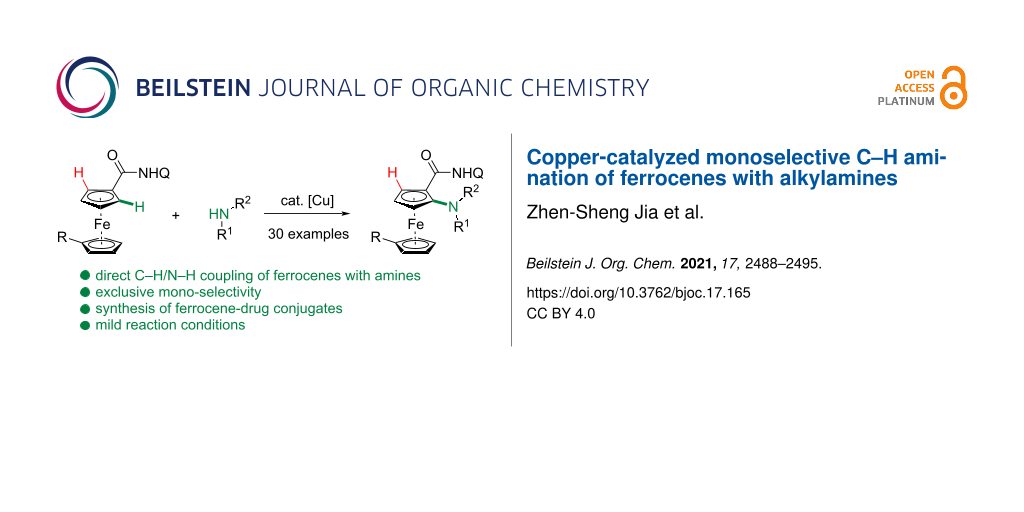
Abstract
A copper-catalyzed mono-selective C–H amination of ferrocenes assisted by 8-aminoquinoline is presented here. A range of amines, including bioactive molecules, were successfully installed to the ortho-position of ferrocene amides with high efficiency under mild conditions. A range of functionalized ferrocenes were compatible to give the aminated products in moderate to good yields. The gram-scale reaction was smoothly conducted and the directing group could be removed easily under basic conditions.

Graphical Abstract
Introduction
Ferrocene-based compounds have broad applications from asymmetric catalysis to medicinal discovery [1-8]. Therefore, the development of efficient methods to access multifunctional ferrocenes has attracted tremendous attention. Conventionally, functionalized ferrocenes were derived via electrophilic aromatic substitution mediated by strong Lewis acids or direct metalation using strong bases, such as alkyllithium reagents [3,9-12]. However, the above protocols generally proceeded under harsh conditions that led to poor functional group tolerance and generated stoichiometric amounts of waste.
Thus far, the transition-metal-catalyzed C–H functionalization strategy has innovated the way to producing ferrocene derivatives [13-16]. Especially, the 3d transition metals, such as Cu, Co and Ni, have been exploited to convert C–H bonds to various functional groups, attributing to the cost-effective and less toxic properties, which render C–H transformations both economically desirable and environmentally benign (Scheme 1a) [17-22]. Early in 2015, the Ackermann group reported the first example of a low-valent Co-catalyzed C–H alkenylation of 2-pyridinylferrocene [23]. In 2017, the Butenschön group reported the ortho-C–H alkylation and arylation of ferrocene derivatives enabled by a combination of Fe or Co catalyst and N-containing directing groups, while an excess of Grignard reagents was used [24,25]. Thereafter, they also reported the Cp*Co-catalyzed ortho-C–H alkenylation of ferrocenes with alkynes [26] and the mono- and di-selectivity could be controlled by the fine-tuned directing groups. Kumar and co-workers developed a Cu-mediated C–H chalcogenation and sulfonation of ferrocenes [27-29]. The use of a bidentate 1,10-phenathroline ligand was critical to achieve mono-selectivity in the chacogenation reactions [28]. Meanwhile, Co(III)-catalyzed ortho-C–H amidation of ferrocene derivatives were also developed by the groups of You [30], Ackermann [31] and Shi [32,33] with 1,4,2-dioxazol-5-ones as versatile amidating reagents. In 2019, the alkynylated ferrocenes were isolated in the formation of alkyne-Cu(I) π-complexes by the Tan group via Cu-mediated C–H alkynylations [34]. Later in 2020, an enantioselective C–H annulation of ferrocenylformamides with alkynes was achieved by the Ye group enabled by Ni-Al bimetallic catalysis and a chiral secondary phosphine oxide (SPO) ligand [35]. Hou et al. also reported the asymmetric C−H alkenylation of quinoline- and pyridine-substituted ferrocenes with alkynes by using an unprecedented half-sandwich Sc catalyst [36]. Very recently, Shi and Zhang demonstrated a Cp*Co-catalyzed ortho-C–H allylation of ferrocenes assisted by thioamide using allyl carbonates and vinylcyclopropanes as allylating partners [37]. Meanwhile, Zhang and co-authors also reported the Co-catalyzed C–H alkoxylation of ferrocenes under nearly room temperature [38].
In comparison, despite the direct C–H amination of arenes with alkylamines has emerged as an efficient strategy to prepare substituted anilines [39-49], the application of this environmentally benign, oxidative coupling strategy to the synthesis of valuable ortho-amino ferrocene derivatives hasn’t been achieved [50], probably ascribing to several challenges. First, unprotected amines are sensitive and unendurable to several oxidants in the presence of transition metals. Second, both amines and the resulting aminated products could coordinate with metal catalysts and cause the deactivation of catalysts. Besides, high reaction temperature could lead to a mixture of byproducts or the decomposition of the ferrocene products. Herein, we described a Cu-catalyzed oxidative C–H/N–H coupling of ferrocenes with free amines to provide mono-aminated ferrocenes exclusively under mild conditions (Scheme 1b). During the preparation of the manuscript of this article, a nice report on Cu-catalyzed C–H amination of ferrocenes directed by 8-aminoquinoline was reported by Fukuzawa and Kanemoto [50]. Notably, our method proceeded under silver-free conditions at relatively lower temperature and in shorter time, providing a complementary alternative to the work of Fukuzawa and Kanemoto.
![[1860-5397-17-165-i1]](/bjoc/content/inline/1860-5397-17-165-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: 3d-Transition-metal-catalyzed C–H functionalization to access functionalized ferrocenes.
Scheme 1: 3d-Transition-metal-catalyzed C–H functionalization to access functionalized ferrocenes.
Results and Discussion
We initiated our study by investigating the C–H amination of ferrocene carboxylic amide 1a with morpholine (2a) using 8-amonoquinoline as directing group [51-56]. The ortho-aminated ferrocenylamide 3a was isolated in 11% yield in the presence of CuI, N-methylmorpholine N-oxide (NMO) and K2CO3 in DMF (Table 1, entry 1). When the reaction was conducted in MeCN, the yield could be improved to 32% (Table 1, entry 4). Further screening of other oxidants revealed that NMO was the optimal (Table 1, entries 9–11). When the reaction was conducted in neat in the presence of 2-pyridone, 3a was obtained in 36% yield (Table 1, entry 12). However, significant decomposition of the aminated product 3a was observed. Consequently, we exclusively evaluated the reaction temperature and time (Table 1, entries 13–16). To our delight, 3a could be obtained in 80% yield under relatively lower temperature (80 °C) and shorter time (4 hours). To note, this reaction showed excellent mono-selectivity and no diaminated ferrocenylamide was detected. The exclusive monoselectivity is most likely originated from the strong coordination of the amino group, which could form a tridentate copper complex and prevent the second C–H amination [34,50].
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-165-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | [Cu] | Solvent | Oxidant | Additive | T (°C) | t (h) | Yieldb |
| 1 | CuI | DMF | NMO | – | 120 | 12 | 11% |
| 2 | CuI | NMP | NMO | – | 120 | 12 | 15% |
| 3 | CuI | DMSO | NMO | – | 120 | 12 | trace |
| 4 | CuI | MeCN | NMO | – | 120 | 12 | 32% |
| 5 | Cu(OAc)2 | MeCN | NMO | – | 120 | 12 | 10% |
| 6 | CuCN | MeCN | NMO | – | 120 | 12 | 12% |
| 7 | CuCl | MeCN | NMO | – | 120 | 12 | 18% |
| 8 | CuTc | MeCN | NMO | – | 120 | 12 | trace |
| 9 | CuI | MeCN | TEMPO | – | 120 | 12 | 23% |
| 10 | CuI | MeCN | MnO2 | – | 120 | 12 | trace |
| 11c | CuI | MeCN | O2 | – | 120 | 12 | 8% |
| 12d | CuI | neat | NMO | 2-pyridone | 120 | 12 | 36% |
| 13d | CuI | neat | NMO | 2-pyridone | 100 | 12 | 46% |
| 14d | CuI | neat | NMO | 2-pyridone | 80 | 12 | 56% |
| 15d | CuI | neat | NMO | 2-pyridone | 80 | 6 | 68% |
| 16d | CuI | neat | NMO | 2-pyridone | 80 | 4 | 80% |
aReactions conditions: 1a (0.1 mmol), 2a (0.3 mmol), [Cu] (20 mol %), oxidant (2.0 equiv), K2CO3 (1.0 equiv) and additive (1.0 equiv) in a sealed tube. bIsolated yield. cOxygen balloon. d5.0 equiv of morpholine.
Having obtained the optimized reaction conditions, we started to investigate the generality of this C–H amination protocol with regard to modified ferrocenes (Scheme 2). Delightfully, a variety of functional groups tailored to the other cyclopentadienyl (Cp) moieties were well tolerated, furnishing the desired ortho-mono-aminating ferrocenes in moderate to good yields. Alkyl substituents on the other Cp ring of ferrocenylamides only showed slightly effects (3b–d). Notably, the terminal alkenyl group of 3e was well tolerated during the amination process. Weakly coordinating carbonyl groups were also tolerated and the desired C–H amination occurred selectively at the ortho position to the N-quinolinyl amides with acceptable yields (3f–l). Notably, free alcohol was also compatible with this protocol, exclusively giving the mono-aminated product in 73% yield (3m) without the observation of any competitive alkoxylation product [38,57,58].
![[1860-5397-17-165-i2]](/bjoc/content/inline/1860-5397-17-165-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of ferrocenes with morpholine.
Scheme 2: Scope of ferrocenes with morpholine.
We then explored the scope of multifarious amines. As displayed in Scheme 3, a range of cyclic amines, such as morpholine 4a,b, piperazine 4c, piperidine 4d–j and thiomorpholine 4l,m, reacted smoothly to give the amination products in 23% to 85% yields. A variety of synthetically useful functionalities, such as ester 4f, cyano 4g and ketal 4i, were well tolerated. Unfortunately, 1,2,3,4-tetrahydroquinoline (2k) was proved unreactive under our conditions, probably due to the steric hindrance. Thiomorpholine (2l) was compatible with this reaction, albeit with significantly dropped yield (29%), largely due to the poison of copper catalyst by thioether. Acyclic amines were also tested and the amination products were obtained in low yields (4n, 18%; 4o, 15%). Unfortunately, primary amines and anilines were completely inert.
![[1860-5397-17-165-i3]](/bjoc/content/inline/1860-5397-17-165-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of various amines with 1a.
Scheme 3: Scope of various amines with 1a.
Encouraged by the above results, we further tried to synthesize ferrocene–drug conjugates (Scheme 4a). Three amines used to treat psychosis were subjected to couple with 1a and the desired conjugates were obtained in good yields (with haloperidol, 4p, 63%; with buspirone, 4q, 60%; with perospirone, 4r, 70%). This protocol was also amendable to gram-scale synthesis, giving 3a in 50% yield (Scheme 4b, 1.32 g). For synthetic utility, the directing group was conveniently removed by refluxing with KOH in EtOH and the benzyl-protected ester 5 was obtained in 75% yield.
![[1860-5397-17-165-i4]](/bjoc/content/inline/1860-5397-17-165-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
We also conducted several deuteration experiments to shed a preliminary insight into the mechanism. No H/D exchange was observed at the ortho-position of 1a with 3.0 equivalents of CD3CO2D under standard conditions (Scheme 5a). Furthermore, a larger value of kinetic isotope effect (KIE = 2.4) was detected (Scheme 5b). These results indicated that the cleavage of C–H bond was most likely involved in the rate-determining step.
![[1860-5397-17-165-i5]](/bjoc/content/inline/1860-5397-17-165-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
To summarize, we have reported a copper-catalyzed direct ortho-C–H/N–H coupling reaction of ferrocenes with alkyl amines directed by 8-aminoquinoline. Fruitful mono-aminated ferrocenes were obtained in moderate to good yields and the mild conditions offered the possibility to the preparation of ferrocene–drug conjugates effectively. Mechanistic studies indicated that the C–H activation step was the rate-determining step.
Supporting Information
| Supporting Information File 1: Full experimental details, compound characterization, and copies of NMR spectra. | ||
| Format: PDF | Size: 12.2 MB | Download |
Funding
Financial support from the National Natural Science Foundation of China (21925109 and 21772170), China Postdoctoral Science Foundation (2021M692771), Center of Chemistry for Frontier Technologies of Zhejiang University, and Open Research Fund of School of Chemistry and Chemical Engineering of Henan Normal University is gratefully acknowledged.
References
-
Hayashi, T.; Togni, A., Eds. Ferrocenes; VCH: Weinheim, Germany, 1995.
Return to citation in text: [1] -
Štěpnička, P., Ed. Ferrocenes; John Wiley & Sons: Chichester, UK, 2008.
Return to citation in text: [1] -
Dai, L.-X.; Hou, X.-L., Eds. Chiral Ferrocenes in Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2010. doi:10.1002/9783527628841
Return to citation in text: [1] [2] -
van Staveren, D. R.; Metzler-Nolte, N. Chem. Rev. 2004, 104, 5931–5986. doi:10.1021/cr0101510
Return to citation in text: [1] -
Gómez Arrayás, R.; Adrio, J.; Carretero, J. C. Angew. Chem., Int. Ed. 2006, 45, 7674–7715. doi:10.1002/anie.200602482
Return to citation in text: [1] -
Hartinger, C. G.; Dyson, P. J. Chem. Soc. Rev. 2009, 38, 391–401. doi:10.1039/b707077m
Return to citation in text: [1] -
Gasser, G.; Ott, I.; Metzler-Nolte, N. J. Med. Chem. 2011, 54, 3–25. doi:10.1021/jm100020w
Return to citation in text: [1] -
Toma, Š.; Csizmadiová, J.; Mečiarová, M.; Šebesta, R. Dalton Trans. 2014, 43, 16557–16579. doi:10.1039/c4dt01784f
Return to citation in text: [1] -
Tsukazaki, M.; Tinkl, M.; Roglans, A.; Chapell, B. J.; Taylor, N. J.; Snieckus, V. J. Am. Chem. Soc. 1996, 118, 685–686. doi:10.1021/ja953246q
Return to citation in text: [1] -
Metallinos, C.; Szillat, H.; Taylor, N. J.; Snieckus, V. Adv. Synth. Catal. 2003, 345, 370–382. doi:10.1002/adsc.200390042
Return to citation in text: [1] -
Butt, N. A.; Liu, D.; Zhang, W. Synlett 2014, 25, 615–630. doi:10.1055/s-0033-1340487
Return to citation in text: [1] -
Schaarschmidt, D.; Lang, H. Organometallics 2013, 32, 5668–5704. doi:10.1021/om400564x
Return to citation in text: [1] -
López, L. A.; López, E. Dalton Trans. 2015, 44, 10128–10135. doi:10.1039/c5dt01373a
Return to citation in text: [1] -
Zhu, D.-Y.; Chen, P.; Xia, J.-B. ChemCatChem 2016, 8, 68–73. doi:10.1002/cctc.201500895
Return to citation in text: [1] -
Gao, D.-W.; Gu, Q.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2017, 50, 351–365. doi:10.1021/acs.accounts.6b00573
Return to citation in text: [1] -
Huang, J.; Gu, Q.; You, S. Chin. J. Org. Chem. 2018, 38, 51–61. doi:10.6023/cjoc201708030
Return to citation in text: [1] -
Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. Chem. Rev. 2019, 119, 2192–2452. doi:10.1021/acs.chemrev.8b00507
Return to citation in text: [1] -
Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel‐Delord, J.; Ackermann, L. Angew. Chem., Int. Ed. 2019, 58, 12803–12818. doi:10.1002/anie.201904214
Return to citation in text: [1] -
Woźniak, Ł.; Cramer, N. Trends Chem. 2019, 1, 471–484. doi:10.1016/j.trechm.2019.03.013
Return to citation in text: [1] -
Rao, W.-H.; Shi, B.-F. Org. Chem. Front. 2016, 3, 1028–1047. doi:10.1039/c6qo00156d
Return to citation in text: [1] -
Liu, J.; Chen, G.; Tan, Z. Adv. Synth. Catal. 2016, 358, 1174–1194. doi:10.1002/adsc.201600031
Return to citation in text: [1] -
Liu, Y.-H.; Xia, Y.-N.; Shi, B.-F. Chin. J. Chem. 2020, 38, 635–662. doi:10.1002/cjoc.201900468
Return to citation in text: [1] -
Moselage, M.; Sauermann, N.; Richter, S. C.; Ackermann, L. Angew. Chem., Int. Ed. 2015, 54, 6352–6355. doi:10.1002/anie.201412319
Return to citation in text: [1] -
Schmiel, D.; Butenschön, H. Eur. J. Org. Chem. 2017, 3041–3048. doi:10.1002/ejoc.201700358
Return to citation in text: [1] -
Schmiel, D.; Butenschön, H. Organometallics 2017, 36, 4979–4989. doi:10.1021/acs.organomet.7b00799
Return to citation in text: [1] -
Schmiel, D.; Gathy, R.; Butenschön, H. Organometallics 2018, 37, 2095–2110. doi:10.1021/acs.organomet.8b00243
Return to citation in text: [1] -
Sattar, M.; Shareef, M.; Patidar, K.; Kumar, S. J. Org. Chem. 2018, 83, 8241–8249. doi:10.1021/acs.joc.8b00974
Return to citation in text: [1] -
Sattar, M.; Patidar, K.; Thorat, R. A.; Kumar, S. J. Org. Chem. 2019, 84, 6669–6678. doi:10.1021/acs.joc.9b00311
Return to citation in text: [1] [2] -
Sattar, M.; Kumar, N.; Yadav, P.; Mandhar, Y.; Kumar, S. Chem. – Asian J. 2019, 14, 4807–4813. doi:10.1002/asia.201901334
Return to citation in text: [1] -
Wang, S.-B.; Gu, Q.; You, S.-L. J. Catal. 2018, 361, 393–397. doi:10.1016/j.jcat.2018.03.007
Return to citation in text: [1] -
Yetra, S. R.; Shen, Z.; Wang, H.; Ackermann, L. Beilstein J. Org. Chem. 2018, 14, 1546–1553. doi:10.3762/bjoc.14.131
Return to citation in text: [1] -
Huang, D.-Y.; Yao, Q.-J.; Zhang, S.; Xu, X.-T.; Zhang, K.; Shi, B.-F. Org. Lett. 2019, 21, 951–954. doi:10.1021/acs.orglett.8b03938
Return to citation in text: [1] -
Liu, Y.-H.; Li, P.-X.; Yao, Q.-J.; Zhang, Z.-Z.; Huang, D.-Y.; Le, M. D.; Song, H.; Liu, L.; Shi, B.-F. Org. Lett. 2019, 21, 1895–1899. doi:10.1021/acs.orglett.9b00511
Return to citation in text: [1] -
Song, Z.; Yu, Y.; Yu, L.; Liu, D.; Wu, Q.; Xia, Z.; Xiao, Y.; Tan, Z. Organometallics 2019, 38, 3349–3357. doi:10.1021/acs.organomet.9b00447
Return to citation in text: [1] [2] -
Chen, H.; Wang, Y.-X.; Luan, Y.-X.; Ye, M. Angew. Chem., Int. Ed. 2020, 59, 9428–9432. doi:10.1002/anie.202001267
Return to citation in text: [1] -
Lou, S.-J.; Zhuo, Q.; Nishiura, M.; Luo, G.; Hou, Z. J. Am. Chem. Soc. 2021, 143, 2470–2476. doi:10.1021/jacs.0c13166
Return to citation in text: [1] -
Zhang, Z.-Z.; Liao, G.; Chen, H.-M.; Shi, B.-F. Org. Lett. 2021, 23, 2626–2631. doi:10.1021/acs.orglett.1c00533
Return to citation in text: [1] -
Zhang, Z.-Z.; Cheng, J.; Yao, Q.-J.; Yue, Q.; Zhou, G. Adv. Synth. Catal. 2021, 363, 3946–3951. doi:10.1002/adsc.202100423
Return to citation in text: [1] [2] -
Kim, H.; Chang, S. ACS Catal. 2016, 6, 2341–2351. doi:10.1021/acscatal.6b00293
Return to citation in text: [1] -
Tran, L. D.; Roane, J.; Daugulis, O. Angew. Chem., Int. Ed. 2013, 52, 6043–6046. doi:10.1002/anie.201300135
Return to citation in text: [1] -
Matsubara, T.; Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2014, 136, 646–649. doi:10.1021/ja412521k
Return to citation in text: [1] -
Yan, Q.; Chen, Z.; Yu, W.; Yin, H.; Liu, Z.; Zhang, Y. Org. Lett. 2015, 17, 2482–2485. doi:10.1021/acs.orglett.5b00990
Return to citation in text: [1] -
Roane, J.; Daugulis, O. J. Am. Chem. Soc. 2016, 138, 4601–4607. doi:10.1021/jacs.6b01117
Return to citation in text: [1] -
Yan, Q.; Xiao, T.; Liu, Z.; Zhang, Y. Adv. Synth. Catal. 2016, 358, 2707–2711. doi:10.1002/adsc.201600282
Return to citation in text: [1] -
Gao, X.; Wang, P.; Zeng, L.; Tang, S.; Lei, A. J. Am. Chem. Soc. 2018, 140, 4195–4199. doi:10.1021/jacs.7b13049
Return to citation in text: [1] -
Zhang, S.-K.; Samanta, R. C.; Sauermann, N.; Ackermann, L. Chem. – Eur. J. 2018, 24, 19166–19170. doi:10.1002/chem.201805441
Return to citation in text: [1] -
Kathiravan, S.; Suriyanarayanan, S.; Nicholls, I. A. Org. Lett. 2019, 21, 1968–1972. doi:10.1021/acs.orglett.9b00003
Return to citation in text: [1] -
Shang, M.; Shao, Q.; Sun, S.-Z.; Chen, Y.-Q.; Xu, H.; Dai, H.-X.; Yu, J.-Q. Chem. Sci. 2017, 8, 1469–1473. doi:10.1039/c6sc03383k
Return to citation in text: [1] -
Wu, P.; Huang, W.; Cheng, T.-J.; Lin, H.-X.; Xu, H.; Dai, H.-X. Org. Lett. 2020, 22, 5051–5056. doi:10.1021/acs.orglett.0c01632
Return to citation in text: [1] -
Kanemoto, K.; Horikawa, N.; Hoshino, S.; Tokoro, Y.; Fukuzawa, S.-i. Org. Lett. 2021, 23, 4966–4970. doi:10.1021/acs.orglett.1c01294
Return to citation in text: [1] [2] [3] -
Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154–13155. doi:10.1021/ja054549f
Return to citation in text: [1] -
Daugulis, O.; Roane, J.; Tran, L. D. Acc. Chem. Res. 2015, 48, 1053–1064. doi:10.1021/ar5004626
Return to citation in text: [1] -
Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M. F.; Wencel-Delord, J.; Besset, T.; Maes, B. U. W.; Schnürch, M. Chem. Soc. Rev. 2018, 47, 6603–6743. doi:10.1039/c8cs00201k
Return to citation in text: [1] -
Zhang, Q.; Shi, B.-F. Chin. J. Chem. 2019, 37, 647–656. doi:10.1002/cjoc.201900090
Return to citation in text: [1] -
Rej, S.; Ano, Y.; Chatani, N. Chem. Rev. 2020, 120, 1788–1887. doi:10.1021/acs.chemrev.9b00495
Return to citation in text: [1] -
Zhang, Q.; Shi, B.-F. Acc. Chem. Res. 2021, 54, 2750–2763. doi:10.1021/acs.accounts.1c00168
Return to citation in text: [1] -
Roane, J.; Daugulis, O. Org. Lett. 2013, 15, 5842–5845. doi:10.1021/ol402904d
Return to citation in text: [1] -
Hu, Y.; Wang, M.; Li, P.; Li, H.; Wang, L. Asian J. Org. Chem. 2019, 8, 171–178. doi:10.1002/ajoc.201800664
Return to citation in text: [1]
| 50. | Kanemoto, K.; Horikawa, N.; Hoshino, S.; Tokoro, Y.; Fukuzawa, S.-i. Org. Lett. 2021, 23, 4966–4970. doi:10.1021/acs.orglett.1c01294 |
| 38. | Zhang, Z.-Z.; Cheng, J.; Yao, Q.-J.; Yue, Q.; Zhou, G. Adv. Synth. Catal. 2021, 363, 3946–3951. doi:10.1002/adsc.202100423 |
| 39. | Kim, H.; Chang, S. ACS Catal. 2016, 6, 2341–2351. doi:10.1021/acscatal.6b00293 |
| 40. | Tran, L. D.; Roane, J.; Daugulis, O. Angew. Chem., Int. Ed. 2013, 52, 6043–6046. doi:10.1002/anie.201300135 |
| 41. | Matsubara, T.; Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2014, 136, 646–649. doi:10.1021/ja412521k |
| 42. | Yan, Q.; Chen, Z.; Yu, W.; Yin, H.; Liu, Z.; Zhang, Y. Org. Lett. 2015, 17, 2482–2485. doi:10.1021/acs.orglett.5b00990 |
| 43. | Roane, J.; Daugulis, O. J. Am. Chem. Soc. 2016, 138, 4601–4607. doi:10.1021/jacs.6b01117 |
| 44. | Yan, Q.; Xiao, T.; Liu, Z.; Zhang, Y. Adv. Synth. Catal. 2016, 358, 2707–2711. doi:10.1002/adsc.201600282 |
| 45. | Gao, X.; Wang, P.; Zeng, L.; Tang, S.; Lei, A. J. Am. Chem. Soc. 2018, 140, 4195–4199. doi:10.1021/jacs.7b13049 |
| 46. | Zhang, S.-K.; Samanta, R. C.; Sauermann, N.; Ackermann, L. Chem. – Eur. J. 2018, 24, 19166–19170. doi:10.1002/chem.201805441 |
| 47. | Kathiravan, S.; Suriyanarayanan, S.; Nicholls, I. A. Org. Lett. 2019, 21, 1968–1972. doi:10.1021/acs.orglett.9b00003 |
| 48. | Shang, M.; Shao, Q.; Sun, S.-Z.; Chen, Y.-Q.; Xu, H.; Dai, H.-X.; Yu, J.-Q. Chem. Sci. 2017, 8, 1469–1473. doi:10.1039/c6sc03383k |
| 49. | Wu, P.; Huang, W.; Cheng, T.-J.; Lin, H.-X.; Xu, H.; Dai, H.-X. Org. Lett. 2020, 22, 5051–5056. doi:10.1021/acs.orglett.0c01632 |
| 1. | Hayashi, T.; Togni, A., Eds. Ferrocenes; VCH: Weinheim, Germany, 1995. |
| 2. | Štěpnička, P., Ed. Ferrocenes; John Wiley & Sons: Chichester, UK, 2008. |
| 3. | Dai, L.-X.; Hou, X.-L., Eds. Chiral Ferrocenes in Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2010. doi:10.1002/9783527628841 |
| 4. | van Staveren, D. R.; Metzler-Nolte, N. Chem. Rev. 2004, 104, 5931–5986. doi:10.1021/cr0101510 |
| 5. | Gómez Arrayás, R.; Adrio, J.; Carretero, J. C. Angew. Chem., Int. Ed. 2006, 45, 7674–7715. doi:10.1002/anie.200602482 |
| 6. | Hartinger, C. G.; Dyson, P. J. Chem. Soc. Rev. 2009, 38, 391–401. doi:10.1039/b707077m |
| 7. | Gasser, G.; Ott, I.; Metzler-Nolte, N. J. Med. Chem. 2011, 54, 3–25. doi:10.1021/jm100020w |
| 8. | Toma, Š.; Csizmadiová, J.; Mečiarová, M.; Šebesta, R. Dalton Trans. 2014, 43, 16557–16579. doi:10.1039/c4dt01784f |
| 23. | Moselage, M.; Sauermann, N.; Richter, S. C.; Ackermann, L. Angew. Chem., Int. Ed. 2015, 54, 6352–6355. doi:10.1002/anie.201412319 |
| 36. | Lou, S.-J.; Zhuo, Q.; Nishiura, M.; Luo, G.; Hou, Z. J. Am. Chem. Soc. 2021, 143, 2470–2476. doi:10.1021/jacs.0c13166 |
| 17. | Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. Chem. Rev. 2019, 119, 2192–2452. doi:10.1021/acs.chemrev.8b00507 |
| 18. | Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel‐Delord, J.; Ackermann, L. Angew. Chem., Int. Ed. 2019, 58, 12803–12818. doi:10.1002/anie.201904214 |
| 19. | Woźniak, Ł.; Cramer, N. Trends Chem. 2019, 1, 471–484. doi:10.1016/j.trechm.2019.03.013 |
| 20. | Rao, W.-H.; Shi, B.-F. Org. Chem. Front. 2016, 3, 1028–1047. doi:10.1039/c6qo00156d |
| 21. | Liu, J.; Chen, G.; Tan, Z. Adv. Synth. Catal. 2016, 358, 1174–1194. doi:10.1002/adsc.201600031 |
| 22. | Liu, Y.-H.; Xia, Y.-N.; Shi, B.-F. Chin. J. Chem. 2020, 38, 635–662. doi:10.1002/cjoc.201900468 |
| 37. | Zhang, Z.-Z.; Liao, G.; Chen, H.-M.; Shi, B.-F. Org. Lett. 2021, 23, 2626–2631. doi:10.1021/acs.orglett.1c00533 |
| 13. | López, L. A.; López, E. Dalton Trans. 2015, 44, 10128–10135. doi:10.1039/c5dt01373a |
| 14. | Zhu, D.-Y.; Chen, P.; Xia, J.-B. ChemCatChem 2016, 8, 68–73. doi:10.1002/cctc.201500895 |
| 15. | Gao, D.-W.; Gu, Q.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2017, 50, 351–365. doi:10.1021/acs.accounts.6b00573 |
| 16. | Huang, J.; Gu, Q.; You, S. Chin. J. Org. Chem. 2018, 38, 51–61. doi:10.6023/cjoc201708030 |
| 34. | Song, Z.; Yu, Y.; Yu, L.; Liu, D.; Wu, Q.; Xia, Z.; Xiao, Y.; Tan, Z. Organometallics 2019, 38, 3349–3357. doi:10.1021/acs.organomet.9b00447 |
| 3. | Dai, L.-X.; Hou, X.-L., Eds. Chiral Ferrocenes in Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2010. doi:10.1002/9783527628841 |
| 9. | Tsukazaki, M.; Tinkl, M.; Roglans, A.; Chapell, B. J.; Taylor, N. J.; Snieckus, V. J. Am. Chem. Soc. 1996, 118, 685–686. doi:10.1021/ja953246q |
| 10. | Metallinos, C.; Szillat, H.; Taylor, N. J.; Snieckus, V. Adv. Synth. Catal. 2003, 345, 370–382. doi:10.1002/adsc.200390042 |
| 11. | Butt, N. A.; Liu, D.; Zhang, W. Synlett 2014, 25, 615–630. doi:10.1055/s-0033-1340487 |
| 12. | Schaarschmidt, D.; Lang, H. Organometallics 2013, 32, 5668–5704. doi:10.1021/om400564x |
| 35. | Chen, H.; Wang, Y.-X.; Luan, Y.-X.; Ye, M. Angew. Chem., Int. Ed. 2020, 59, 9428–9432. doi:10.1002/anie.202001267 |
| 28. | Sattar, M.; Patidar, K.; Thorat, R. A.; Kumar, S. J. Org. Chem. 2019, 84, 6669–6678. doi:10.1021/acs.joc.9b00311 |
| 31. | Yetra, S. R.; Shen, Z.; Wang, H.; Ackermann, L. Beilstein J. Org. Chem. 2018, 14, 1546–1553. doi:10.3762/bjoc.14.131 |
| 34. | Song, Z.; Yu, Y.; Yu, L.; Liu, D.; Wu, Q.; Xia, Z.; Xiao, Y.; Tan, Z. Organometallics 2019, 38, 3349–3357. doi:10.1021/acs.organomet.9b00447 |
| 50. | Kanemoto, K.; Horikawa, N.; Hoshino, S.; Tokoro, Y.; Fukuzawa, S.-i. Org. Lett. 2021, 23, 4966–4970. doi:10.1021/acs.orglett.1c01294 |
| 27. | Sattar, M.; Shareef, M.; Patidar, K.; Kumar, S. J. Org. Chem. 2018, 83, 8241–8249. doi:10.1021/acs.joc.8b00974 |
| 28. | Sattar, M.; Patidar, K.; Thorat, R. A.; Kumar, S. J. Org. Chem. 2019, 84, 6669–6678. doi:10.1021/acs.joc.9b00311 |
| 29. | Sattar, M.; Kumar, N.; Yadav, P.; Mandhar, Y.; Kumar, S. Chem. – Asian J. 2019, 14, 4807–4813. doi:10.1002/asia.201901334 |
| 32. | Huang, D.-Y.; Yao, Q.-J.; Zhang, S.; Xu, X.-T.; Zhang, K.; Shi, B.-F. Org. Lett. 2019, 21, 951–954. doi:10.1021/acs.orglett.8b03938 |
| 33. | Liu, Y.-H.; Li, P.-X.; Yao, Q.-J.; Zhang, Z.-Z.; Huang, D.-Y.; Le, M. D.; Song, H.; Liu, L.; Shi, B.-F. Org. Lett. 2019, 21, 1895–1899. doi:10.1021/acs.orglett.9b00511 |
| 38. | Zhang, Z.-Z.; Cheng, J.; Yao, Q.-J.; Yue, Q.; Zhou, G. Adv. Synth. Catal. 2021, 363, 3946–3951. doi:10.1002/adsc.202100423 |
| 57. | Roane, J.; Daugulis, O. Org. Lett. 2013, 15, 5842–5845. doi:10.1021/ol402904d |
| 58. | Hu, Y.; Wang, M.; Li, P.; Li, H.; Wang, L. Asian J. Org. Chem. 2019, 8, 171–178. doi:10.1002/ajoc.201800664 |
| 26. | Schmiel, D.; Gathy, R.; Butenschön, H. Organometallics 2018, 37, 2095–2110. doi:10.1021/acs.organomet.8b00243 |
| 50. | Kanemoto, K.; Horikawa, N.; Hoshino, S.; Tokoro, Y.; Fukuzawa, S.-i. Org. Lett. 2021, 23, 4966–4970. doi:10.1021/acs.orglett.1c01294 |
| 24. | Schmiel, D.; Butenschön, H. Eur. J. Org. Chem. 2017, 3041–3048. doi:10.1002/ejoc.201700358 |
| 25. | Schmiel, D.; Butenschön, H. Organometallics 2017, 36, 4979–4989. doi:10.1021/acs.organomet.7b00799 |
| 30. | Wang, S.-B.; Gu, Q.; You, S.-L. J. Catal. 2018, 361, 393–397. doi:10.1016/j.jcat.2018.03.007 |
| 51. | Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154–13155. doi:10.1021/ja054549f |
| 52. | Daugulis, O.; Roane, J.; Tran, L. D. Acc. Chem. Res. 2015, 48, 1053–1064. doi:10.1021/ar5004626 |
| 53. | Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M. F.; Wencel-Delord, J.; Besset, T.; Maes, B. U. W.; Schnürch, M. Chem. Soc. Rev. 2018, 47, 6603–6743. doi:10.1039/c8cs00201k |
| 54. | Zhang, Q.; Shi, B.-F. Chin. J. Chem. 2019, 37, 647–656. doi:10.1002/cjoc.201900090 |
| 55. | Rej, S.; Ano, Y.; Chatani, N. Chem. Rev. 2020, 120, 1788–1887. doi:10.1021/acs.chemrev.9b00495 |
| 56. | Zhang, Q.; Shi, B.-F. Acc. Chem. Res. 2021, 54, 2750–2763. doi:10.1021/acs.accounts.1c00168 |
© 2021 Jia et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)