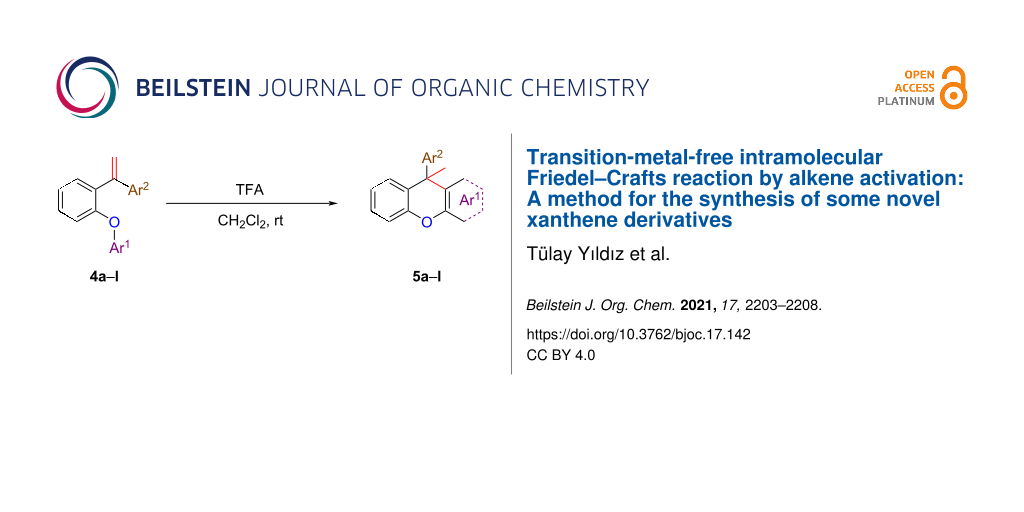
Abstract
In this work, new derivatives (substituted 9-methyl-9-arylxanthenes) of xanthene compounds (5a–l) of possible biological significance were synthesized by developing a new synthesis method. In order to obtain xanthene derivatives, the original alkene compounds to be used as the starting materials were synthesized in four steps using appropriate reactions. A cyclization reaction by intramolecular Friedel–Crafts alkylation was carried out in order to synthesize the desired xanthene derivatives using the alkenes as starting compounds. The intramolecular Friedel–Crafts reaction was catalyzed by trifluoroacetic acid (TFA) and provided some novel substituted 9-methyl-9-arylxanthenes with good yields at room temperature within 6–24 hours. As a result, an alkene compound was used for activation with TFA in the synthesis of xanthene through intramolecular Friedel–Crafts alkylation for the first time.

Graphical Abstract
Introduction
The interest in xanthenes has increased remarkably in recent years due to their wide range of biological and pharmacological properties. Xanthenes are important biologically active oxygen-containing heterocyclic compounds. These types of compounds have some biological and pharmacological properties, such as anticarcinogenic [1,2], antiviral [3], antibacterial [4,5], and anti-inflammatory [6]. Because of their pharmaceutical activity, they are used in photodynamic therapy [7-9]. In addition, they have an important place in the dye industry due to their photochemical and photophysical properties. They are also used in pH-sensitive fluorescent materials [10] and laser technologies [11-14]. Moreover, xanthenes are present in some natural sources, such as santalin pigments found in many varieties of plants [15,16].
There have been many studies on the synthesis of xanthene derivatives to date because of their important biological and fluorescent uses. To summarize the main syntheses of these studies: in particular, transition metal-catalyzed cascade benzylation–cyclization [17], cyclization of polycyclic aryl triflate esters [18], reaction of β-naphthol and aldehydes [19,20] or inter- or intramolecular coupling of arynes by aldehydes or phenols [21-24], and Lewis acid-catalyzed cyclization of salicylaldehydes and cyclohexenones or tetralones [25]. Some other new and prominent synthesis methods of xanthenes are the tandem arylation/Friedel–Crafts reaction of o-hydroxy bisbenzylic alcohols with diaryliodonium salts [26], the Sc(OTf)3-catalyzed domino reaction [27], and the iodine-catalyzed nucleophilic substitution reaction of xanthen-9-ol [28]. As an example of an intramolecular hydroarylation of an olefin, 9,10-dihydroacridines, which are N derivatives of xanthenes, were synthesized using the combination of hexafluoroisopropanol and triflimide as a catalyst [29].
One of the most effective C–C bond establishing reactions, which ensures an efficient synthetic way to a plenty of functionalized aryl compounds, is the Friedel–Crafts cyclization reaction [30-34]. Therefore, there has been an increase in arylation methods using Friedel–Crafts alkylation (FCA) protocols without transition metals [35,36]. Environmentally friendly methods, in which metal-free catalysts are used, have come to the fore and are gaining importance because there are many disadvantages of organometallic chemistry and using transition metal catalysts, which need expensive and toxic chemicals. With the increasing interest in these methods, new organocatalysts and reagents that are less toxic, easier to use, readily accessible, and cheap have been developed [37]. In particular, the synthesis of arenes using the FCA methods with π-activated alcohols and organocatalysts has begun to be preferred over using conventional reagents, such as organohalogens and transition metal catalysts, which are toxic and require working under harsher conditions [38]. After these developments, the intramolecular FCA method using π-activated alcohols has frequently been used for xanthene synthesis. Some of these methods are the stereoselective synthesis of 9-vinyl-substituted unsymmetrical xanthenes and thioxanthenes by intramolecular FCA reaction [39], Lewis acid-catalyzed intramolecular FCA [40], and the synthesis of xanthenes and thioxanthenes by intramolecular FCA catalyzed with organic Brønsted superacid, which are our works [41,42]. According to the literature, there are also hydroarylation methods with FCA, which are made by unactivated alkenes instead of π-activated alcohols. In these studies, generally gold(III) [43], iridium(III) [44,45], iron(III) [46], or bismuth(III) [47,48] were used as catalysts for the intermolecular hydroarylation of unactivated alkenes. Organic Brønsted acids were also used as catalysts in a smaller number of studies [49,50].
In this work, we searched for some organic Brønsted acids and Lewis acids as catalysts (Table 1) to develop an intramolecular FCA protocol with activating alkenes effectively and economically in order to obtain some originally substituted arylxanthenes under mild conditions for the first time. We found that, among these acids, trifluoroacetic acid (TFA) was the best and most appropriate catalyst for this reaction. According to the literature, different from our work, TFA was reported as a catalyst for FCA with 6-acetoxy-4-alkenylarenes and benzyl alcohols in some previous studies [51,52].
As a continuation of our series of works to develop organic Brønsted acid-catalyzed cyclization reactions [41,42], herein we report a highly efficient intramolecular FCA of appropriate unactivated alkenes with a polyaromatic structure in order to synthesize xanthene derivatives. We developed a new intramolecular FCA method by activating alkenes working under mild reaction conditions and have widened the substrate scope of alkenes to those containing varied electronic and steric properties.
Results and Discussion
Our starting alkenes 4a–l are original and were synthesized in four steps involving coupling, Grignard, oxidation, and Wittig reactions. We synthesized the novel unactivated alkenes 4a–l containing three aryl groups as the starting materials. The synthesis of 4a is demonstrated in Scheme 1. First, 2-phenoxybenzaldehyde (1a) was synthesized by coupling reaction of phenol with commercial 2-fluorobenzaldehyde. This reaction was carried out with very high yield by refluxing the reactants in the presence of K2CO3 in DMF.
![[1860-5397-17-142-i1]](/bjoc/content/inline/1860-5397-17-142-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 4a: (i) phenol, K2CO3, DMF, reflux, 2 h, 91%; (ii) PhMgBr, dry THF, 0 °C, 2 h, 86%; (iii) PCC, CH2Cl2, rt, 2 h, 80%; (iv) Me(Ph)3PBr, t-BuOK, NaH, dry THF, rt, 3 h, 85%.
Scheme 1: Synthesis of 4a: (i) phenol, K2CO3, DMF, reflux, 2 h, 91%; (ii) PhMgBr, dry THF, 0 °C, 2 h, 86%; (i...
2-Phenoxybenzaldehyde (1a) was converted into secondary alcohol derivative 2a by adding a phenyl group using a Grignard reaction. This reaction was carried out with high yield by adding the freshly prepared Grignard compound of phenyl bromide to 2-phenoxybenzaldehyde. As a result, phenoxy secondary alcohol 2a containing three aromatic rings was obtained. In the third step, 2a was oxidized and the ketone derivative 3a was obtained in high yield. The oxidation reaction was carried out using PCC in DCM at room temperature. In the fourth and final step, phenoxydiphenylalkene derivative 4a was prepared by a Wittig reaction, which was carried out using methyltriphenylphosphonium bromide with ketone 3a, in basic medium, at room temperature, and dry THF.
After the structures of the starting compounds were elucidated, the method development trials for the synthesis of xanthene derivatives were carried out. For this purpose, catalyst researches were carried out using compound 4a. An intramolecular Friedel–Crafts reaction was tried by activating the alkene with various organic Brønsted acids and Lewis acids (Table 1). In the reaction, iron(III) chloride hexahydrate, trifluoroacetic acid (TFA), N-trifylphosphoramide (NTPA), benzoic acid, diphenyl phosphate (DPP), malonic acid, chloroacetic acid, copper(II) triflate, acetic acid, and p-toluenesulfonic acid (p-TSA) were used as catalysts. TFA gave the best yield of these catalysts with 78% (Table 1, entry 2). The second-best yield was 65% when FeCl3·6H2O was used (Table 1, entry 1).
Table 1: Screening of catalysts for intramolecular FCA of 1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-142-i3.svg?max-width=637&scale=1.0)
|
||
| Entry | Catalyst (10 mol %) | Conv. (%)b |
| 1 | FeCl3·6H2O | 65 |
| 2 | TFA | 78 |
| 3 | NTPA | 10 |
| 4 | benzoic acid | 0 |
| 5 | DPP | 2 |
| 6 | malonic acid | 0 |
| 7 | chloroacetic acid | 14 |
| 8 | Cu(OTf)2 | 12 |
| 9 | AcOH | 0 |
| 10 | p-TSA | 3 |
aConditions: 4a (0.1 mmol) and catalyst (10 mol %) in CHCl3 (1 mL) were stirred at room temperature for 24 hours. bConversions were determined with GC–MS.
Then the solvent was investigated. Toluene, methyl alcohol, ethyl acetate, THF, DMF, dichloromethane, chloroform, acetone, and acetonitrile were tested as solvents. As a result, it was determined that the best conversion was with dichloromethane (Table 2). Later, quantity and time experiments were performed (Table 2, entries 10–15) and, at the end of these trials, it was determined that the reaction was completed with >99% conversion in 6 hours with 10 mol % catalyst at room temperature (Table 2, entry 14).
Table 2: Exploration of solvents for intramolecular FCA of 1a.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-142-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Cat. amount (mol %) | Solvent | Time | Conv.b |
| 1 | 10 | CHCl3 | 24 h | 78 |
| 2 | 10 | acetone | 24 h | 75 |
| 3 | 10 | toluene | 24 h | 70 |
| 4 | 10 | CH2Cl2 | 24 h | >99 |
| 5 | 10 | THF | 24 h | 45 |
| 6 | 10 | CH3CN | 24 h | 10 |
| 7 | 10 | MeOH | 24 h | 0 |
| 8 | 10 | EtAc | 24 h | 35 |
| 9 | 10 | DMF | 24 h | 0 |
| 10 | 10 | CH2Cl2 | 1 h | 32 |
| 11 | 10 | CH2Cl2 | 2 h | 53 |
| 12 | 10 | CH2Cl2 | 3 h | 70 |
| 13 | 10 | CH2Cl2 | 4 h | 85 |
| 14 | 10 | CH2Cl2 | 6 h | >99 |
| 15 | 5 | CH2Cl2 | 6 h | 95 |
aConditions: 4a (0.1 mmol) and TFA in dry solvent (1 mL) were stirred at room temperature. bConversions were determined with GC–MS.
After determining the most suitable conditions for the intramolecular Friedel–Crafts reaction with alkene activation, the synthesis of the new xanthene derivatives was performed according to this method. The synthesized xanthene derivatives with their isolated yields are shown in Figure 1. Compounds 5b–l were synthesized for the first time in this study. The first synthesis of 5a was prepared by reduction of the corresponding xanthydrol [53] and was also synthesized recently by a different method from our group [29].
![[1860-5397-17-142-1]](/bjoc/content/figures/1860-5397-17-142-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Scope of substrates for intramolecular FCA by activation of 4a–l and their isolated yields. aConditions: 4a–l (0.1 mmol) and TFA (10 mol %) in dry CH2Cl2 (1 mL) were stirred at room temperature for 1–24 hours. bYield of isolated product.
Figure 1: Scope of substrates for intramolecular FCA by activation of 4a–l and their isolated yields. aCondit...
Although in the reactions for FCA reagents and strong inorganic acids, such as AlCl3, H2SO4, or H3PO4, which have generally corrosive properties, were used, in this study, an intramolecular ring closure reaction was carried out under easy operating conditions with an organic Brønsted acid catalyst with high yields. So, the xanthene synthesis with alkene activation was performed for the first time using TFA. The reasonable mechanism of this reaction is delineated in Scheme 2.
![[1860-5397-17-142-i2]](/bjoc/content/inline/1860-5397-17-142-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible reaction mechanism for the cyclization reaction of alkene 4a.
Scheme 2: Plausible reaction mechanism for the cyclization reaction of alkene 4a.
Despite this reaction occurring by the classical Friedel–Crafts mechanism, we believe that o-quinone methide is formed as an intermediate. Because of its very reactive structure, most of the xanthene synthesis is based on the o-quinone methide intermediate [54-58]. The carbocation formed by the activation of an alkene with acid turns into an intermediate o-quinone methide, resulting in a successful cyclization.
As seen in the mechanism, the acid catalyst adds to the vinyl group, allowing the formation of a tertiary carbocation. The carbocation is then transformed into the o-quinone methide intermediate, which undergoes cyclization to yield 9-methyl-9-arylxanthene by aromatization. When the yields of the synthesized compounds are examined, it is seen that the yields are high when there is no substituent at the ring to which the carbocation is attached or when there is an electron-donating group, such as a methoxy or methyl group (Figure 1). When there are electron-withdrawing groups, such as chlorine or cyano groups, in the ring, the yield is slightly reduced because they deactivate the ring in the transition state. The lowest yield was observed for compound 5j to which a cyclopentyl group is attached. Since the cyclopentyl group in this compound is directly attached to the carbon from which the carbocation is formed, byproducts are formed and the yield is reduced since conversions can occur.
Conclusion
In this study, a novel transition metal-free FCA method for the synthesis of 9-methyl-9-arylxanthenes has been developed. For this method, starting compounds with alkene structures suitable for our targets were synthesized. Among the synthesis methods of xanthene derivatives in the literature, the intramolecular Friedel–Crafts method, in which alkenes are activated, has not been used before. Remarkably, our mild reaction conditions using highly effective, inexpensive, and non-toxic reagents are very suitable for the efficient synthesis of xanthenes. The substituted 9-methyl-9-arylxanthenes 5a–l may be used in the pharmaceutical chemistry field as native and bioactive products, and their biochemical potentials and efficiencies will be investigated in the coming years because of their potential biological uses, such as antileukemic, antifungal, antimycobacterial, antitumor, antioxidant, and anticarcinogenic.
Experimental
General information
The majority of the chemicals used in this work were commercially available from Merck or Aldrich. The starting compounds 1a–l were prepared by Ullmann coupling of 2-fluorobenzaldehyde and substituted phenols. Compounds 2a–l were synthesized by Grignard reaction of 1a–l and aryl(or alkyl)magnesium bromides. Then 3a–l were prepared by oxidation of 2a–l using PCC. The final alkene compounds 4a–l were obtained by Wittig reaction using Me(Ph)3PBr, t-BuOK, and NaH. All substrates were purified by crystallization or column chromatography and were characterized by IR and GC–MS. All novel pruducts were characterized by IR, 1H NMR, 13C NMR, elemental analysis and GC–MS. The reactions were monitored by TLC using silica gel plates and the products were purified by flash column chromatography on silica gel (Merck; 230–400 mesh), eluting with hexane/ethyl acetate (v/v 9:1). NMR spectra were recorded at 500 MHz for 1H and 125 MHz for 13C using Me4Si as the internal standard in CDCl3. GC–MS were recorded on a Shimadzu/ QP2010 Plus spectrometer. IR spectra were recorded on a Mattson 1000 spectrometer. Melting points were determined with a Büchi Melting Point B-540 apparatus.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Song, Y.; Yang, Y.; You, J.; Liu, B.; Wu, L.; Hou, Y.; Wang, W.; Zhu, J. Chem. Pharm. Bull. 2013, 61, 167–175. doi:10.1248/cpb.c12-00723
Return to citation in text: [1] -
Lin, C.-N.; Liou, S.-J.; Lee, T.-H.; Chuang, Y.-C.; Won, S.-J. J. Pharm. Pharmacol. 1996, 48, 539–544. doi:10.1111/j.2042-7158.1996.tb05970.x
Return to citation in text: [1] -
Poupelin, J. P.; Saint-Ruf, G.; Foussard-Blanpin, O.; Narcisse, G.; Uchida-Ernouf, G.; Lacroix, R. Eur. J. Med. Chem. 1978, 13, 67.
Return to citation in text: [1] -
Barmak, A.; Niknam, K.; Mohebbi, G.; Pournabi, H. Microb. Pathog. 2019, 130, 95–99. doi:10.1016/j.micpath.2019.03.002
Return to citation in text: [1] -
Akbari, A.; Hosseini-Nia, A. J. Saudi Chem. Soc. 2017, 21 (Suppl. 1), S7–S11. doi:10.1016/j.jscs.2013.09.009
Return to citation in text: [1] -
Lambert, R. W.; Martin, J. A.; Merrett, J. H.; Parkes, K. E. B.; Thomas, G. J. PCT Int. Appl. WO9706178, 1997.
Return to citation in text: [1] -
Banerjee, A.; Mukherjee, A. K. Stain Technol. 1981, 56, 83–85. doi:10.3109/10520298109067286
Return to citation in text: [1] -
Ion, R.-M. Prog. Catal. 1997, 2, 55–64.
Return to citation in text: [1] -
Ion, R. M.; Planner, A.; Wiktorowicz, K.; Frackowiak, D. Acta Biochim. Pol. 1998, 45, 833–845. doi:10.18388/abp.1998_4279
Return to citation in text: [1] -
Knight, C. G.; Stephens, T. Biochem. J. 1989, 258, 683–687. doi:10.1042/bj2580683
Return to citation in text: [1] -
Gonçalves, M. S. T. Chem. Rev. 2009, 109, 190–212. doi:10.1021/cr0783840
Return to citation in text: [1] -
Feringa, B. L. J. Org. Chem. 2007, 72, 6635–6652. doi:10.1021/jo070394d
Return to citation in text: [1] -
Tisseh, Z. N.; Azimi, S. C.; Mirzaei, P.; Bazgir, A. Dyes Pigm. 2008, 79, 273–275. doi:10.1016/j.dyepig.2008.04.001
Return to citation in text: [1] -
Hirata, R.; Torii, A.; Kawano, K.; Futaki, S.; Imayoshi, A.; Tsubaki, K. Tetrahedron 2018, 74, 3608–3615. doi:10.1016/j.tet.2018.05.019
Return to citation in text: [1] -
Kinjo, J.; Uemura, H.; Nohara, T.; Yamashita, M.; Marubayashi, N.; Yoshihira, K. Tetrahedron Lett. 1995, 36, 5599–5602. doi:10.1016/00404-0399(50)1071o-
Return to citation in text: [1] -
Robertson, L. P.; Lucantoni, L.; Duffy, S.; Avery, V. M.; Carroll, A. R. J. Nat. Prod. 2019, 82, 1019–1023. doi:10.1021/acs.jnatprod.8b00956
Return to citation in text: [1] -
Xu, X.; Xu, X.; Li, H.; Xie, X.; Li, Y. Org. Lett. 2010, 12, 100–103. doi:10.1021/ol902489a
Return to citation in text: [1] -
Wang, J.-Q.; Harvey, R. G. Tetrahedron 2002, 58, 5927–5931. doi:10.1016/s0040-4020(02)00534-3
Return to citation in text: [1] -
Su, W.; Yang, D.; Jin, C.; Zhang, B. Tetrahedron Lett. 2008, 49, 3391–3394. doi:10.1016/j.tetlet.2008.03.124
Return to citation in text: [1] -
Naidu, K. R. M.; Krishna, B. S.; Kumar, M. A.; Arulselvan, P.; Khalivulla, S. I.; Lasekan, O. Molecules 2012, 17, 7543–7555. doi:10.3390/molecules17067543
Return to citation in text: [1] -
Zhao, J.; Larock, R. C. Org. Lett. 2005, 7, 4273–4275. doi:10.1021/ol0517731
Return to citation in text: [1] -
Okuma, K.; Nojima, A.; Matsunaga, N.; Shioji, K. Org. Lett. 2009, 11, 169–171. doi:10.1021/ol802597x
Return to citation in text: [1] -
Yoshida, H.; Watanabe, M.; Fukushima, H.; Ohshita, J.; Kunai, A. Org. Lett. 2004, 6, 4049–4051. doi:10.1021/ol048298b
Return to citation in text: [1] -
Zhao, J.; Larock, R. C. J. Org. Chem. 2007, 72, 583–588. doi:10.1021/jo0620718
Return to citation in text: [1] -
Böβ, E.; Hillringhaus, T.; Nitsch, J.; Klussmann, M. Org. Biomol. Chem. 2011, 9, 1744. doi:10.1039/c0ob00954g
Return to citation in text: [1] -
Mao, S.; Hua, Z.; Wu, X.; Yang, Y.; Han, J.; Wang, L. ChemistrySelect 2016, 1, 403–407. doi:10.1002/slct.201600036
Return to citation in text: [1] -
Singh, R.; Panda, G. Org. Biomol. Chem. 2010, 8, 1097. doi:10.1039/b919666h
Return to citation in text: [1] -
Miao, W.; Ye, P.; Bai, M.; Yang, Z.; Duan, S.; Duan, H.; Wang, X. RSC Adv. 2020, 10, 25165–25169. doi:10.1039/d0ra05217e
Return to citation in text: [1] -
Wang, S.; Force, G.; Carpentier, J.-F.; Sarazin, Y.; Bour, C.; Gandon, V.; Lebœuf, D. Org. Lett. 2021, 23, 2565–2570. doi:10.1021/acs.orglett.1c00487
Return to citation in text: [1] [2] -
Zhu, Y.-Q.; Dong, L. J. Org. Chem. 2015, 80, 9973–9979. doi:10.1021/acs.joc.5b01517
Return to citation in text: [1] -
Liu, C.-R.; Yang, F.-L.; Jin, Y.-Z.; Ma, X.-T.; Cheng, D.-J.; Li, N.; Tian, S.-K. Org. Lett. 2010, 12, 3832–3835. doi:10.1021/ol101524w
Return to citation in text: [1] -
Kozak, J. A.; Patrick, B. O.; Dake, G. R. J. Org. Chem. 2010, 75, 8585–8590. doi:10.1021/jo102036n
Return to citation in text: [1] -
Wang, S.; Zhu, Y.; Wang, Y.; Lu, P. Org. Lett. 2009, 11, 2615–2618. doi:10.1021/ol901033h
Return to citation in text: [1] -
Zhou, X.; Zhang, H.; Xie, X.; Li, Y. J. Org. Chem. 2008, 73, 3958–3960. doi:10.1021/jo800232p
Return to citation in text: [1] -
Shirakawa, E.; Itoh, K.-i.; Higashino, T.; Hayashi, T. J. Am. Chem. Soc. 2010, 132, 15537–15539. doi:10.1021/ja1080822
Return to citation in text: [1] -
Yanagisawa, S.; Ueda, K.; Taniguchi, T.; Itami, K. Org. Lett. 2008, 10, 4673–4676. doi:10.1021/ol8019764
Return to citation in text: [1] -
Deng, X.; Liang, J. T.; Liu, J.; McAllister, H.; Schubert, C.; Mani, N. S. Org. Process Res. Dev. 2007, 11, 1043–1050. doi:10.1021/op700183q
Return to citation in text: [1] -
Bandini, M.; Tragni, M. Org. Biomol. Chem. 2009, 7, 1501. doi:10.1039/b823217b
Return to citation in text: [1] -
Prajapati, A.; Kumar, M.; Thakuria, R.; Basak, A. K. Tetrahedron Lett. 2020, 61, 152347. doi:10.1016/j.tetlet.2020.152347
Return to citation in text: [1] -
Das, S. K.; Singh, R.; Panda, G. Eur. J. Org. Chem. 2009, 4757–4761. doi:10.1002/ejoc.200900676
Return to citation in text: [1] -
Yıldız, T.; Küçük, H. B. RSC Adv. 2017, 7, 16644–16649. doi:10.1039/c6ra27094h
Return to citation in text: [1] [2] -
Yildiz, T. Synth. Commun. 2018, 48, 2177–2188. doi:10.1080/00397911.2018.1482351
Return to citation in text: [1] [2] -
Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. J. Organomet. Chem. 2009, 694, 494–501. doi:10.1016/j.jorganchem.2008.07.035
Return to citation in text: [1] -
Matsumoto, T.; Periana, R. A.; Taube, D. J.; Yoshida, H. J. Mol. Catal. A: Chem. 2002, 180, 1–18. doi:10.1016/s1381-1169(01)00390-9
Return to citation in text: [1] -
Bhalla, G.; Oxgaard, J.; Goddard, W. A.; Periana, R. A. Organometallics 2005, 24, 3229–3232. doi:10.1021/om0501733
Return to citation in text: [1] -
Kischel, J.; Jovel, I.; Mertins, K.; Zapf, A.; Beller, M. Org. Lett. 2006, 8, 19–22. doi:10.1021/ol0523143
Return to citation in text: [1] -
Rueping, M.; Nachtsheim, B. J.; Scheidt, T. Org. Lett. 2006, 8, 3717–3719. doi:10.1021/ol0612962
Return to citation in text: [1] -
Sun, H.-B.; Li, B.; Hua, R.; Yin, Y. Eur. J. Org. Chem. 2006, 4231–4236. doi:10.1002/ejoc.200600390
Return to citation in text: [1] -
Bonderoff, S. A.; West, F. G.; Tremblay, M. Tetrahedron Lett. 2012, 53, 4600–4603. doi:10.1016/j.tetlet.2012.06.082
Return to citation in text: [1] -
Fleischer, I.; Pospech, J. RSC Adv. 2015, 5, 493–496. doi:10.1039/c4ra13647k
Return to citation in text: [1] -
Ma, S.; Zhang, J. Tetrahedron 2003, 59, 6273–6283. doi:10.1016/s0040-4020(03)01026-3
Return to citation in text: [1] -
Ladd, A. L.; Gribble, G. W. Biomed. J. Sci. Tech. Res. 2018, 12, 9258–9262. doi:10.26717/bjstr.2018.12.002257
Return to citation in text: [1] -
Lambelet, P.; Lucken, E. A. C. J. Chem. Soc., Perkin Trans. 2 1975, 1652. doi:10.1039/p29750001652
Return to citation in text: [1] -
Karthick, M.; Konikkara Abi, E.; Someshwar, N.; Anthony, S. P.; Ramanathan, C. R. Org. Biomol. Chem. 2020, 18, 8653–8667. doi:10.1039/d0ob01868f
Return to citation in text: [1] -
Huang, C.-G.; Shukla, D.; Wan, P. J. Org. Chem. 1991, 56, 5437–5442. doi:10.1021/jo00018a044
Return to citation in text: [1] -
Yoshida, H.; Watanabe, M.; Fukushima, H.; Ohshita, J.; Kunai, A. Org. Lett. 2004, 6, 4049–4051. doi:10.1021/ol048298b
Return to citation in text: [1] -
Hsiao, C.-C.; Liao, H.-H.; Rueping, M. Angew. Chem., Int. Ed. 2014, 53, 13258–13263. doi:10.1002/anie.201406587
Return to citation in text: [1] -
Hsiao, C.-C.; Raja, S.; Liao, H.-H.; Atodiresei, I.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 5762–5765. doi:10.1002/anie.201409850
Return to citation in text: [1]
| 47. | Rueping, M.; Nachtsheim, B. J.; Scheidt, T. Org. Lett. 2006, 8, 3717–3719. doi:10.1021/ol0612962 |
| 48. | Sun, H.-B.; Li, B.; Hua, R.; Yin, Y. Eur. J. Org. Chem. 2006, 4231–4236. doi:10.1002/ejoc.200600390 |
| 49. | Bonderoff, S. A.; West, F. G.; Tremblay, M. Tetrahedron Lett. 2012, 53, 4600–4603. doi:10.1016/j.tetlet.2012.06.082 |
| 50. | Fleischer, I.; Pospech, J. RSC Adv. 2015, 5, 493–496. doi:10.1039/c4ra13647k |
| 51. | Ma, S.; Zhang, J. Tetrahedron 2003, 59, 6273–6283. doi:10.1016/s0040-4020(03)01026-3 |
| 52. | Ladd, A. L.; Gribble, G. W. Biomed. J. Sci. Tech. Res. 2018, 12, 9258–9262. doi:10.26717/bjstr.2018.12.002257 |
| 1. | Song, Y.; Yang, Y.; You, J.; Liu, B.; Wu, L.; Hou, Y.; Wang, W.; Zhu, J. Chem. Pharm. Bull. 2013, 61, 167–175. doi:10.1248/cpb.c12-00723 |
| 2. | Lin, C.-N.; Liou, S.-J.; Lee, T.-H.; Chuang, Y.-C.; Won, S.-J. J. Pharm. Pharmacol. 1996, 48, 539–544. doi:10.1111/j.2042-7158.1996.tb05970.x |
| 7. | Banerjee, A.; Mukherjee, A. K. Stain Technol. 1981, 56, 83–85. doi:10.3109/10520298109067286 |
| 8. | Ion, R.-M. Prog. Catal. 1997, 2, 55–64. |
| 9. | Ion, R. M.; Planner, A.; Wiktorowicz, K.; Frackowiak, D. Acta Biochim. Pol. 1998, 45, 833–845. doi:10.18388/abp.1998_4279 |
| 6. | Lambert, R. W.; Martin, J. A.; Merrett, J. H.; Parkes, K. E. B.; Thomas, G. J. PCT Int. Appl. WO9706178, 1997. |
| 28. | Miao, W.; Ye, P.; Bai, M.; Yang, Z.; Duan, S.; Duan, H.; Wang, X. RSC Adv. 2020, 10, 25165–25169. doi:10.1039/d0ra05217e |
| 4. | Barmak, A.; Niknam, K.; Mohebbi, G.; Pournabi, H. Microb. Pathog. 2019, 130, 95–99. doi:10.1016/j.micpath.2019.03.002 |
| 5. | Akbari, A.; Hosseini-Nia, A. J. Saudi Chem. Soc. 2017, 21 (Suppl. 1), S7–S11. doi:10.1016/j.jscs.2013.09.009 |
| 25. | Böβ, E.; Hillringhaus, T.; Nitsch, J.; Klussmann, M. Org. Biomol. Chem. 2011, 9, 1744. doi:10.1039/c0ob00954g |
| 3. | Poupelin, J. P.; Saint-Ruf, G.; Foussard-Blanpin, O.; Narcisse, G.; Uchida-Ernouf, G.; Lacroix, R. Eur. J. Med. Chem. 1978, 13, 67. |
| 26. | Mao, S.; Hua, Z.; Wu, X.; Yang, Y.; Han, J.; Wang, L. ChemistrySelect 2016, 1, 403–407. doi:10.1002/slct.201600036 |
| 17. | Xu, X.; Xu, X.; Li, H.; Xie, X.; Li, Y. Org. Lett. 2010, 12, 100–103. doi:10.1021/ol902489a |
| 19. | Su, W.; Yang, D.; Jin, C.; Zhang, B. Tetrahedron Lett. 2008, 49, 3391–3394. doi:10.1016/j.tetlet.2008.03.124 |
| 20. | Naidu, K. R. M.; Krishna, B. S.; Kumar, M. A.; Arulselvan, P.; Khalivulla, S. I.; Lasekan, O. Molecules 2012, 17, 7543–7555. doi:10.3390/molecules17067543 |
| 29. | Wang, S.; Force, G.; Carpentier, J.-F.; Sarazin, Y.; Bour, C.; Gandon, V.; Lebœuf, D. Org. Lett. 2021, 23, 2565–2570. doi:10.1021/acs.orglett.1c00487 |
| 15. | Kinjo, J.; Uemura, H.; Nohara, T.; Yamashita, M.; Marubayashi, N.; Yoshihira, K. Tetrahedron Lett. 1995, 36, 5599–5602. doi:10.1016/00404-0399(50)1071o- |
| 16. | Robertson, L. P.; Lucantoni, L.; Duffy, S.; Avery, V. M.; Carroll, A. R. J. Nat. Prod. 2019, 82, 1019–1023. doi:10.1021/acs.jnatprod.8b00956 |
| 21. | Zhao, J.; Larock, R. C. Org. Lett. 2005, 7, 4273–4275. doi:10.1021/ol0517731 |
| 22. | Okuma, K.; Nojima, A.; Matsunaga, N.; Shioji, K. Org. Lett. 2009, 11, 169–171. doi:10.1021/ol802597x |
| 23. | Yoshida, H.; Watanabe, M.; Fukushima, H.; Ohshita, J.; Kunai, A. Org. Lett. 2004, 6, 4049–4051. doi:10.1021/ol048298b |
| 24. | Zhao, J.; Larock, R. C. J. Org. Chem. 2007, 72, 583–588. doi:10.1021/jo0620718 |
| 54. | Karthick, M.; Konikkara Abi, E.; Someshwar, N.; Anthony, S. P.; Ramanathan, C. R. Org. Biomol. Chem. 2020, 18, 8653–8667. doi:10.1039/d0ob01868f |
| 55. | Huang, C.-G.; Shukla, D.; Wan, P. J. Org. Chem. 1991, 56, 5437–5442. doi:10.1021/jo00018a044 |
| 56. | Yoshida, H.; Watanabe, M.; Fukushima, H.; Ohshita, J.; Kunai, A. Org. Lett. 2004, 6, 4049–4051. doi:10.1021/ol048298b |
| 57. | Hsiao, C.-C.; Liao, H.-H.; Rueping, M. Angew. Chem., Int. Ed. 2014, 53, 13258–13263. doi:10.1002/anie.201406587 |
| 58. | Hsiao, C.-C.; Raja, S.; Liao, H.-H.; Atodiresei, I.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 5762–5765. doi:10.1002/anie.201409850 |
| 11. | Gonçalves, M. S. T. Chem. Rev. 2009, 109, 190–212. doi:10.1021/cr0783840 |
| 12. | Feringa, B. L. J. Org. Chem. 2007, 72, 6635–6652. doi:10.1021/jo070394d |
| 13. | Tisseh, Z. N.; Azimi, S. C.; Mirzaei, P.; Bazgir, A. Dyes Pigm. 2008, 79, 273–275. doi:10.1016/j.dyepig.2008.04.001 |
| 14. | Hirata, R.; Torii, A.; Kawano, K.; Futaki, S.; Imayoshi, A.; Tsubaki, K. Tetrahedron 2018, 74, 3608–3615. doi:10.1016/j.tet.2018.05.019 |
| 41. | Yıldız, T.; Küçük, H. B. RSC Adv. 2017, 7, 16644–16649. doi:10.1039/c6ra27094h |
| 42. | Yildiz, T. Synth. Commun. 2018, 48, 2177–2188. doi:10.1080/00397911.2018.1482351 |
| 10. | Knight, C. G.; Stephens, T. Biochem. J. 1989, 258, 683–687. doi:10.1042/bj2580683 |
| 18. | Wang, J.-Q.; Harvey, R. G. Tetrahedron 2002, 58, 5927–5931. doi:10.1016/s0040-4020(02)00534-3 |
| 53. | Lambelet, P.; Lucken, E. A. C. J. Chem. Soc., Perkin Trans. 2 1975, 1652. doi:10.1039/p29750001652 |
| 35. | Shirakawa, E.; Itoh, K.-i.; Higashino, T.; Hayashi, T. J. Am. Chem. Soc. 2010, 132, 15537–15539. doi:10.1021/ja1080822 |
| 36. | Yanagisawa, S.; Ueda, K.; Taniguchi, T.; Itami, K. Org. Lett. 2008, 10, 4673–4676. doi:10.1021/ol8019764 |
| 29. | Wang, S.; Force, G.; Carpentier, J.-F.; Sarazin, Y.; Bour, C.; Gandon, V.; Lebœuf, D. Org. Lett. 2021, 23, 2565–2570. doi:10.1021/acs.orglett.1c00487 |
| 30. | Zhu, Y.-Q.; Dong, L. J. Org. Chem. 2015, 80, 9973–9979. doi:10.1021/acs.joc.5b01517 |
| 31. | Liu, C.-R.; Yang, F.-L.; Jin, Y.-Z.; Ma, X.-T.; Cheng, D.-J.; Li, N.; Tian, S.-K. Org. Lett. 2010, 12, 3832–3835. doi:10.1021/ol101524w |
| 32. | Kozak, J. A.; Patrick, B. O.; Dake, G. R. J. Org. Chem. 2010, 75, 8585–8590. doi:10.1021/jo102036n |
| 33. | Wang, S.; Zhu, Y.; Wang, Y.; Lu, P. Org. Lett. 2009, 11, 2615–2618. doi:10.1021/ol901033h |
| 34. | Zhou, X.; Zhang, H.; Xie, X.; Li, Y. J. Org. Chem. 2008, 73, 3958–3960. doi:10.1021/jo800232p |
| 44. | Matsumoto, T.; Periana, R. A.; Taube, D. J.; Yoshida, H. J. Mol. Catal. A: Chem. 2002, 180, 1–18. doi:10.1016/s1381-1169(01)00390-9 |
| 45. | Bhalla, G.; Oxgaard, J.; Goddard, W. A.; Periana, R. A. Organometallics 2005, 24, 3229–3232. doi:10.1021/om0501733 |
| 46. | Kischel, J.; Jovel, I.; Mertins, K.; Zapf, A.; Beller, M. Org. Lett. 2006, 8, 19–22. doi:10.1021/ol0523143 |
| 41. | Yıldız, T.; Küçük, H. B. RSC Adv. 2017, 7, 16644–16649. doi:10.1039/c6ra27094h |
| 42. | Yildiz, T. Synth. Commun. 2018, 48, 2177–2188. doi:10.1080/00397911.2018.1482351 |
| 43. | Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. J. Organomet. Chem. 2009, 694, 494–501. doi:10.1016/j.jorganchem.2008.07.035 |
| 39. | Prajapati, A.; Kumar, M.; Thakuria, R.; Basak, A. K. Tetrahedron Lett. 2020, 61, 152347. doi:10.1016/j.tetlet.2020.152347 |
| 40. | Das, S. K.; Singh, R.; Panda, G. Eur. J. Org. Chem. 2009, 4757–4761. doi:10.1002/ejoc.200900676 |
| 37. | Deng, X.; Liang, J. T.; Liu, J.; McAllister, H.; Schubert, C.; Mani, N. S. Org. Process Res. Dev. 2007, 11, 1043–1050. doi:10.1021/op700183q |
| 38. | Bandini, M.; Tragni, M. Org. Biomol. Chem. 2009, 7, 1501. doi:10.1039/b823217b |
© 2021 Yıldız et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)