D.; Bouit, P.-A.; Hissler, M. J. Mater. Chem. C 2016, 4, 3686–3698. doi:10.1039/c6tc00590j |
| 25. | Duffy, M. P.; Delaunay, W.; Bouit, P.-A.; Hissler, M. Chem. Soc. Rev. 2016, 45, 5296–5310. doi:10.1039/c6cs00257a |
| 26. | Regulska, E.; Romero-Nieto, C. Dalton Trans. 2018, 47, 10344–10359. doi:10.1039/c8dt01485j |
| 27. | Yamada, M.; Yamashita, M.; Suyama, T.; Yamashita, J.; Asai, K.; Niimi, T.; Ozaki, N.; Fujie, M.; Maddali, K.; Nakamura, S.; Ohnishi, K. Bioorg. Med. Chem. Lett. 2010, 20, 5943–5946. doi:10.1016/j.bmcl.2010.01.061 |
| 29. | Yamaoka, M.; Yamashita, M.; Yamada, M.; Fujie, M.; Kiyofuji, K.; Ozaki, N.; Asai, K.; Niimi, T.; Suyama, T.; Yamashita, J.; Sawada, A.; Makita, R.; Sugiyama, M.; Toda, M.; Nakamura, S.; Ohnishi, K. Pure Appl. Chem. 2012, 84, 37–48. doi:10.1351/pac-con-11-10-09 |
| 34. | Páv, O.; Zborníková, E.; Buděšínský, M.; Rosenberg, I. Tetrahedron 2013, 69, 9120–9129. doi:10.1016/j.tet.2013.08.021 |
| 35. | Páv, O.; Buděšínský, M.; Rosenberg, I. Tetrahedron 2017, 73, 5220–5228. doi:10.1016/j.tet.2017.07.020 |
| 18. | Zhao, W.; Yan, P. K.; Radosevich, A. T. J. Am. Chem. Soc. 2015, 137, 616–619. doi:10.1021/ja511889y |
| 22. | Keglevich, G.; Fekete, M.; Chuluunbaatar, T.; Dobó, A.; Harmat, V.; Tőke, L. J. Chem. Soc., Perkin Trans. 1 2000, 4451–4455. doi:10.1039/b005380p |
| 23. | O'Brien, C. J.; Tellez, J. L.; Nixon, Z. S.; Kang, L. J.; Carter, A. L.; Kunkel, S. R.; Przeworski, K. C.; Chass, G. A. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. doi:10.1002/anie.200902525 |
| 73. | Caldin, E. F. Chem. Rev. 1969, 69, 135–156. doi:10.1021/cr60257a006 |
| 74. | Bell, R. P. Chem. Soc. Rev. 1974, 3, 513–544. doi:10.1039/cs9740300513 |
| 16. | Voituriez, A.; Saleh, N. Tetrahedron Lett. 2016, 57, 4443–4451. doi:10.1016/j.tetlet.2016.08.036 |
| 17. | Lao, Z.; Toy, P. H. Beilstein J. Org. Chem. 2016, 12, 2577–2587. doi:10.3762/bjoc.12.253 |
| 18. | Zhao, W.; Yan, P. K.; Radosevich, A. T. J. Am. Chem. Soc. 2015, 137, 616–619. doi:10.1021/ja511889y |
| 19. | Schoene, J.; Bel Abed, H.; Schmieder, P.; Christmann, M.; Nazaré, M. Chem. – Eur. J. 2018, 24, 9090–9100. doi:10.1002/chem.201800763 |
| 20. | Han, X.; Saleh, N.; Retailleau, P.; Voituriez, A. Org. Lett. 2018, 20, 4584–4588. doi:10.1021/acs.orglett.8b01870 |
| 21. | Cai, L.; Zhang, K.; Chen, S.; Lepage, R. J.; Houk, K. N.; Krenske, E. H.; Kwon, O. J. Am. Chem. Soc. 2019, 141, 9537–9542. doi:10.1021/jacs.9b04803 |
| 31. | Yudelevich, V. I.; Belakhov, V. V.; Belyaeva, T. N.; Sergienko, L. V.; Komarov, E. V.; Ionin, B. I.; Mikhailova, M. A.; Araviiskii, R. A. Pharm. Chem. J. 1983, 17, 866–872. doi:10.1007/bf00765969 |
| 32. | Machetti, F.; Anichini, B.; Cicchi, S.; Brandi, A.; Wieczorek, W.; Pietrusiewicz, K. M.; Gehret, J.-C. J. Heterocycl. Chem. 1996, 33, 1091–1098. doi:10.1002/jhet.5570330416 |
| 56. | Wu, X.; O’Brien, P.; Ellwood, S.; Secci, F.; Kelly, B. Org. Lett. 2013, 15, 192–195. doi:10.1021/ol303253h |
| 57. | Carr, D. J.; Kudavalli, J. S.; Dunne, K. S.; Müller-Bunz, H.; Gilheany, D. G. J. Org. Chem. 2013, 78, 10500–10505. doi:10.1021/jo401318g |
| 35. | Páv, O.; Buděšínský, M.; Rosenberg, I. Tetrahedron 2017, 73, 5220–5228. doi:10.1016/j.tet.2017.07.020 |
| 51. | Trevitt, M.; Gouverneur, V. Tetrahedron Lett. 1999, 40, 7333–7336. doi:10.1016/s0040-4039(99)01534-8 |
| 52. | Bujard, M.; Gouverneur, V.; Mioskowski, C. J. Org. Chem. 1999, 64, 2119–2123. doi:10.1021/jo981795j |
| 53. | McReynolds, M. D.; Dougherty, J. M.; Hanson, P. R. Chem. Rev. 2004, 104, 2239–2258. doi:10.1021/cr020109k |
| 54. | Harvey, J. S.; Malcolmson, S. J.; Dunne, K. S.; Meek, S. J.; Thompson, A. L.; Schrock, R. R.; Hoveyda, A. H.; Gouverneur, V. Angew. Chem., Int. Ed. 2009, 48, 762–766. doi:10.1002/anie.200805066 |
| 34. | Páv, O.; Zborníková, E.; Buděšínský, M.; Rosenberg, I. Tetrahedron 2013, 69, 9120–9129. doi:10.1016/j.tet.2013.08.021 |
| 35. | Páv, O.; Buděšínský, M.; Rosenberg, I. Tetrahedron 2017, 73, 5220–5228. doi:10.1016/j.tet.2017.07.020 |
| 55. | Montchamp, J.-L.; Tian, F.; Frost, J. W. J. Org. Chem. 1995, 60, 6076–6081. doi:10.1021/jo00124a018 |
| 59. | Quin, L. D.; Gratz, J. P.; Barket, T. P. J. Org. Chem. 1968, 33, 1034–1041. doi:10.1021/jo01267a020 |
| 62. | Kiss, N. Z.; Ludányi, K.; Drahos, L.; Keglevich, G. Synth. Commun. 2009, 39, 2392–2404. doi:10.1080/00397910802654880 |
| 64. | Keglevich, G.; Rádai, Z.; Harsági, N.; Szigetvári, Á.; Kiss, N. Z. Heteroat. Chem. 2017, 28, e21394. doi:10.1002/hc.21394 |
| 68. | Stankevič, M.; Pisklak, J.; Włodarczyk, K. Tetrahedron 2016, 72, 810–824. doi:10.1016/j.tet.2015.12.043 |
| 65. | Herbay, R.; Bagi, P.; Mucsi, Z.; Mátravölgyi, B.; Drahos, L.; Fogassy, E.; Keglevich, G. Tetrahedron Lett. 2017, 58, 458–461. doi:10.1016/j.tetlet.2016.12.059 |
| 65. | Herbay, R.; Bagi, P.; Mucsi, Z.; Mátravölgyi, B.; Drahos, L.; Fogassy, E.; Keglevich, G. Tetrahedron Lett. 2017, 58, 458–461. doi:10.1016/j.tetlet.2016.12.059 |
| 62. | Kiss, N. Z.; Ludányi, K.; Drahos, L.; Keglevich, G. Synth. Commun. 2009, 39, 2392–2404. doi:10.1080/00397910802654880 |
| 63. | Bálint, E.; Jablonkai, E.; Bálint, M.; Keglevich, G. Heteroat. Chem. 2010, 21, 211–214. doi:10.1002/hc.20596 |
| 64. | Keglevich, G.; Rádai, Z.; Harsági, N.; Szigetvári, Á.; Kiss, N. Z. Heteroat. Chem. 2017, 28, e21394. doi:10.1002/hc.21394 |
| 65. | Herbay, R.; Bagi, P.; Mucsi, Z.; Mátravölgyi, B.; Drahos, L.; Fogassy, E.; Keglevich, G. Tetrahedron Lett. 2017, 58, 458–461. doi:10.1016/j.tetlet.2016.12.059 |
| 66. | Bagi, P.; Herbay, R.; Ábrányi-Balogh, P.; Mátravölgyi, B.; Fogassy, E.; Keglevich, G. Tetrahedron 2018, 74, 5850–5857. doi:10.1016/j.tet.2018.07.058 |
| 67. | Herbay, R.; Péczka, N.; Györke, G.; Bagi, P.; Fogassy, E.; Keglevich, G. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 610–613. doi:10.1080/10426507.2018.1553043 |
| 58. | Hunger, K.; Hasserodt, U.; Korte, F. Tetrahedron 1964, 20, 1593–1604. doi:10.1016/s0040-4020(01)99156-2 |
| 59. | Quin, L. D.; Gratz, J. P.; Barket, T. P. J. Org. Chem. 1968, 33, 1034–1041. doi:10.1021/jo01267a020 |
| 60. | Hintermann, L.; Schmitz, M. Adv. Synth. Catal. 2008, 350, 1469–1473. doi:10.1002/adsc.200800226 |
| 61. | Wu, Y.; Singh, R. P.; Deng, L. J. Am. Chem. Soc. 2011, 133, 12458–12461. doi:10.1021/ja205674x |
| 58. | Hunger, K.; Hasserodt, U.; Korte, F. Tetrahedron 1964, 20, 1593–1604. doi:10.1016/s0040-4020(01)99156-2 |
| 59. | Quin, L. D.; Gratz, J. P.; Barket, T. P. J. Org. Chem. 1968, 33, 1034–1041. doi:10.1021/jo01267a020 |
© 2020 Bagi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)