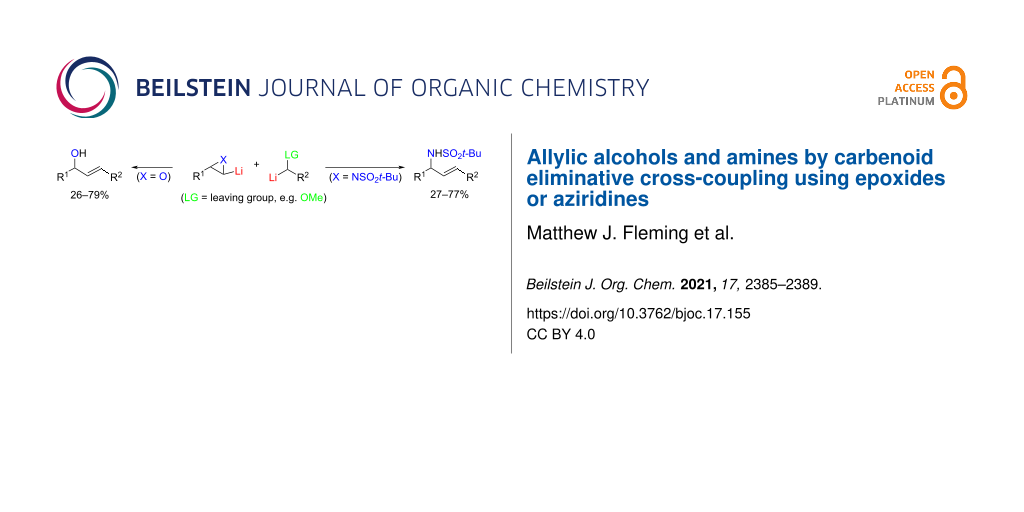
Abstract
α-Lithiated terminal epoxides and N-(tert-butylsulfonyl)aziridines undergo eliminative cross-coupling with α-lithio ethers, to give convergent access to allylic alcohols and allylic amines, respectively. The process can be considered as proceeding by selective strain-relieving attack (ring-opening) of the lithiated three-membered heterocycle by the lithio ether and then selective β-elimination of lithium alkoxide.

Graphical Abstract
Introduction
Methods for the convergent generation of alkenes can be of significant utility in organic synthesis [1]. A relatively under-examined approach is through the interaction of two carbenoids [2]. Dimerisation of carbenoids may compete with a desired carbenoid transformation although its value has been demonstrated in, for example, our studies on lithium 2,2,6,6-tetramethylpiperidide (1, LTMP)-induced syntheses of 2-ene-1,4-diols and 2-ene-1,4-diamines from terminal epoxides [3] and aziridines [4,5], respectively (Scheme 1). The eliminative cross-coupling of carbenoids can provide a way to unsymmetrical alkenes, provided the differential reactivity of the two carbenoids is suitably matched [2]. In the current letter, we report preliminary results on the latter strategy to form alkenes which possess an allylic heteroatom (hydroxy, amino) functionality (Scheme 2).
![[1860-5397-17-155-i1]](/bjoc/content/inline/1860-5397-17-155-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Dimerisation of α-lithio epoxides or aziridines [3-5].
Scheme 1: Dimerisation of α-lithio epoxides or aziridines [3-5].
![[1860-5397-17-155-i2]](/bjoc/content/inline/1860-5397-17-155-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed eliminative cross-coupling of carbenoids to allylic alcohols (X = O) or allylic amines (X = NSO2t-Bu).
Scheme 2: Proposed eliminative cross-coupling of carbenoids to allylic alcohols (X = O) or allylic amines (X ...
Results and Discussion
Our studies began (Scheme 3) by reaction of BuLi (4 equiv) with a mixture of stannane 4 [6] (2 equiv) and tetramethylpiperidine (TMP, 2 equiv), to generate methoxymethyllithium and LTMP, followed by addition of terminal epoxide 5. This led to the desired allylic alcohol 6 (38%), likely via the selective (ring strain-relieving) 1,2-metalate rearrangement outlined in Scheme 2 (2→3, X = O, LG = OMe), then preferential β-elimination [7,8] of lithium methoxide rather than dilithium oxide. However, also isolated was dodecanal (50%), which arises from hydrolysis during work-up of the enamine that is formed from trapping of the lithiated epoxide by LTMP [9,10]. Omitting LTMP gave a significantly improved yield of the allylic alcohol 6 (79%, using BuLi and stannane 4 (3 equiv each)). This latter result suggests that methoxymethyllithium is capable of deprotonating terminal epoxide 5, and this occurs in preference to direct attack at the (unlithiated) epoxide 5. In contrast, no reaction was observed with a 2,2-disubstituted epoxide: 1-oxaspiro[2.11]tetradecane (9) [11] being recovered (90%) under the reaction conditions.
![[1860-5397-17-155-i3]](/bjoc/content/inline/1860-5397-17-155-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Allylic alcohol 6 by one-carbon homologation from epoxide 5.
Scheme 3: Allylic alcohol 6 by one-carbon homologation from epoxide 5.
The one-carbon homologation of an epoxide to an allylic alcohol (cf Scheme 3) can also be achieved using excess dimethylsulfonium methylide [12,13], although non-terminal alkenes have not been shown to be directly accessible by higher homologation. To examine the latter in the context of the current chemistry, α-methoxyhexyllithium derived from stannane 7 [14,15] was reacted with terminal epoxide 5, which gave the allylic alcohol 8 (79%, E/Z = 73:27, Scheme 4). This organolithium also proved reactive with 2,2-disubstituted epoxide 9, giving allylic tertiary alcohol 10 (72%, E/Z = 82:18).
![[1860-5397-17-155-i4]](/bjoc/content/inline/1860-5397-17-155-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Internal allylic alcohols from epoxides and stannane 7.
Scheme 4: Internal allylic alcohols from epoxides and stannane 7.
A trisubstituted alkene 12 (30%) could be formed from terminal epoxide 5, using cyclopropylstannane 11 [16] (Scheme 5); in this case the presence of LTMP was also necessary as epoxide 5 was recovered (>80%) in its absence.
![[1860-5397-17-155-i5]](/bjoc/content/inline/1860-5397-17-155-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cyclopropylidene synthesis from epoxide 5.
Scheme 5: Cyclopropylidene synthesis from epoxide 5.
A silyl-stabilised methoxymethyllithium, available by direct lithiation of (methoxymethyl)trimethylsilane (13) [17], gave vinylsilane 14 (26%, E/Z = 81:19) on reaction with terminal epoxide 5 in the presence of LTMP (Scheme 6); the allylic alcohol 6 was also isolated (20%), suggesting that in our hands lithium–trimethylsilyl exchange competes with lithiation of (methoxymethyl)trimethylsilane (13).
![[1860-5397-17-155-i6]](/bjoc/content/inline/1860-5397-17-155-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Access to allylic alcohol 8 was also achievable (55%, E/Z = 56:44) in a tin-free process using a sulfonyl leaving group, via α-lithiation of sulfone 15 [18] and in the presence of LTMP (Scheme 7). γ-Hydroxysulfone 16 was formed competitively (44%, dr = 50:50), by direct addition of the lithiated sulfone to (unlithiated) epoxide 5 and was formed quantitatively (dr = 57:43) if the LTMP was omitted.
![[1860-5397-17-155-i7]](/bjoc/content/inline/1860-5397-17-155-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Allylic alcohol 8 from epoxide 5 and sulfone 15.
Scheme 7: Allylic alcohol 8 from epoxide 5 and sulfone 15.
Analogous chemistry to that described above (Scheme 3 and Scheme 4) was found to be possible with a terminal aziridine 17, providing access to the corresponding N-Bus-protected allylic amines 18 [19] and 19 (Scheme 8). In these cases, the amines are formed by preferential β-elimination [20,21] of lithium methoxide rather than BusNLi2.
![[1860-5397-17-155-i8]](/bjoc/content/inline/1860-5397-17-155-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Allylic amines from aziridine 17.
Scheme 8: Allylic amines from aziridine 17.
Synthesis of cyclopropylidene 21 (Scheme 9), suggests a terminal N-Bus-aziridine is capable of being deprotonated by the α-lithio cyclopropane from stannane 11; this contrasts with cross-coupling using the same carbenoid and epoxide 5 (Scheme 5), where the presence of LTMP also proved necessary.
![[1860-5397-17-155-i9]](/bjoc/content/inline/1860-5397-17-155-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Cyclopropylidene synthesis from aziridine 20.
Scheme 9: Cyclopropylidene synthesis from aziridine 20.
A cinnamylamine 23 could be obtained in a tin-free process (Scheme 10), which utilises the increased acidity of a benzylic ether 22. In this case, the presence of LTMP was necessary as only γ-amino ether 25 was observed in its absence. It was also important to carry out the reaction at −78 °C to avoid a 1,2-Wittig rearrangement of the lithiated benzyl ether [22]; this restricts the reaction to N-Bus-aziridines, as epoxides are not deprotonated by LTMP at such low temperatures. Alongside the cinnamylamine 23, small amounts of the aziridine-derived carbenoid dimerisation product, 2-ene-1,4-diamine 24 [5], were observed. While the reaction profile was not altered on a solvent switch to hexane (23 (62%, E/Z = 61:39); 24 (16%)), the yield of cinnamylamine 23 was slightly improved in hexane (69%, E/Z = 62:38) and the amount of dimer 24 curtailed (8%) by reducing the amount of LTMP from 2 to 1.2 equiv.
![[1860-5397-17-155-i10]](/bjoc/content/inline/1860-5397-17-155-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Cinnamylamine 23 synthesis from aziridine 17.
Scheme 10: Cinnamylamine 23 synthesis from aziridine 17.
The viability of a benzyl ether (Scheme 10) in the carbenoid eliminative cross-coupling offered a straightforward way to probe any effect of the size of the leaving group on stereoselectivity. However, neither isopropyl or neopentyl benzylic ethers 26 and 27 [23,24] led to a significant change in the E/Z ratio for cinnamylamine 23 (Scheme 11).
![[1860-5397-17-155-i11]](/bjoc/content/inline/1860-5397-17-155-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Cinnamylamine 23 synthesis from isopropyl or neopentyl benzylic ethers 26 and 27.
Scheme 11: Cinnamylamine 23 synthesis from isopropyl or neopentyl benzylic ethers 26 and 27.
Conclusion
In summary, we report a new, convergent access to allylic alcohols and amines. The process proceeds by selective cross-coupling of α-lithio terminal epoxides or N-Bus-aziridines with α-lithio ethers. Where 1,2-disubstituted alkenes are generated the E/Z stereoselectivity is modest, and preliminary results suggest the size of the leaving group does not play a significant role. However, the geometry of alkene formation might be controllable by using enantiomerically pure coupling partners [2]. Such terminal epoxides and aziridines are readily available [3,5], while the corresponding α-lithio ethers can be accessed from enantioenriched α-stannyl ethers [25]. The enantiopure variants await future investigation.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterisation data for all new compounds. | ||
| Format: PDF | Size: 617.0 KB | Download |
References
-
Williams, J. M. J., Ed. Preparation of Alkenes, A Practical Approach; Oxford University Press: Oxford, UK, 1996.
Return to citation in text: [1] -
Blakemore, P. R.; Hoffmann, R. W. Angew. Chem., Int. Ed. 2018, 57, 390–407. doi:10.1002/anie.201707026
Return to citation in text: [1] [2] [3] -
Hodgson, D. M.; Bray, C. D.; Kindon, N. D. Org. Lett. 2005, 7, 2305–2308. doi:10.1021/ol050402h
Return to citation in text: [1] [2] [3] -
Hodgson, D. M.; Miles, S. M. Angew. Chem., Int. Ed. 2006, 45, 935–938. doi:10.1002/anie.200503303
Return to citation in text: [1] [2] -
Hodgson, D. M.; Humphreys, P. G.; Miles, S. M.; Brierley, C. A. J.; Ward, J. G. J. Org. Chem. 2007, 72, 10009–10021. doi:10.1021/jo701901t
Return to citation in text: [1] [2] [3] [4] -
Kaufman, T. S. Synlett 1997, 1377–1378. doi:10.1055/s-1997-1064
Return to citation in text: [1] -
Dechoux, L.; Doris, E.; Mioskowski, C. Chem. Commun. 1996, 549–550. doi:10.1039/cc9960000549
Return to citation in text: [1] -
Hodgson, D. M.; Stent, M. A. H.; Wilson, F. X. Synthesis 2002, 1445–1453. doi:10.1055/s-2002-33112
Return to citation in text: [1] -
Hodgson, D. M.; Bray, C. D.; Kindon, N. D. J. Am. Chem. Soc. 2004, 126, 6870–6871. doi:10.1021/ja031770o
Return to citation in text: [1] -
Hodgson, D. M.; Bray, C. D.; Kindon, N. D.; Reynolds, N. J.; Coote, S. J.; Um, J. M.; Houk, K. N. J. Org. Chem. 2009, 74, 1019–1028. doi:10.1021/jo802016t
Return to citation in text: [1] -
Michnick, T. J.; Matteson, D. S. Synlett 1991, 631–632. doi:10.1055/s-1991-20821
Return to citation in text: [1] -
Alcaraz, L.; Harnett, J. J.; Mioskowski, C.; Martel, J. P.; Le Gall, T.; Shin, D.-S.; Falck, J. R. Tetrahedron Lett. 1994, 35, 5449–5452. doi:10.1016/s0040-4039(00)73522-2
Return to citation in text: [1] -
Alcaraz, L.; Cridland, A.; Kinchin, E. Org. Lett. 2001, 3, 4051–4053. doi:10.1021/ol016782y
Return to citation in text: [1] -
Still, W. C. J. Am. Chem. Soc. 1978, 100, 1481–1487. doi:10.1021/ja00473a025
Return to citation in text: [1] -
Linderman, R. J.; Siedlecki, J. M. J. Org. Chem. 1996, 61, 6492–6493. doi:10.1021/jo961161h
Return to citation in text: [1] -
Gadwood, R. C.; Rubino, M. R.; Nagarajan, S. C.; Michel, S. T. J. Org. Chem. 1985, 50, 3255–3260. doi:10.1021/jo00218a003
Return to citation in text: [1] -
Magnus, P.; Roy, G. Organometallics 1982, 1, 553–559. doi:10.1021/om00063a027
Return to citation in text: [1] -
Orita, A.; Yoshioka, N.; Struwe, P.; Braier, A.; Beckmann, A.; Otera, J. Chem. – Eur. J. 1999, 5, 1355–1363. doi:10.1002/(sici)1521-3765(19990401)5:4<1355::aid-chem1355>3.0.co;2-0
Return to citation in text: [1] -
Hodgson, D. M.; Fleming, M. J.; Stanway, S. J. Org. Lett. 2005, 7, 3295–3298. doi:10.1021/ol051124p
Return to citation in text: [1] -
Hodgson, D. M.; Štefane, B.; Miles, T. J.; Witherington, J. Chem. Commun. 2004, 2234–2235. doi:10.1039/b409486g
Return to citation in text: [1] -
Hodgson, D. M.; Štefane, B.; Miles, T. J.; Witherington, J. J. Org. Chem. 2006, 71, 8510–8515. doi:10.1021/jo0615201
Return to citation in text: [1] -
Azzena, U.; Pilo, L.; Sechi, A. Tetrahedron 1998, 54, 12389–12398. doi:10.1016/s0040-4020(98)00758-3
Return to citation in text: [1] -
Kim, J. D.; Han, G.; Jeong, L. S.; Park, H.-J.; Zee, O. P.; Jung, Y. H. Tetrahedron 2002, 58, 4395–4402. doi:10.1016/s0040-4020(02)00413-1
Return to citation in text: [1] -
Caubere, P.; Moreau, J. Tetrahedron 1970, 26, 2637–2647. doi:10.1016/s0040-4020(01)92838-8
Return to citation in text: [1] -
Clayden, J. Organolithiums: Selectivity for Synthesis; Pergamon Press: Oxford, UK, 2002; pp 178–180.
Return to citation in text: [1]
| 5. | Hodgson, D. M.; Humphreys, P. G.; Miles, S. M.; Brierley, C. A. J.; Ward, J. G. J. Org. Chem. 2007, 72, 10009–10021. doi:10.1021/jo701901t |
| 20. | Hodgson, D. M.; Štefane, B.; Miles, T. J.; Witherington, J. Chem. Commun. 2004, 2234–2235. doi:10.1039/b409486g |
| 21. | Hodgson, D. M.; Štefane, B.; Miles, T. J.; Witherington, J. J. Org. Chem. 2006, 71, 8510–8515. doi:10.1021/jo0615201 |
| 22. | Azzena, U.; Pilo, L.; Sechi, A. Tetrahedron 1998, 54, 12389–12398. doi:10.1016/s0040-4020(98)00758-3 |
| 1. | Williams, J. M. J., Ed. Preparation of Alkenes, A Practical Approach; Oxford University Press: Oxford, UK, 1996. |
| 2. | Blakemore, P. R.; Hoffmann, R. W. Angew. Chem., Int. Ed. 2018, 57, 390–407. doi:10.1002/anie.201707026 |
| 18. | Orita, A.; Yoshioka, N.; Struwe, P.; Braier, A.; Beckmann, A.; Otera, J. Chem. – Eur. J. 1999, 5, 1355–1363. doi:10.1002/(sici)1521-3765(19990401)5:4<1355::aid-chem1355>3.0.co;2-0 |
| 4. | Hodgson, D. M.; Miles, S. M. Angew. Chem., Int. Ed. 2006, 45, 935–938. doi:10.1002/anie.200503303 |
| 5. | Hodgson, D. M.; Humphreys, P. G.; Miles, S. M.; Brierley, C. A. J.; Ward, J. G. J. Org. Chem. 2007, 72, 10009–10021. doi:10.1021/jo701901t |
| 19. | Hodgson, D. M.; Fleming, M. J.; Stanway, S. J. Org. Lett. 2005, 7, 3295–3298. doi:10.1021/ol051124p |
| 3. | Hodgson, D. M.; Bray, C. D.; Kindon, N. D. Org. Lett. 2005, 7, 2305–2308. doi:10.1021/ol050402h |
| 16. | Gadwood, R. C.; Rubino, M. R.; Nagarajan, S. C.; Michel, S. T. J. Org. Chem. 1985, 50, 3255–3260. doi:10.1021/jo00218a003 |
| 2. | Blakemore, P. R.; Hoffmann, R. W. Angew. Chem., Int. Ed. 2018, 57, 390–407. doi:10.1002/anie.201707026 |
| 17. | Magnus, P.; Roy, G. Organometallics 1982, 1, 553–559. doi:10.1021/om00063a027 |
| 9. | Hodgson, D. M.; Bray, C. D.; Kindon, N. D. J. Am. Chem. Soc. 2004, 126, 6870–6871. doi:10.1021/ja031770o |
| 10. | Hodgson, D. M.; Bray, C. D.; Kindon, N. D.; Reynolds, N. J.; Coote, S. J.; Um, J. M.; Houk, K. N. J. Org. Chem. 2009, 74, 1019–1028. doi:10.1021/jo802016t |
| 12. | Alcaraz, L.; Harnett, J. J.; Mioskowski, C.; Martel, J. P.; Le Gall, T.; Shin, D.-S.; Falck, J. R. Tetrahedron Lett. 1994, 35, 5449–5452. doi:10.1016/s0040-4039(00)73522-2 |
| 13. | Alcaraz, L.; Cridland, A.; Kinchin, E. Org. Lett. 2001, 3, 4051–4053. doi:10.1021/ol016782y |
| 3. | Hodgson, D. M.; Bray, C. D.; Kindon, N. D. Org. Lett. 2005, 7, 2305–2308. doi:10.1021/ol050402h |
| 5. | Hodgson, D. M.; Humphreys, P. G.; Miles, S. M.; Brierley, C. A. J.; Ward, J. G. J. Org. Chem. 2007, 72, 10009–10021. doi:10.1021/jo701901t |
| 7. | Dechoux, L.; Doris, E.; Mioskowski, C. Chem. Commun. 1996, 549–550. doi:10.1039/cc9960000549 |
| 8. | Hodgson, D. M.; Stent, M. A. H.; Wilson, F. X. Synthesis 2002, 1445–1453. doi:10.1055/s-2002-33112 |
| 14. | Still, W. C. J. Am. Chem. Soc. 1978, 100, 1481–1487. doi:10.1021/ja00473a025 |
| 15. | Linderman, R. J.; Siedlecki, J. M. J. Org. Chem. 1996, 61, 6492–6493. doi:10.1021/jo961161h |
| 25. | Clayden, J. Organolithiums: Selectivity for Synthesis; Pergamon Press: Oxford, UK, 2002; pp 178–180. |
| 23. | Kim, J. D.; Han, G.; Jeong, L. S.; Park, H.-J.; Zee, O. P.; Jung, Y. H. Tetrahedron 2002, 58, 4395–4402. doi:10.1016/s0040-4020(02)00413-1 |
| 24. | Caubere, P.; Moreau, J. Tetrahedron 1970, 26, 2637–2647. doi:10.1016/s0040-4020(01)92838-8 |
| 3. | Hodgson, D. M.; Bray, C. D.; Kindon, N. D. Org. Lett. 2005, 7, 2305–2308. doi:10.1021/ol050402h |
| 4. | Hodgson, D. M.; Miles, S. M. Angew. Chem., Int. Ed. 2006, 45, 935–938. doi:10.1002/anie.200503303 |
| 5. | Hodgson, D. M.; Humphreys, P. G.; Miles, S. M.; Brierley, C. A. J.; Ward, J. G. J. Org. Chem. 2007, 72, 10009–10021. doi:10.1021/jo701901t |
| 11. | Michnick, T. J.; Matteson, D. S. Synlett 1991, 631–632. doi:10.1055/s-1991-20821 |
| 2. | Blakemore, P. R.; Hoffmann, R. W. Angew. Chem., Int. Ed. 2018, 57, 390–407. doi:10.1002/anie.201707026 |
© 2021 Fleming and Hodgson; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)