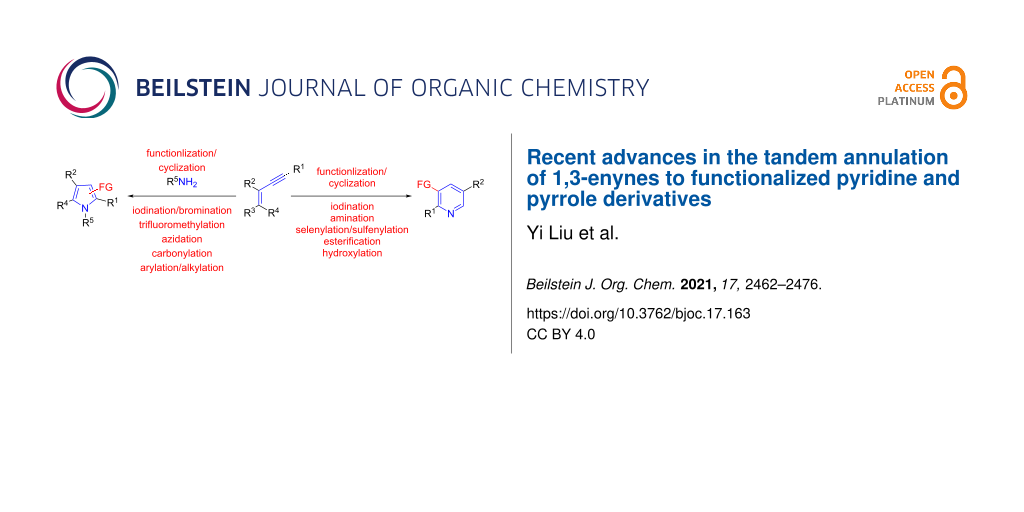
Abstract
Great progress has been made in the tandem annulation of enynes in the past few years. This review only presents the corresponding reactions of 1,3-enyne structural motifs to provide the functionalized pyridine and pyrrole derivatives. The functionalization reactions cover iodination, bromination, trifluoromethylation, azidation, carbonylation, arylation, alkylation, selenylation, sulfenylation, amidation, esterification, and hydroxylation. We also briefly introduce the applications of the products and the reaction mechanisms for the synthesis of corresponding N-heterocycles.

Graphical Abstract
Introduction
The pyridine moiety is an important class of six-membered N-heterocycles that is widely found in many natural products, pharmaceuticals, and bioactive molecules. For instance, some pyridine derivatives have been used for therapy of HIV, cancer, inflammation, microbial infection and so on [1-5]. In addition, it is also an important synthetic unit, which is frequently used as catalyst or ligand in organic chemistry [6-10]. Therefore, the development of efficient methods for the synthesis of pyridine derivatives has attracted considerable attention [11-14]. The industrial synthetic methods of pyridines mainly involve: i) extraction from coal tar; ii) condensation of ammonia, formaldehyde, and acetaldehyde; and iii) preparation from furfural and ammonia. In addition, Hantzsch pyridine synthesis from ethyl acetoacetate, formaldehyde, and ammonia is a commonly used laboratory synthetic method. Recently, extensive and efficient methods for the construction of pyridine derivatives have been developed through the intramolecular or intermolecular tandem addition annulation/functionalization of alkynes with some N-containing compounds, such as nitriles, oximes, and imines [15-19].
The pyrrole structural motif is also an invaluable five-membered N-heterocycle that is widely used in pharmaceuticals, photoelectric materials, and functional materials [20-23]. Many pyrrole derivatives play a significant role in the life science and medicine domains due to the good bioactivities, such as antitumor, anti-HIV, and anti-HSV-1 activity [24-29]. In industry, pyrrole mainly comes from the extraction of coal tar, the condensation reaction of furan and ammonia under high temperature, or the cascade cyclization reaction of acetylene, formaldehyde, and ammonia. In the laboratory, there are many efficient methods for the synthesis of pyrrole derivatives: i) Knorr reaction: the condensation of α-aminoketones or α-amino esters in the presence of zinc powder and sodium acetate; ii) Paal–Knorr reaction: the condensation of 1,4-dicarbonyl compounds and amines, catalyzed by formic acid in anhydrous alcohol; iii) Hantzsch reaction: the condensation of α-halogenated carbonyl compounds, β-dicarbonyl compounds and amines; and iv) the latest developed multicomponent tandem reactions and transition metal-catalyzed coupling reactions [30-39].
Recently, substantial achievements have been made using azides as a powerful nitrogen source for the synthesis of various N-heterocycles, such as isoquinolines, quinolines, pyridines, pyrroles, indoles, azoles, and azepines [40-45]. 1,3-Enyne, as a powerful Michael acceptor, is a wonderful synthon for the synthesis of N-heterocycles via tandem addition and annulation. Recently, there have been several elegant reviews covering the 1,3-enynes chemistry [46-48]. For instance, Procter and co-workers reviewed the copper-catalyzed functionalization of enynes [46]. In 2020, the Wang group reviewed the development of 2-activated 1,3-enyne in enantioselective synthesis [47]. Further, the Liu group reviewed the synthesis of allenes via transition metal-catalyzed 1,4-functionalizations of unactivated 1,3-enynes [48]. In this review, we will highlight the recent advances in the tandem annulation reactions of 1,3-enyne structural motifs for the construction of functionalized pyridines and pyrroles.
Review
Synthesis of pyridines via tandem annulation of 1,3-enynes
In 2015, Reddy and co-workers reported the synthesis of substituted pyridines via Lewis acid-mediated aza-annulation of 2-en-4-ynyl azides 1 (Scheme 1) [49]. They discovered that Ag-mediated intramolecular annulation of 2-en-4-ynyl azides 1 could provide the corresponding 3,6-disubstituted pyridines 2 in 60–88% yield in the presence of TFA (2.0 equiv). The reaction substrates, 2-en-4-ynyl azides 1, derived from MBH acetates of acetylenic aldehydes, could tolerate various substituted aryl, indolyl, and alkyl (such as n-propyl and n-hexyl) groups under the standard conditions. 2-En-4-ynyl azides 1 bearing electron-donating substituents (such as methyl and methoxy groups) obviously worked better than those with electron-withdrawing (such as nitro, cyano, acetyl, and trifluoromethyl) groups. Meanwhile, they also found that the aza-annulation could be carried out under iodine-mediated electrophilic annulation reaction conditions to give 5-iodo-3,6-disubstituted pyridines 3 as the major products, occasionally with a small amount of 2-acylated pyrroles 4.
![[1860-5397-17-163-i1]](/bjoc/content/inline/1860-5397-17-163-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Ag/I2-mediated electrophilic annulation of 2-en-4-ynyl azides 1.
Scheme 1: Ag/I2-mediated electrophilic annulation of 2-en-4-ynyl azides 1.
The proposed mechanism for the Ag-catalyzed aza-annulation of (E)-2-en-4-ynyl azides 1 was considered to involve 6-endo-dig cyclization to give a corresponding N-N2-substituted 1,2-dihydropyridine silver salt 5. This was protonated by TFA and the following species neutralized by base to provide a final 3,6-disubstituted pyridine product 2 (Scheme 2). However, an iodonium ion 6 was formed as a key intermediate in I2-mediated aza-annulations. Subsequently, the iodonium ion 6 proceeds through a 6-endo-dig cyclization to form the 5-iodopyridine 3. On the other side, the iodonium ion 6 may undergo 5-exo-dig cyclization to yield the 2-acylpyrrole 4. Normally, (E)-2-en-4-yn-1-azides 1 with electron-rich substituent groups favorably give the 5-iodopyridine 3, while for substrates containing electron-poor groups, the 2-acylpyrrole 4 is favored (Scheme 3).
![[1860-5397-17-163-i2]](/bjoc/content/inline/1860-5397-17-163-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The proposed mechanism of Ag-catalyzed aza-annulation.
Scheme 2: The proposed mechanism of Ag-catalyzed aza-annulation.
![[1860-5397-17-163-i3]](/bjoc/content/inline/1860-5397-17-163-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The proposed mechanism of I2-mediated aza-annulation.
Scheme 3: The proposed mechanism of I2-mediated aza-annulation.
Then, the Reddy group developed a copper-catalyzed aminative aza-annulation of enynyl azides with N-fluorobenzenesulfonimide (NFSI) to provide amino-substituted nicotinate derivatives 8 in good to excellent yield (Scheme 4) [50]. The investigation showed that the electronic effect of the residue R on the substrates influences the results significantly. (E)-2-en-4-ynyl azides 1 bearing electron-donating groups had better reactivity, with a higher yield and a shorter reaction time. In addition, substrates 1 with aliphatic groups (such as R = n-propyl, n-pentyl, and n-hexyl) were also tolerated under standard conditions, with an excellent yield.
![[1860-5397-17-163-i4]](/bjoc/content/inline/1860-5397-17-163-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Copper-catalyzed amination of (E)-2-en-4-ynyl azides 1.
Scheme 4: Copper-catalyzed amination of (E)-2-en-4-ynyl azides 1.
The previous literature and control experiments showed that this aminative aza-annulation reaction may undergo a free-radical addition pathway. Firstly, NFSI oxidizes Cu(I) to form bissulfonylamidyl radical 10. Secondly, intermolecular nitrogen free-radical addition to the alkyne provides the vinyl radical 11. Then, there may be two possible pathways. Path a: vinyl radical 11 is trapped by Cu(II) to deliver the Cu(III) species 12, which undergoes intramolecular annulation and reductive elimination to afford the desired product 8 and regenerate the Cu(I) catalyst. Path b: vinyl radical intermediate 11 is oxidized by Cu(II) to give the cationic vinyl species 14. Finally, the intramolecular nucleophilic attack by azide and the following deprotonation by a fluoride anion provide the final product 8 (Scheme 5).
![[1860-5397-17-163-i5]](/bjoc/content/inline/1860-5397-17-163-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The proposed mechanism of copper-catalyzed amination.
Scheme 5: The proposed mechanism of copper-catalyzed amination.
The derivatization of sulfonated aminonicotinates 8 could easily be achieved. Desulfonylation of aminonicotinate 8b proceeded smoothly in the presence of triflic acid (2.0 equiv) in DCE at 90 °C to provide the desulfonated 5-amino-substituted nicotinate 15 in 77% yield. Furthermore, treatment of aminonicotinate 8b with KOH (8.0 equiv) in MeOH, or with NiCl2(dppp) (5 mol %) and K3PO4 (4.0 equiv) in 1,4-dioxane afforded 5-(phenylsulfonamido)-6-(p-tolyl)nicotinic acid 16 (in 90% yield) and monodesulfonated nicotinate 17 (in 70% yield), respectively (Scheme 6).
![[1860-5397-17-163-i6]](/bjoc/content/inline/1860-5397-17-163-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: The derivatization of sulfonated aminonicotinates.
Scheme 6: The derivatization of sulfonated aminonicotinates.
Selenyl- and sulfenylpyridine derivatives are gaining prominence due to the prospective biological activities. They could be used for treatment of HIV, cancer, inflammation, and microbial infection. Therefore, the synthesis of selenyl- and sulfenylpyridines has attracted considerable attention. In 2019, the Reddy group reported a copper-catalyzed aza-annulation of enynyl azides 1 for the synthesis of 5-selenyl- and sulfenylpyridine derivatives 18 and 19 (Scheme 7) [51]. Diorganyl dichalcogenides (R1XXR1, X = Se, S) were used as selenyl and sulfenyl sources, respectively. The method was performed under open atmosphere to provide the target products in good to excellent yield. In comparison, the selenoamination of (E)-2-en-4-ynyl azides 1 showed higher reactivity and could be carried out at 0 °C in 1 h to give the selenyl-substituted nicotinates 18 in excellent yield. Electron-donating and electron-withdrawing group-substituted diaryl diselenides, 1,2-di(thiophen-2-yl)diselane, and dimethyl diselenide were compatible to give the corresponding products 18. The sulfenylamination of (E)-2-en-4-ynyl azides 1 could also be carried out at 90 °C in 8 h to provide the 5-sulfenyl-substituted nicotinates 19 efficiently.
![[1860-5397-17-163-i7]](/bjoc/content/inline/1860-5397-17-163-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Copper-catalyzed chalcogenoamination of (E)-2-en-4-ynyl azides 1.
Scheme 7: Copper-catalyzed chalcogenoamination of (E)-2-en-4-ynyl azides 1.
Based on previous literature and control experiments, the possible mechanism is outlined in Scheme 8. First, the Cu-complex-polarized X−X bond can promote the electrophilic addition onto the alkyne to generate intermediate 20. Then, the intramolecular nucleophilic attack by azide and the following deprotonation give the final product 18 or 19, respectively.
![[1860-5397-17-163-i8]](/bjoc/content/inline/1860-5397-17-163-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: The possible mechanism of chalcogenoamination.
Scheme 8: The possible mechanism of chalcogenoamination.
5‑Selenyl- and 5-sulfenyl-substituted nicotinates can carry out versatile transformations, which have potential application in pharmaceutical, agrochemical, and organic synthetic chemistry. For example, 5-selenyl- and 5-sulfenyl-appended nicotinates 18c and 19c could be oxidized by mCPBA to the corresponding selenoxide, sulfoxide, and sulfone derivatives 22, 24, and 25, respectively. In addition, 5‑selenyl-substituted nicotinate 18c could be converted to the corresponding acid 23 in 94% yield in the presence of KOH (2.0 equiv) in MeOH (Scheme 9).
![[1860-5397-17-163-i9]](/bjoc/content/inline/1860-5397-17-163-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: The derivatization of 5‑selenyl- and 5-sulfenyl-substituted nicotinates.
Scheme 9: The derivatization of 5‑selenyl- and 5-sulfenyl-substituted nicotinates.
In 2011, Lee and co-workers reported a one-pot method for the construction of polysubstituted pyridines 29 via tandem sequential reactions of nitriles 26, Reformatsky reagents 27, and 1,3-enynes 28 (Scheme 10) [52]. The tandem reaction involved a regio- and chemoselective addition of the Blaise reaction intermediate to 1,3-enyne, and the following sequential processes: isomerization, cyclization, and aromatization. Both carbocyclic and acyclic 1,3-enyenes 28 were compatible to give the corresponding esterified pyridines 29 in moderate to high yield. It is worth noting that 1,3-enynes 28 bearing internal alkyne moieties were not tolerated as substrates.
![[1860-5397-17-163-i10]](/bjoc/content/inline/1860-5397-17-163-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: The tandem reaction of nitriles, Reformatsky reagents, and 1,3-enynes.
Scheme 10: The tandem reaction of nitriles, Reformatsky reagents, and 1,3-enynes.
In 2016, Aïssa and co-workers reported a nickel-catalyzed [4 + 2]-cycloaddition of 3-azetidinones 30 with 1,3-enynes 31 for the synthesis of 3‑hydroxy-4,5-alkyl-substituted pyridines 33 (Scheme 11) [53]. The transformation involved a two-step sequence of successive reactions: Firstly, the nickel-catalyzed [4 + 2]-cycloaddition of 1,3-enynes 31 and N-Ts-substituted 3-azetidinone 30 afforded dihydropyridinones 32 in good yield. The next step involved the hydrogenation of dihydropyridinones 32 and a following desulfonylation and aromatization to give pyridine derivatives 33 in moderate to good yield.
![[1860-5397-17-163-i11]](/bjoc/content/inline/1860-5397-17-163-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Nickel-catalyzed [4 + 2]-cycloaddition of 3-azetidinones with 1,3-enynes.
Scheme 11: Nickel-catalyzed [4 + 2]-cycloaddition of 3-azetidinones with 1,3-enynes.
Synthesis of pyrroles via tandem annulation of 1,3-enynes
Recently, great achievements have been made in electrophilic iodocyclization of alkynes for the synthesis of five- or six-membered cyclic compounds [54]. Various efficient synthetic methods have been developed for the synthesis of halogenated pyrroles, which are widely presented in many pharmacologically active natural products, bioactive molecules, and organic building blocks. Based on the previous studies on heterocycle synthesis, Punniyamurthy and co-workers designed the electrophilic iodocyclization of 2-nitro-1,3-enynes 34 for the synthesis of pyrrole derivatives. In 2013, they reported an efficient route to pentasubstituted pyrroles from 2-nitro-1,3-enynes 34, amines, and iodine under mild conditions (Scheme 12) [55]. The reaction was performed in CH2Cl2 at ambient conditions in the presence of 2.0 equiv of K2CO3. Substrates bearing electron-donating or electron-withdrawing groups were compatible under standard conditions to give the highly substituted pyrroles in moderate to good yield. Aliphatic amines were also tolerated, providing the desired products in only moderate yield. The plausible mechanism involves a tandem base-promoted aza-Michael addition, 1,2-iodocyclization, and iodine-mediated oxidative aromatization.
![[1860-5397-17-163-i12]](/bjoc/content/inline/1860-5397-17-163-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Electrophilic iodocyclization of 2-nitro-1,3-enynes to pyrroles.
Scheme 12: Electrophilic iodocyclization of 2-nitro-1,3-enynes to pyrroles.
In 2017, Zhang and co-workers reported a silver-catalyzed tandem reaction of 2-trifluoromethyl-1,3-enynes 36 with primary amines, affording various trifluoromethyl-substituted 3-pyrrolines [56]. Subsequently, they also developed a novel route for the synthesis of halogenated trifluoromethylated pyrroles 37 and 38 by sequential intermolecular hydroamination reaction of 2-trifluoromethyl-1,3-enynes 36 with aliphatic primary amines and the following NXS-mediated oxidative cyclization (Scheme 13) [57]. The method tolerated various substituted benzylamines, 2-phenylethanamines, isopropylamine, and other aliphatic chain-like amines. Furthermore, both furan-2-ylmethanamine and thiophen-2-ylmethanamine were reacted smoothly with NIS under standard conditions, while they did not react well with NBS. Notably, under the same reaction conditions, the desired products of the iodination and bromoniation reactions were trifluoromethylated monoiodopyrroles 37 and dibromopyrroles 38, respectively.
![[1860-5397-17-163-i13]](/bjoc/content/inline/1860-5397-17-163-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Electrophilic halogenation of 2-trifluoromethyl-1,3-enynes to pyrroles.
Scheme 13: Electrophilic halogenation of 2-trifluoromethyl-1,3-enynes to pyrroles.
Subsequently, Punniyamurthy and co-workers also described the copper-catalyzed cascade cyclization of 2-nitro-1,3-enynes 34 to tetrasubstituted pyrroles 39 (Scheme 14) [58]. Through screening the conditions, the Cu(OTf)2-promoted (5 mol %) annulation addition reaction of 2-nitro-1,3-enynes 34 and amines was carried out smoothly in THF at room temperature under air. The protocol showed broad substrate scope, and various different aromatic substrates (R1, R2, and R3 = aryl) reacted well. However, no target product was observed when aliphatic amine was used as substrate under standard conditions. The proposed catalytic cycle included aza-Michael addition of arylamines, Lewis acid copper(II)-catalyzed intramolecular 5-endo-dig cyclization, protonation, and oxidation to provide the final products, tetrasubstituted pyrroles 39.
![[1860-5397-17-163-i14]](/bjoc/content/inline/1860-5397-17-163-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Copper-catalyzed cascade cyclization of 2-nitro-1,3-enynes with amines.
Scheme 14: Copper-catalyzed cascade cyclization of 2-nitro-1,3-enynes with amines.
The introduction of a trifluoromethyl group into organic molecules can efficiently modify the physical, chemical, and biological properties of the compounds. A trifluoromethyl-substituted pyrrole unit is widely present in many natural compounds and pharmaceuticals with high biological activity. Based on our previous study on the construction of trifluoromethylated coumarins [59], we recently developed a Rh-catalyzed approach to trifluoromethyl-substituted pyrroles using the Togni reagent II as trifluoromethyl source. It involves a three-component cascade reaction of 1,3-enynes, anilines, and Togni reagent II to afford fully substituted pyrrole derivatives in DMF at room temperature (Scheme 15) [60]. The reaction was promoted by Cu(OAc)2·H2O (2.0 equiv) and Ca(OH)2 (2.0 equiv), providing the desired products in moderate to good yield. Various substituted 1,3-enynes with methyl, methoxy, fluoro, and chloro groups could react with p-toluidine under standard conditions in moderate yield (40–71%). Notably, aromatic amines bearing electron-donating or electron-withdrawing groups were compatible. When 2-naphthylamine was used under standard conditions, α-trifluoromethyl-substituted 2-naphthylamine was obtained as major product (51% yield), and only a trace amount of the desired product was observed. Control experiments indicated that the possible reaction mechanism may proceed through a Cu(II)/Rh(III)-promoted radical process.
![[1860-5397-17-163-i15]](/bjoc/content/inline/1860-5397-17-163-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Tandem cyclization of 2-nitro-1,3-enynes, Togni reagent II, and amines.
Scheme 15: Tandem cyclization of 2-nitro-1,3-enynes, Togni reagent II, and amines.
Aryl azides are versatile intermediates, which were widely used in synthetic and medicinal chemistry as well as material and biological sciences. Recently, great attention has been paid to synthesize organic azido compounds via various transformations. In 2020, we reported an efficient method for the synthesis of fully substituted azidopyrroles 41 via Cu- and Mn-co-mediated aerobic oxidative cyclization/azidation reaction of 2-nitro-1,3-enynes 34 with amines, and trimethylsilyl azide (TMSN3, Scheme 16) [61]. The reaction could be carried out efficiently in the presence of 2.0 equiv of Cu(OAc)2·H2O. Interestingly, the addition of 10 mol % MnCl2 could promote the reaction more smoothly. A wide range of substituted aromatic amines were reacted well, while amines substituted with strongly electron-withdrawing (such as nitro and trifluoromethyl) groups, heteroaryl amines, and aliphatic amines were not compatible. Control experiments showed that the addition of 2.0 equiv of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) under standard conditions could inhibit the formation of target product. In contrast, the compound TEMPO–N3 was detected by GC–MS analysis. Based on the radical trapping experiment and previous reports, the reaction may undergo a radical process.
![[1860-5397-17-163-i16]](/bjoc/content/inline/1860-5397-17-163-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Tandem cyclization of 2-nitro-1,3-enynes, TMSN3, and amines.
Scheme 16: Tandem cyclization of 2-nitro-1,3-enynes, TMSN3, and amines.
2-Carbonylpyrrole is a key subunit of many bioactive natural products with potential biological activities or pharmacological activities. For instance, longanlactone, zomepirac (Zomax), ketorolac and pollenopyrroside A are pyrrole derivatives bearing a 2-carbonyl group. Therefore, the synthesis of such kinds of pyrrole derivatives is highly valuable. In 2017, Baire and Gandhi reported an Ag-catalyzed cascade cyclization of 6-hydroxyhex-2-en-4-ynals 42 and primary amines to give the 2-(α-hydroxyacyl)pyrroles 43 in moderate to good yield (Scheme 17) [62]. The proposed mechanism involves the condensation of amine and aldehyde to give the imine 44 and the AgNO3-promoted 5-exo-dig cyclization of imine to form a zwitter ion intermediate 45.
![[1860-5397-17-163-i17]](/bjoc/content/inline/1860-5397-17-163-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Cascade cyclization of 6-hydroxyhex-2-en-4-ynals to pyrroles.
Scheme 17: Cascade cyclization of 6-hydroxyhex-2-en-4-ynals to pyrroles.
In 2017, the Reddy group also reported a method for the construction of 2-carbonylpyrroles 46 through Au/Ag-catalyzed intramolecular oxidative aza-annulation of 1,3-enynyl azides 1 (Scheme 18) [63]. The method is very applicable, and various aryl-substituted enynyl azides bearing electron-donating or -withdrawing (such as methyl, methoxy, chloro, cyano, nitro, acyl, and trifluoromethyl) groups all worked smoothly to deliver the corresponding 2-carbonylpyrroles 46 in good to excellent yield. Aliphatic enynyl azide (R = 1-hexyl) was also tolerated efficiently under the standard conditions to afford the desired product 46k in 64% yield. In addition, tert-butyldimethylsilyl (TBS)-substituted enynyl azide provided the target product 46l in 34% yield.
![[1860-5397-17-163-i18]](/bjoc/content/inline/1860-5397-17-163-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Au/Ag-catalyzed oxidative aza-annulation of 1,3-enynyl azides.
Scheme 18: Au/Ag-catalyzed oxidative aza-annulation of 1,3-enynyl azides.
The transformation involves a sequence of C−N/C−O bond formation, and the corresponding plausible mechanism is shown in Scheme 19. Firstly, Au-coordinated alkyne undergoes regioselective hydration to form intermediate 48. Then, intramolecular nucleophilic attack by azide occurs to give 2-carbonyl intermediate 49. Subsequently, intermediate 49 will undergo aromatization as well as the release of a nitrogen molecule to form the desired product 46.
![[1860-5397-17-163-i19]](/bjoc/content/inline/1860-5397-17-163-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: The plausible mechanism of Au/Ag-catalyzed oxidative aza-annulation.
Scheme 19: The plausible mechanism of Au/Ag-catalyzed oxidative aza-annulation.
In 2018, Ding and co-workers reported the synthesis of 2-tetrazolyl-substituted 3-acylpyrroles 53 via sequential Ugi-azide/Ag-catalyzed oxidative cycloisomerization reactions in good yield (Scheme 20) [64]. Firstly, The Ugi-azide reaction products 52 were obtained efficiently through the cascade reactions of enynals 51, primary amines, aliphatic isocyanides, and trimethylsilyl azide. The following reaction involves Ag-catalyzed intramolecular 5-endo-dig cyclization and base (DMAP)-promoted oxidative isomerization. The presence of DMAP is necessary for this transformation.
![[1860-5397-17-163-i20]](/bjoc/content/inline/1860-5397-17-163-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of 2-tetrazolyl-substituted 3-acylpyrroles from enynals.
Scheme 20: Synthesis of 2-tetrazolyl-substituted 3-acylpyrroles from enynals.
Recently, copper hydride (CuH) catalysis has been a wonderful procedure for olefin hydrofunctionalization via the formation of nucleophilic alkylcopper intermediate. In 2016, Buchwald and co-workers described a CuH-catalyzed asymmetric addition of olefin to ketones [65]. Then, they also reported another CuH-catalyzed coupling reaction of 1,3-enynes 54 and nitrile to prepare polysubstituted pyrroles 55 (Scheme 21) [66]. The substrates 54 could be easily prepared by Sonogashira coupling of terminal alkynes and vinyl halides. It is worth mentioning that the addition of the bisphosphine ligand DTBM-SEGPHOS (56) was very important to promote the transformation efficiently. The reaction showed a broad substrate scope, with aromatic and aliphatic substrates 54 (R1 = aryl, heterocycle, and alkyl) being good coupling partners, providing the corresponding 2,3-dialkyl-5-aryl-substituted pyrroles 55 in moderate to good yield. In addition, the method could tolerate a wide range of functional groups, such as phenolic hydroxy, aryl bromide, ester, terminal olefin, aryl chloride, and silyl-protected alcohol moieties. Furthermore, both aromatic and aliphatic nitriles performed well in the reaction with 1,3-enynes 54, providing moderate yield and regioselectivity. The CuH-catalyzed intramolecular coupling of enyne containing a nitrile group worked smoothly and gave a moderate yield under standard conditions at a decreased concentration.
![[1860-5397-17-163-i21]](/bjoc/content/inline/1860-5397-17-163-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: CuH-catalyzed coupling reaction of 1,3-enynes and nitriles to pyrroles.
Scheme 21: CuH-catalyzed coupling reaction of 1,3-enynes and nitriles to pyrroles.
A plausible mechanism according to previously reported methods is proposed in Scheme 22. Firstly, the hydrocupration of enyne 54 with LCuH 57 provides propargylcopper intermediate 58. The 1,3-isomerization of 58 and the following nitrile addition produces imine intermediate 60, which subsequently undergoes intramolecular cyclization, a 1,5-hydrogen shift and σ-bond metathesis with hydrosilane to give the silylated pyrrole product 63 and the LCuH catalyst 57. In addition, the intermediate 58 might go through isomerization to form imine intermediate 64, which undergoes intramolecular cyclization to provide the minor regioisomer 67 (inner cycle in Scheme 22).
![[1860-5397-17-163-i22]](/bjoc/content/inline/1860-5397-17-163-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: The mechanism of CuH-catalyzed coupling of 1,3-enynes and nitriles to pyrroles.
Scheme 22: The mechanism of CuH-catalyzed coupling of 1,3-enynes and nitriles to pyrroles.
Conclusion
1,3-Enynes, one of the most significant classes of Michael acceptors for the construction of N-heterocycles, have been widely used in organic synthesis. We herein reviewed the recent advances in the development of tandem cyclization reactions of 1,3-enynes in the presence of electrophiles or Lewis acid catalysts to form pyridines and pyrroles. Series of iodinated, aminated, selenylated, sulfenylated, esterified, and hydroxylated pyridine derivatives have been prepared based on 1,3-enynes. In addition, we also reviewed the tandem cyclization of 1,3-enynes to realize various functionalizations of pyrrole derivatives, such as iodination, bromination, trifluoromethylation, azidation, carbonylation, arylation, and alkylation. The proposed mechanism generally involves two kinds of intramolecular cyclizations: one is 6-endo-dig cyclization to promote the formation of pyridine ring derivatives and the other is 5-exo-dig cyclization to afford the pyrrole derivatives.
Considering the good biological activities and the wide applicability in synthetic organic chemistry, biopharmaceuticals, and materials, it is desirable to develop more efficient methods for the synthesis of diverse functionalized pyridine and pyrrole derivatives from easily available 1,3-enynes. Therefore, the significant challenges will focus on the following aspects in the future: i) development of more functionalizations of pyridines and pyrroles (such as fluorination, trifluoromethylthiolation, olefination, alkynylation, boronization, phosphorization, etc); ii) discovery of new transformations of 1,3-enynes to other N-heterocycles; and iii) more extensive investigations into the mechanism.
References
-
O’Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d
Return to citation in text: [1] -
Narendar, P.; Parthiban, J.; Anbalagan, N.; Gunasekaran, V.; Leonard, J. T. Biol. Pharm. Bull. 2003, 26, 182–187. doi:10.1248/bpb.26.182
Return to citation in text: [1] -
Bagley, M. C.; Dale, J. W.; Merritt, E. A.; Xiong, X. Chem. Rev. 2005, 105, 685–714. doi:10.1021/cr0300441
Return to citation in text: [1] -
Metobo, S. E.; Jin, H.; Tsiang, M.; Kim, C. U. Bioorg. Med. Chem. Lett. 2006, 16, 3985–3988. doi:10.1016/j.bmcl.2006.05.018
Return to citation in text: [1] -
Kishbaugh, T. L. S. Curr. Top. Med. Chem. 2016, 16, 3274–3302. doi:10.2174/1568026616666160506145141
Return to citation in text: [1] -
Gibson, V. C.; Redshaw, C.; Solan, G. A. Chem. Rev. 2007, 107, 1745–1776. doi:10.1021/cr068437y
Return to citation in text: [1] -
Peloquin, D. M.; Schmedake, T. A. Coord. Chem. Rev. 2016, 323, 107–119. doi:10.1016/j.ccr.2016.02.005
Return to citation in text: [1] -
Rajput, A.; Mukherjee, R. Coord. Chem. Rev. 2013, 257, 350–368. doi:10.1016/j.ccr.2012.03.024
Return to citation in text: [1] -
Ding, Q.; Ye, S.; Cheng, G.; Wang, P.; Farmer, M. E.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 417–425. doi:10.1021/jacs.6b11097
Return to citation in text: [1] -
Liu, J.; Ding, Q.; Fang, W.; Wu, W.; Zhang, Y.; Peng, Y. J. Org. Chem. 2018, 83, 13211–13216. doi:10.1021/acs.joc.8b01933
Return to citation in text: [1] -
Wei, Y.; Yoshikai, N. J. Am. Chem. Soc. 2013, 135, 3756–3759. doi:10.1021/ja312346s
Return to citation in text: [1] -
Michlik, S.; Kempe, R. Angew. Chem., Int. Ed. 2013, 52, 6326–6329. doi:10.1002/anie.201301919
Return to citation in text: [1] -
Loy, N. S. Y.; Singh, A.; Xu, X.; Park, C.-M. Angew. Chem., Int. Ed. 2013, 52, 2212–2216. doi:10.1002/anie.201209301
Return to citation in text: [1] -
Yamamoto, S.-i.; Okamoto, K.; Murakoso, M.; Kuninobu, Y.; Takai, K. Org. Lett. 2012, 14, 3182–3185. doi:10.1021/ol301273j
Return to citation in text: [1] -
Xu, F.; Wang, C.; Wang, D.; Li, X.; Wan, B. Chem. – Eur. J. 2013, 19, 2252–2255. doi:10.1002/chem.201203909
Return to citation in text: [1] -
Martin, R. M.; Bergman, R. G.; Ellman, J. A. J. Org. Chem. 2012, 77, 2501–2507. doi:10.1021/jo202280e
Return to citation in text: [1] -
Wang, C.; Li, X.; Wu, F.; Wan, B. Angew. Chem., Int. Ed. 2011, 50, 7162–7166. doi:10.1002/anie.201102001
Return to citation in text: [1] -
D’Souza, B. R.; Lane, T. K.; Louie, J. Org. Lett. 2011, 13, 2936–2939. doi:10.1021/ol2009939
Return to citation in text: [1] -
Hyster, T. K.; Rovis, T. Chem. Commun. 2011, 47, 11846–11848. doi:10.1039/c1cc15248c
Return to citation in text: [1] -
Reyes, J. C. P.; Romo, D. Angew. Chem., Int. Ed. 2012, 51, 6870–6873. doi:10.1002/anie.201200959
Return to citation in text: [1] -
Fan, H.; Peng, J.; Hamann, M. T.; Hu, J.-F. Chem. Rev. 2008, 108, 264–287. doi:10.1021/cr078199m
Return to citation in text: [1] -
Khajuria, R.; Dham, S.; Kapoor, K. K. RSC Adv. 2016, 6, 37039–37066. doi:10.1039/c6ra03411j
Return to citation in text: [1] -
Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. RSC Adv. 2015, 5, 15233–15266. doi:10.1039/c4ra15710a
Return to citation in text: [1] -
Jung, E.-K.; Leung, E.; Barker, D. Bioorg. Med. Chem. Lett. 2016, 26, 3001–3005. doi:10.1016/j.bmcl.2016.05.026
Return to citation in text: [1] -
Lucas, X.; Wohlwend, D.; Hügle, M.; Schmidtkunz, K.; Gerhardt, S.; Schüle, R.; Jung, M.; Einsle, O.; Günther, S. Angew. Chem., Int. Ed. 2013, 52, 14055–14059. doi:10.1002/anie.201307652
Return to citation in text: [1] -
Young, I. S.; Thornton, P. D.; Thompson, A. Nat. Prod. Rep. 2010, 27, 1801–1839. doi:10.1039/c0np00014k
Return to citation in text: [1] -
Walsh, C. T.; Garneau-Tsodikova, S.; Howard-Jones, A. R. Nat. Prod. Rep. 2006, 23, 517–531. doi:10.1039/b605245m
Return to citation in text: [1] -
Ren, Y.; Jiao, X.; Zhang, L. Saudi J. Biol. Sci. 2018, 25, 469–473. doi:10.1016/j.sjbs.2017.11.043
Return to citation in text: [1] -
Wójcicka, A.; Redzicka, A. Pharmaceuticals 2021, 14, 354. doi:10.3390/ph14040354
Return to citation in text: [1] -
Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2014, 43, 4633–4657. doi:10.1039/c3cs60015g
Return to citation in text: [1] -
Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2010, 39, 4402–4421. doi:10.1039/b917644f
Return to citation in text: [1] -
Xu, X.; Chen, J.; Ke, J.; Zhang, K.; Wu, P.; Wang, S. Chin. J. Org. Chem. 2021, 41, 206–228. doi:10.6023/cjoc202005018
Return to citation in text: [1] -
Menéndez, J.; Leonardi, M.; Estévez, V.; Villacampa, M. Synthesis 2019, 51, 816–828. doi:10.1055/s-0037-1610320
Return to citation in text: [1] -
Balakrishna, A.; Aguiar, A.; Sobral, P. J. M.; Wani, M. Y.; Almeida e Silva, J.; Sobral, A. J. F. N. Catal. Rev.: Sci. Eng. 2019, 61, 84–110. doi:10.1080/01614940.2018.1529932
Return to citation in text: [1] -
Gulevich, A. V.; Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Chem. Rev. 2013, 113, 3084–3213. doi:10.1021/cr300333u
Return to citation in text: [1] -
Nakamura, I.; Yamamoto, Y. Chem. Rev. 2004, 104, 2127–2198. doi:10.1021/cr020095i
Return to citation in text: [1] -
Yoshikai, N.; Wei, Y. Asian J. Org. Chem. 2013, 2, 466–478. doi:10.1002/ajoc.201300016
Return to citation in text: [1] -
Kawakita, K.; Beaumier, E. P.; Kakiuchi, Y.; Tsurugi, H.; Tonks, I. A.; Mashima, K. J. Am. Chem. Soc. 2019, 141, 4194–4198. doi:10.1021/jacs.8b13390
Return to citation in text: [1] -
Li, M.-B.; Grape, E. S.; Bäckvall, J.-E. ACS Catal. 2019, 9, 5184–5190. doi:10.1021/acscatal.9b01041
Return to citation in text: [1] -
Wang, P.; Xiao, J.; Leng, X.; Deng, L. Chin. J. Org. Chem. 2019, 39, 2243–2250. doi:10.6023/cjoc201904040
Return to citation in text: [1] -
Bläsing, K.; Bresien, J.; Labbow, R.; Michalik, D.; Schulz, A.; Thomas, M.; Villinger, A. Angew. Chem., Int. Ed. 2019, 58, 6540–6544. doi:10.1002/anie.201902226
Return to citation in text: [1] -
Anker, M. D.; Lein, M.; Coles, M. P. Chem. Sci. 2019, 10, 1212–1218. doi:10.1039/c8sc04078h
Return to citation in text: [1] -
Song, X.-R.; Qiu, Y.-F.; Liu, X.-Y.; Liang, Y.-M. Org. Biomol. Chem. 2016, 14, 11317–11331. doi:10.1039/c6ob01965j
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5
Return to citation in text: [1] -
Dherbassy, Q.; Manna, S.; Talbot, F. J. T.; Prasitwatcharakorn, W.; Perry, G. J. P.; Procter, D. J. Chem. Sci. 2020, 11, 11380–11393. doi:10.1039/d0sc04012f
Return to citation in text: [1] [2] -
Bao, X.; Ren, J.; Yang, Y.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2020, 18, 7977–7986. doi:10.1039/d0ob01614d
Return to citation in text: [1] [2] -
Fu, L.; Greßies, S.; Chen, P.; Liu, G. Chin. J. Chem. 2020, 38, 91–100. doi:10.1002/cjoc.201900277
Return to citation in text: [1] [2] -
Raji Reddy, C.; Panda, S. A.; Reddy, M. D. Org. Lett. 2015, 17, 896–899. doi:10.1021/ol503752k
Return to citation in text: [1] -
Reddy, C. R.; Prajapti, S. K.; Ranjan, R. Org. Lett. 2018, 20, 3128–3131. doi:10.1021/acs.orglett.8b01228
Return to citation in text: [1] -
Reddy, C. R.; Ranjan, R.; Prajapti, S. K. Org. Lett. 2019, 21, 623–626. doi:10.1021/acs.orglett.8b03695
Return to citation in text: [1] -
Chun, Y. S.; Lee, J. H.; Kim, J. H.; Ko, Y. O.; Lee, S.-g. Org. Lett. 2011, 13, 6390–6393. doi:10.1021/ol202691b
Return to citation in text: [1] -
Barday, M.; Ho, K. Y. T.; Halsall, C. T.; Aïssa, C. Org. Lett. 2016, 18, 1756–1759. doi:10.1021/acs.orglett.6b00451
Return to citation in text: [1] -
Sajid, M. A.; Khan, Z. A.; Shahzad, S. A.; Naqvi, S. A. R.; Usman, M. Mol. Diversity 2020, 24, 295–317. doi:10.1007/s11030-019-09930-x
Return to citation in text: [1] -
Bharathiraja, G.; Sakthivel, S.; Sengoden, M.; Punniyamurthy, T. Org. Lett. 2013, 15, 4996–4999. doi:10.1021/ol402305b
Return to citation in text: [1] -
Zhou, X.; Huang, C.; Zeng, Y.; Xiong, J.; Xiao, Y.; Zhang, J. Chem. Commun. 2017, 53, 1084–1087. doi:10.1039/c6cc09595j
Return to citation in text: [1] -
Huang, C.; Zeng, Y.; Cheng, H.; Hu, A.; Liu, L.; Xiao, Y.; Zhang, J. Org. Lett. 2017, 19, 4968–4971. doi:10.1021/acs.orglett.7b02427
Return to citation in text: [1] -
Bharathiraja, G.; Sengoden, M.; Kannan, M.; Punniyamurthy, T. Org. Biomol. Chem. 2015, 13, 2786–2792. doi:10.1039/c4ob02508c
Return to citation in text: [1] -
Li, Y.; Lu, Y.; Qiu, G.; Ding, Q. Org. Lett. 2014, 16, 4240–4243. doi:10.1021/ol501939m
Return to citation in text: [1] -
Ge, J.; Ding, Q.; Wang, X.; Peng, Y. J. Org. Chem. 2020, 85, 7658–7665. doi:10.1021/acs.joc.9b03470
Return to citation in text: [1] -
Ge, J.; Ding, Q.; Yang, M.; He, T.; Peng, Y. Org. Biomol. Chem. 2020, 18, 8908–8915. doi:10.1039/d0ob01927e
Return to citation in text: [1] -
Gandhi, S.; Baire, B. ChemistrySelect 2017, 2, 3964–3968. doi:10.1002/slct.201700514
Return to citation in text: [1] -
Reddy, C. R.; Panda, S. A.; Ramaraju, A. J. Org. Chem. 2017, 82, 944–949. doi:10.1021/acs.joc.6b02468
Return to citation in text: [1] -
Kong, H.-H.; Pan, H.-L.; Ding, M.-W. J. Org. Chem. 2018, 83, 12921–12930. doi:10.1021/acs.joc.8b01984
Return to citation in text: [1] -
Yang, Y.; Perry, I. B.; Lu, G.; Liu, P.; Buchwald, S. L. Science 2016, 353, 144–150. doi:10.1126/science.aaf7720
Return to citation in text: [1] -
Zhou, Y.; Zhou, L.; Jesikiewicz, L. T.; Liu, P.; Buchwald, S. L. J. Am. Chem. Soc. 2020, 142, 9908–9914. doi:10.1021/jacs.0c03859
Return to citation in text: [1]
| 64. | Kong, H.-H.; Pan, H.-L.; Ding, M.-W. J. Org. Chem. 2018, 83, 12921–12930. doi:10.1021/acs.joc.8b01984 |
| 65. | Yang, Y.; Perry, I. B.; Lu, G.; Liu, P.; Buchwald, S. L. Science 2016, 353, 144–150. doi:10.1126/science.aaf7720 |
| 66. | Zhou, Y.; Zhou, L.; Jesikiewicz, L. T.; Liu, P.; Buchwald, S. L. J. Am. Chem. Soc. 2020, 142, 9908–9914. doi:10.1021/jacs.0c03859 |
| 1. | O’Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d |
| 2. | Narendar, P.; Parthiban, J.; Anbalagan, N.; Gunasekaran, V.; Leonard, J. T. Biol. Pharm. Bull. 2003, 26, 182–187. doi:10.1248/bpb.26.182 |
| 3. | Bagley, M. C.; Dale, J. W.; Merritt, E. A.; Xiong, X. Chem. Rev. 2005, 105, 685–714. doi:10.1021/cr0300441 |
| 4. | Metobo, S. E.; Jin, H.; Tsiang, M.; Kim, C. U. Bioorg. Med. Chem. Lett. 2006, 16, 3985–3988. doi:10.1016/j.bmcl.2006.05.018 |
| 5. | Kishbaugh, T. L. S. Curr. Top. Med. Chem. 2016, 16, 3274–3302. doi:10.2174/1568026616666160506145141 |
| 20. | Reyes, J. C. P.; Romo, D. Angew. Chem., Int. Ed. 2012, 51, 6870–6873. doi:10.1002/anie.201200959 |
| 21. | Fan, H.; Peng, J.; Hamann, M. T.; Hu, J.-F. Chem. Rev. 2008, 108, 264–287. doi:10.1021/cr078199m |
| 22. | Khajuria, R.; Dham, S.; Kapoor, K. K. RSC Adv. 2016, 6, 37039–37066. doi:10.1039/c6ra03411j |
| 23. | Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. RSC Adv. 2015, 5, 15233–15266. doi:10.1039/c4ra15710a |
| 51. | Reddy, C. R.; Ranjan, R.; Prajapti, S. K. Org. Lett. 2019, 21, 623–626. doi:10.1021/acs.orglett.8b03695 |
| 15. | Xu, F.; Wang, C.; Wang, D.; Li, X.; Wan, B. Chem. – Eur. J. 2013, 19, 2252–2255. doi:10.1002/chem.201203909 |
| 16. | Martin, R. M.; Bergman, R. G.; Ellman, J. A. J. Org. Chem. 2012, 77, 2501–2507. doi:10.1021/jo202280e |
| 17. | Wang, C.; Li, X.; Wu, F.; Wan, B. Angew. Chem., Int. Ed. 2011, 50, 7162–7166. doi:10.1002/anie.201102001 |
| 18. | D’Souza, B. R.; Lane, T. K.; Louie, J. Org. Lett. 2011, 13, 2936–2939. doi:10.1021/ol2009939 |
| 19. | Hyster, T. K.; Rovis, T. Chem. Commun. 2011, 47, 11846–11848. doi:10.1039/c1cc15248c |
| 52. | Chun, Y. S.; Lee, J. H.; Kim, J. H.; Ko, Y. O.; Lee, S.-g. Org. Lett. 2011, 13, 6390–6393. doi:10.1021/ol202691b |
| 11. | Wei, Y.; Yoshikai, N. J. Am. Chem. Soc. 2013, 135, 3756–3759. doi:10.1021/ja312346s |
| 12. | Michlik, S.; Kempe, R. Angew. Chem., Int. Ed. 2013, 52, 6326–6329. doi:10.1002/anie.201301919 |
| 13. | Loy, N. S. Y.; Singh, A.; Xu, X.; Park, C.-M. Angew. Chem., Int. Ed. 2013, 52, 2212–2216. doi:10.1002/anie.201209301 |
| 14. | Yamamoto, S.-i.; Okamoto, K.; Murakoso, M.; Kuninobu, Y.; Takai, K. Org. Lett. 2012, 14, 3182–3185. doi:10.1021/ol301273j |
| 49. | Raji Reddy, C.; Panda, S. A.; Reddy, M. D. Org. Lett. 2015, 17, 896–899. doi:10.1021/ol503752k |
| 6. | Gibson, V. C.; Redshaw, C.; Solan, G. A. Chem. Rev. 2007, 107, 1745–1776. doi:10.1021/cr068437y |
| 7. | Peloquin, D. M.; Schmedake, T. A. Coord. Chem. Rev. 2016, 323, 107–119. doi:10.1016/j.ccr.2016.02.005 |
| 8. | Rajput, A.; Mukherjee, R. Coord. Chem. Rev. 2013, 257, 350–368. doi:10.1016/j.ccr.2012.03.024 |
| 9. | Ding, Q.; Ye, S.; Cheng, G.; Wang, P.; Farmer, M. E.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 417–425. doi:10.1021/jacs.6b11097 |
| 10. | Liu, J.; Ding, Q.; Fang, W.; Wu, W.; Zhang, Y.; Peng, Y. J. Org. Chem. 2018, 83, 13211–13216. doi:10.1021/acs.joc.8b01933 |
| 50. | Reddy, C. R.; Prajapti, S. K.; Ranjan, R. Org. Lett. 2018, 20, 3128–3131. doi:10.1021/acs.orglett.8b01228 |
| 46. | Dherbassy, Q.; Manna, S.; Talbot, F. J. T.; Prasitwatcharakorn, W.; Perry, G. J. P.; Procter, D. J. Chem. Sci. 2020, 11, 11380–11393. doi:10.1039/d0sc04012f |
| 47. | Bao, X.; Ren, J.; Yang, Y.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2020, 18, 7977–7986. doi:10.1039/d0ob01614d |
| 48. | Fu, L.; Greßies, S.; Chen, P.; Liu, G. Chin. J. Chem. 2020, 38, 91–100. doi:10.1002/cjoc.201900277 |
| 47. | Bao, X.; Ren, J.; Yang, Y.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2020, 18, 7977–7986. doi:10.1039/d0ob01614d |
| 40. | Wang, P.; Xiao, J.; Leng, X.; Deng, L. Chin. J. Org. Chem. 2019, 39, 2243–2250. doi:10.6023/cjoc201904040 |
| 41. | Bläsing, K.; Bresien, J.; Labbow, R.; Michalik, D.; Schulz, A.; Thomas, M.; Villinger, A. Angew. Chem., Int. Ed. 2019, 58, 6540–6544. doi:10.1002/anie.201902226 |
| 42. | Anker, M. D.; Lein, M.; Coles, M. P. Chem. Sci. 2019, 10, 1212–1218. doi:10.1039/c8sc04078h |
| 43. | Song, X.-R.; Qiu, Y.-F.; Liu, X.-Y.; Liang, Y.-M. Org. Biomol. Chem. 2016, 14, 11317–11331. doi:10.1039/c6ob01965j |
| 44. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 45. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5 |
| 48. | Fu, L.; Greßies, S.; Chen, P.; Liu, G. Chin. J. Chem. 2020, 38, 91–100. doi:10.1002/cjoc.201900277 |
| 30. | Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2014, 43, 4633–4657. doi:10.1039/c3cs60015g |
| 31. | Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2010, 39, 4402–4421. doi:10.1039/b917644f |
| 32. | Xu, X.; Chen, J.; Ke, J.; Zhang, K.; Wu, P.; Wang, S. Chin. J. Org. Chem. 2021, 41, 206–228. doi:10.6023/cjoc202005018 |
| 33. | Menéndez, J.; Leonardi, M.; Estévez, V.; Villacampa, M. Synthesis 2019, 51, 816–828. doi:10.1055/s-0037-1610320 |
| 34. | Balakrishna, A.; Aguiar, A.; Sobral, P. J. M.; Wani, M. Y.; Almeida e Silva, J.; Sobral, A. J. F. N. Catal. Rev.: Sci. Eng. 2019, 61, 84–110. doi:10.1080/01614940.2018.1529932 |
| 35. | Gulevich, A. V.; Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Chem. Rev. 2013, 113, 3084–3213. doi:10.1021/cr300333u |
| 36. | Nakamura, I.; Yamamoto, Y. Chem. Rev. 2004, 104, 2127–2198. doi:10.1021/cr020095i |
| 37. | Yoshikai, N.; Wei, Y. Asian J. Org. Chem. 2013, 2, 466–478. doi:10.1002/ajoc.201300016 |
| 38. | Kawakita, K.; Beaumier, E. P.; Kakiuchi, Y.; Tsurugi, H.; Tonks, I. A.; Mashima, K. J. Am. Chem. Soc. 2019, 141, 4194–4198. doi:10.1021/jacs.8b13390 |
| 39. | Li, M.-B.; Grape, E. S.; Bäckvall, J.-E. ACS Catal. 2019, 9, 5184–5190. doi:10.1021/acscatal.9b01041 |
| 24. | Jung, E.-K.; Leung, E.; Barker, D. Bioorg. Med. Chem. Lett. 2016, 26, 3001–3005. doi:10.1016/j.bmcl.2016.05.026 |
| 25. | Lucas, X.; Wohlwend, D.; Hügle, M.; Schmidtkunz, K.; Gerhardt, S.; Schüle, R.; Jung, M.; Einsle, O.; Günther, S. Angew. Chem., Int. Ed. 2013, 52, 14055–14059. doi:10.1002/anie.201307652 |
| 26. | Young, I. S.; Thornton, P. D.; Thompson, A. Nat. Prod. Rep. 2010, 27, 1801–1839. doi:10.1039/c0np00014k |
| 27. | Walsh, C. T.; Garneau-Tsodikova, S.; Howard-Jones, A. R. Nat. Prod. Rep. 2006, 23, 517–531. doi:10.1039/b605245m |
| 28. | Ren, Y.; Jiao, X.; Zhang, L. Saudi J. Biol. Sci. 2018, 25, 469–473. doi:10.1016/j.sjbs.2017.11.043 |
| 29. | Wójcicka, A.; Redzicka, A. Pharmaceuticals 2021, 14, 354. doi:10.3390/ph14040354 |
| 46. | Dherbassy, Q.; Manna, S.; Talbot, F. J. T.; Prasitwatcharakorn, W.; Perry, G. J. P.; Procter, D. J. Chem. Sci. 2020, 11, 11380–11393. doi:10.1039/d0sc04012f |
| 55. | Bharathiraja, G.; Sakthivel, S.; Sengoden, M.; Punniyamurthy, T. Org. Lett. 2013, 15, 4996–4999. doi:10.1021/ol402305b |
| 53. | Barday, M.; Ho, K. Y. T.; Halsall, C. T.; Aïssa, C. Org. Lett. 2016, 18, 1756–1759. doi:10.1021/acs.orglett.6b00451 |
| 54. | Sajid, M. A.; Khan, Z. A.; Shahzad, S. A.; Naqvi, S. A. R.; Usman, M. Mol. Diversity 2020, 24, 295–317. doi:10.1007/s11030-019-09930-x |
| 62. | Gandhi, S.; Baire, B. ChemistrySelect 2017, 2, 3964–3968. doi:10.1002/slct.201700514 |
| 63. | Reddy, C. R.; Panda, S. A.; Ramaraju, A. J. Org. Chem. 2017, 82, 944–949. doi:10.1021/acs.joc.6b02468 |
| 60. | Ge, J.; Ding, Q.; Wang, X.; Peng, Y. J. Org. Chem. 2020, 85, 7658–7665. doi:10.1021/acs.joc.9b03470 |
| 61. | Ge, J.; Ding, Q.; Yang, M.; He, T.; Peng, Y. Org. Biomol. Chem. 2020, 18, 8908–8915. doi:10.1039/d0ob01927e |
| 58. | Bharathiraja, G.; Sengoden, M.; Kannan, M.; Punniyamurthy, T. Org. Biomol. Chem. 2015, 13, 2786–2792. doi:10.1039/c4ob02508c |
| 59. | Li, Y.; Lu, Y.; Qiu, G.; Ding, Q. Org. Lett. 2014, 16, 4240–4243. doi:10.1021/ol501939m |
| 56. | Zhou, X.; Huang, C.; Zeng, Y.; Xiong, J.; Xiao, Y.; Zhang, J. Chem. Commun. 2017, 53, 1084–1087. doi:10.1039/c6cc09595j |
| 57. | Huang, C.; Zeng, Y.; Cheng, H.; Hu, A.; Liu, L.; Xiao, Y.; Zhang, J. Org. Lett. 2017, 19, 4968–4971. doi:10.1021/acs.orglett.7b02427 |
© 2021 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)