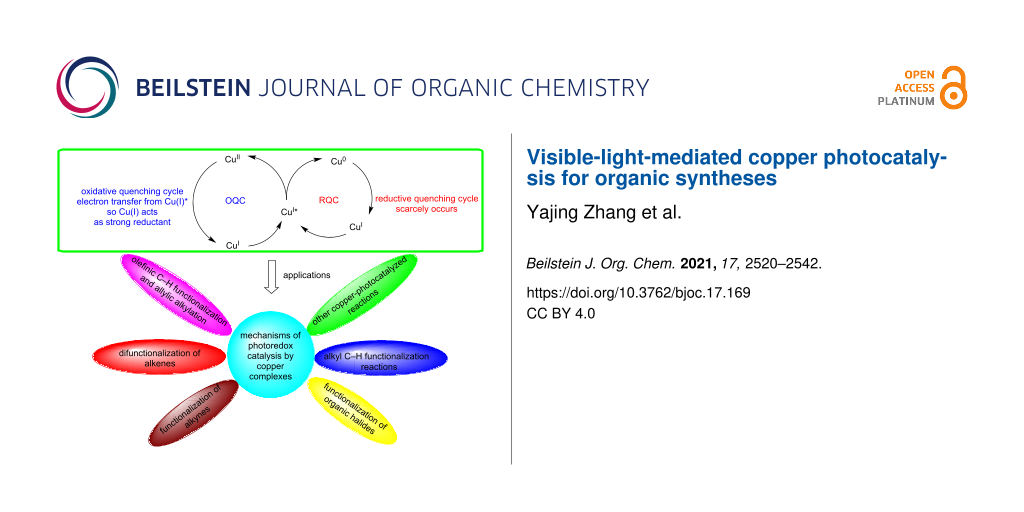
Abstract
Photoredox catalysis has been applied to renewable energy and green chemistry for many years. Ruthenium and iridium, which can be used as photoredox catalysts, are expensive and scarce in nature. Thus, the further development of catalysts based on these transition metals is discouraged. Alternative photocatalysts based on copper complexes are widely investigated, because they are abundant and less expensive. This review discusses the scope and application of photoinduced copper-based catalysis along with recent progress in this field. The special features and mechanisms of copper photocatalysis and highlights of the applications of the copper complexes to photocatalysis are reported. Copper-photocatalyzed reactions, including alkene and alkyne functionalization, organic halide functionalization, and alkyl C–H functionalization that have been reported over the past 5 years, are included.

Graphical Abstract
Introduction
Solar light is an inexhaustible and free energy source for green plants and bacteria. Photosynthetic organisms absorb solar energy and convert it into chemical energy via photosynthesis [1]. Photochemical reactions mimic natural photosynthesis, and photoredox catalysis plays a key role in energy-transfer processes [2-5]. Over the past decades, photoredox catalysis has attracted an increasing amount of attention [6-9], and a series of organic dyes and metal complexes have been investigated [10-12]. Photoredox catalysts have been initially applied to organic reactions, but they are now used for complicated organic processes [13]. As photocatalysts, organic dyes have the advantages of having a low price and not containing metals; however, they suffer from relatively poor photostability [14-16]. Transition-metal-photoredox catalysts, such as ruthenium and iridium polypyridyl complexes, exhibit high redox potentials, long excited state lifetimes, and strong absorption [17-20]. However, high cost and their scarcity discourage development of ruthenium and iridium-based catalysts [21]. Copper salts have become popular materials for photoredox catalysts due to their abundance, low cost, and ability to provide strong photoexcited reducing power [21-24]. In this review, the different catalysis mechanisms between ruthenium-based catalysts and copper-based catalysts are discussed, and the strong reduction ability of copper complexes is explained. Subsequently, mechanisms of the photoredox catalysis by CuI and CuII are summarized, and the copper-catalyzed reactions, including alkene functionalization, alkyne functionalization, organic halides functionalization, and alkyl C–H functionalization, are highlighted.
Review
1. Special features of photoredox-catalyzed processes by copper complexes
To understand photoredox-catalyzed processes, a discussion of the general mechanism of [Ru(bpy)3]2+ is needed [25-27]. When the photocatalyst RuII is irradiated by light, an electron is transferred from the frontier metal d orbital (t2g orbital) to the ligand-centered π* orbital (RuII*). A metal-to-ligand charge transfer (MLCT) results in the excited singlet state. Through rapid intersystem crossing (ISC), the singlet state is transformed to the lowest-energy triplet MLCT state, which has a sufficient lifetime for initiating single-electron transfer. In the triplet species, the electron in the higher singly occupied molecular orbital (SOMO) is transferred from RuII* to an external acceptor (A), thereby yielding oxidized RuIII, which subsequently accepts an electron from an external donor (D) to form the ground-state catalyst RuII. This type of reaction mechanism is an oxidative quenching cycle (OQC). Alternatively, the lower energy SOMO of the excited state RuII* can accept an electron from an external donor, which is referred to as a reductive quenching cycle (RQC; Scheme 1).
![[1860-5397-17-169-i1]](/bjoc/content/inline/1860-5397-17-169-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photoredox catalysis mechanism of [Ru(bpy)3]2+.
Scheme 1: Photoredox catalysis mechanism of [Ru(bpy)3]2+.
Compared with the photoredox mechanism of ruthenium-based catalysts, copper complexes show unique features [22,28]. Under irradiation, the copper complex CuI is converted to the excited state CuI*, which transfers electrons to an acceptor A or receives electrons from a donor D. In the OQC pathway, the excited state CuI* transfers an electron to the acceptor A and is oxidized to CuII. Subsequently, CuII accepts an electron from the donor D to form the ground-state CuI (Scheme 2). However, reports that the excited state CuI* receives electrons from donors are relatively scarce in the literature. Thus, the RQC pathway rarely occurs for CuI-photocatalyzed reactions. Yet, CuI complexes have the potential to replace ruthenium or iridium-based photocatalysts in reductive photoredox reactions due to their strong reduction ability [22,29]. For example, [Cu(dap)2]Cl (*Cu+/Cu2+ = −1.43 V) provides a stronger reducing power than [Ru(bpy)3]Cl (*Ru2+/Ru3+ = −0.81 V) and [Ir{dF(CF3)ppy}2(dtbbpy)]Cl (*Ir2+/Ir3+ = −0.89 V) [28,30]. Nevertheless, upon absorbing a photon, CuI undergoes a reorganization from a tetrahedral geometry to a square-planar geometry, thereby resulting in a shorter excited state lifetime compared with ruthenium and iridium-based photocatalysts and thus limiting the application of CuI complexes to visible-light-mediated organic syntheses [22,31].
![[1860-5397-17-169-i2]](/bjoc/content/inline/1860-5397-17-169-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Photoredox catalysis mechanism of CuI.
Scheme 2: Photoredox catalysis mechanism of CuI.
Homoleptic CuI bisphenanthroline complexes were designed with cooperative steric hindrance based on bulky substituents at the 2,9-position of the phenanthroline moiety [32,33]. Alternatively, heteroleptic CuI complexes with phenanthroline and bulky chelating phosphine ligands were also synthesized [30,34,35]. The photophysical properties are dramatically modified by the homoleptic and heteroleptic CuI complexes [22,31,36]. The introduction of bulky ligand substituents might efficiently prevent the reorganization of the excited state. Thus, changing the nature of the chelating ligand can improve the photostability and lifetime of the excited state to meet the requirements of a given photochemical process. The different ligands and CuI complexes are shown in Scheme 3 [21,30]. The catalysis mechanisms of these CuI complexes are discussed in the following sections.
![[1860-5397-17-169-i3]](/bjoc/content/inline/1860-5397-17-169-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
2. Mechanisms underlying the photoredox catalysis of copper complexes
The mechanisms underlying the photoredox catalysis of CuI complexes with different ligands were investigated by Reiser’s group [21]. Other studies have since provided more information on the photoredox mechanisms underlying the catalysis of copper complexes [37]. In general, redox-active copper complexes include CuI and CuII complexes. The mechanisms underlying photoredox catalysis of CuI complexes have special features and include ligand exchange and rebound mechanisms [38]. CuII complexes provide new avenues for photoredox catalysis, since CuII can undergo ligand exchange/light accelerated homolysis processes, which accelerates homolysis to produce CuI species and radical intermediates. These intermediates can initiate productive organic transformations [39].
2.1 Visible-light-mediated Cu(I) catalytic cycle
Upon the absorption of a photon (Scheme 4), CuILn forms a singlet MLCT state, which subsequently yields the excited triplet state CuI*Ln via rapid ISC. The excited CuI*Ln species has a lifetime to finish the chemical processes. A radical mechanism is proposed in Scheme 4. In path a (a ligand transfer cycle), CuI* is oxidized by an electrophilic reagent (haloalkane) to form CuII and the radical species R• in a single-electron transfer (SET) process. Subsequently, CuII undergoes ligand exchange with a nucleophilic reagent (Nu) to produce the CuII−Nu species. The reorganization of CuII–Nu is trapped by the radical intermediate R• to generate the final product (R–Nu) with concomitant regeneration of the CuI catalyst. Alternatively, in path b (a rebound cycle), CuI* is trapped after a SET by the radical intermediate to generate a CuIII species, which undergoes ligand exchange with the nucleophile and reductive elimination to produce the target product and the regenerated CuI catalyst [37,38,40].
![[1860-5397-17-169-i4]](/bjoc/content/inline/1860-5397-17-169-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanism of CuI-based photocatalysis.
Scheme 4: Mechanism of CuI-based photocatalysis.
2.2 Visible-light-mediated Cu(I)–substrate catalytic cycle
Upon the irradiation of LnCuINu, an electron is transferred from the metal center to the ligand, thereby generating the excited state LnCuINu*. The excited state species can be oxidized by the electrophilic reagent (haloalkane, RX) to form the intermediate [LnCuIINu]X. The desired product Nu–R can be obtained through an inner-sphere pathway between [LnCuIINu]X and the radical R• [41,42] (Scheme 5A). Alternatively, a photosensitizer generated a radical via reduction or oxidation, and is not engaged in the key bond construction. [LCuI] is photoexcited to generate LnCuI*, which transfers an electron to the haloalkane, thereby resulting in the formation of [LCuII]X and R•. Then, the radical R• is trapped by a second copper complex [LnCuIINu]X, which mediates Nu–R bond formation in an out-of-cage process (Scheme 5B).
![[1860-5397-17-169-i5]](/bjoc/content/inline/1860-5397-17-169-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanisms of CuI–substrate complexes.
Scheme 5: Mechanisms of CuI–substrate complexes.
2.3 Visible-light-mediated Cu(II) catalytic cycle
The precatalyst LnCuII (A) undergoes ligand exchange with substrate X to form a catalytically active species, namely, Ln−1CuIIX. Upon irradiation, Ln−1CuIIX is converted to a photoexcited species Ln−1CuII*X, which undergoes homolytic dissociation to produce Ln−1CuI and radical X•. The radical X• can add to the substrate Y to obtain the stable radical X–Y•. Subsequently, Ln−1CuI transfers one electron to X–Y• and accepts one ligand to regenerate the intermediate LnCuII and the final product [39,43] (Scheme 6).
![[1860-5397-17-169-i6]](/bjoc/content/inline/1860-5397-17-169-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Mechanism of CuII-base photocatalysis.
Scheme 6: Mechanism of CuII-base photocatalysis.
Copper photocatalysis is a powerful tool that can be used to construct carbon–heteroatom and carbon–carbon bonds and can be applied to radical chemistry. This review discusses copper-catalyzed reactions including alkene and alkyne, organic halide, and alkyl C–H functionalization.
3. Visible-light-mediated copper-catalyzed alkene and alkyne functionalization
3.1 Olefinic C–H functionalization and allylic alkylation
Under mild conditions, copper salts are able to catalyze olefinic C–H functionalization or allylic alkylation, thus allow introducing alkenyl or allyl groups into organic molecules. Alkenylation and allylation reactions have been extensively investigated under thermal conditions. However, only few studies included visible-light catalysis. In 2012, Reiser’s group [44] reported the allylation of α-haloketones 1 with olefins under irradiation (λ = 530 nm) in the presence of [Cu(dap)2Cl] (dap = 2,9-di(p-anisyl)-1,10-phenanthroline) as the catalyst. They conducted control experiments to establish that [Cu(dap)2Cl] and visible light are necessary for this transformation. In 2013, Ollivier and co-workers [45] successfully applied the same strategy to the allylation of diphenyliodonium 2. In 2017, Liu’s group [46] reported the copper salt-catalyzed cyclization of vinyl azides 3 with ammonium thiocyanate to generate 4-alkyl/aryl-2-aminothiazoles. Mechanistic experiments demonstrated that the photocatalyst formed in situ from Cu(OAc)2 and ammonium thiocyanate promoted the intermolecular cyclization (Scheme 7).
![[1860-5397-17-169-i7]](/bjoc/content/inline/1860-5397-17-169-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Olefinic C–H functionalization and allylic alkylation.
Scheme 7: Olefinic C–H functionalization and allylic alkylation.
3.2 Difunctionalization of alkenes
The 1,2-difunctionalization of alkenes is a versatile strategy for the construction of complex molecules. The primary process involved in the 1,2-difunctionalization of alkenes catalyzed by copper complexes is an atom-transfer radical addition (ATRA). Copper complexes or copper-based photoredox-active complexes formed in situ serve as photocatalysts to transfer electrons to suitable radical precursors. The detailed catalytic cycle is presented in section 3.1 and involves ligand exchange and rebound mechanisms.
A comprehensive survey of copper photocatalysts was initiated from Reiser’s group. In 2015, Reiser and co-workers [47] reported the [Cu(dap)2]Cl-catalyzed trifluoromethylchlorosulfonylation of unactivated alkenes 4 under photochemical conditions. When used in place of [Cu(dap)2]Cl, ruthenium-based, iridium-based, and eosin Y catalysts promoted the trifluoromethylchlorination of alkenes with the extrusion of SO2 (e.g., product 6). Studies were performed to elucidate the strikingly different reactions that occurred with these different photoredox catalysts. The catalytic cycle of [Cu(dap)2]Cl is shown in Scheme 8. In this catalytic cycle, [Cu(dap)2]Cl excited by irradiation with visible light reacts with triflyl chloride in a SET process to generate a trifluoromethyl radical and LnCuIISO2Cl (intermediate A in Scheme 8). The formed trifluoromethyl radical adds to the alkene moiety to deliver a new alkyl radical, which is trapped by the LnCuII-SO2Cl species. Free SO2Cl– decomposes rapidly to SO2 and Cl–. However, in this transformation, SO2Cl– is stabilized by the copper complex. The alkyl radical reacts with LnCuII-SO2Cl to deliver the target product 5. A mechanistic study demonstrated that [Cu(dap)2]Cl can coordinate with the reactive intermediate SO2Cl and suppresses the extrusion of SO2. Thus, [Cu(dap)2]Cl achieves a unique transformation under visible-light irradiation by acting as a catalyst for single-electron reduction and as an intermediate stabilizing agent (Scheme 8). In 2016, the same group [48] reported the [Cu(dap)2]Cl-catalyzed cyclization of α,ω-alkenols and trifluoromethylsulfonyl chloride to form sultones (Scheme 8).
![[1860-5397-17-169-i8]](/bjoc/content/inline/1860-5397-17-169-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Cross-coupling of unactivated alkenes and CF3SO2Cl.
Scheme 8: Cross-coupling of unactivated alkenes and CF3SO2Cl.
Intrigued by this unique transformation, Reiser’s group [49] extended this protocol to the chlorosulfonylation of alkenes and alkynes in 2019. Under visible light irradiation and in the presence of [Cu(dap)2]Cl, the reaction of p-toluenesulfonyl chloride (7) with alkenes gave an excellent yield of the chlorosulfonylated products 8 and 9, whereas replacing the copper catalyst by ruthenium-based, iridium-based, and eosin Y catalysts afforded the desired products only in trace amount. Unexpectedly, the corresponding CuII complex, Cu(dap)Cl2, also produced the desired product with good yield. Based on the literature [28,50], Cu(dap)Cl2 acted as a potential precatalyst in this photoreaction. Upon irradiation, the CuII complex undergoes homolytic cleavage of a Cu–Cl bond forming CuI as the catalytically active species; thus, the CuII complex is the precatalyst and provides a more efficient transformation than CuI. Under optimized conditions, the substrate scope was examined and determined to include activated olefins, unactivated olefins, and arylalkynes. In parallel, Hu and co-workers [51] reported the photoinduced, copper-catalyzed chlorosulfonylation of alkenes and alkynes under irradiation with blue LEDs. Reiser and co-workers [52,53] unexpectedly observed the iodoperfluoroalkylation of alkenes and perfluoroalkyl iodides 10 in the presence of [Cu(dap)2]Cl. Consistent with the previous report, the desired products 11 were not obtained with ruthenium-based, iridium-based, and eosin Y catalysts (Scheme 9), which was due to the ability of copper to stabilize and interact with radical intermediates in its coordination sphere. Mechanistic studies revealed that the iodoperfluoroalkylation of alkenes and alkynes involved a rebound or ligand transfer cycle (section 3.1). In 2017, Wang and co-workers [54] discovered the photoinduced, copper-catalyzed cyanofluoroalkylation of alkenes and fluoroalkyl iodides 12. The reaction was initiated by the reduction of CuII with tertiary amines, which formed CuICN and an amine radical cation [55]. Under irradiation by ultraviolet light, CuICN was excited and transformed to its triplet state CuICN*, in which the fluoroalkyl iodides were reduced to Rf• and I−. Subsequently, the radical Rf• attacks the alkene forming a new alkyl radical species. This radical species is then trapped by CuII(CN)n to generate a CuIII intermediate, which undergoes reductive elimination to form the desired product 13 (Scheme 9). In 2019, the same group [56] applied this protocol to the asymmetric cyanofluoroalkylation of alkenes. Under visible-light irradiation, the Cu-based catalyst plays a dual role as both the photosensitizer for the SET and the catalyst for asymmetric control (Scheme 9).
![[1860-5397-17-169-i9]](/bjoc/content/inline/1860-5397-17-169-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Chlorosulfonylation/cyanofluoroalkylation of alkenes.
Scheme 9: Chlorosulfonylation/cyanofluoroalkylation of alkenes.
In addition to perfluoroalkyl iodides, this protocol was further extended to alkyl halides, trifluoromethylthiolate, amines, cycloketone oxime esters, and carboxylic acid N-hydroxyphthalimide esters (NHPI). In 2018, Peters and Fu [57] explored the copper-catalyzed three-component coupling of alkyl halides 14, olefins, and trifluoromethylthiolate 15. Mechanistic studies demonstrated that the photoexcited CuI/binap/SCF3 complex generated in situ engages in electron transfer with the alkyl halides, thereby providing an alkyl radical and the CuII/binap/SCF3 species. Subsequently, the alkyl radical reacts with the olefin generating a new alkyl radical, which is trapped by CuII/binap/SCF3 to provide the coupling product (Scheme 10). In 2019, Zhang and co-workers [58] reported the photoinduced copper-catalyzed carboamination of alkenes that involved organic halides 16, alkenes, and amines 17, 18 (Scheme 10 and Scheme 11). Based on previous mechanistic studies [41], the authors found that the photoexcited ligand–CuI−amido species transferred electrons to alkyl halides to produce alkyl radicals, which reacted with alkenes and amines to generate the three-component coupling products. In the absence of organic halide, the copper salts catalyzed the hydroamination of the alkene [59]. Mechanistic studies showed that the copper–amido complex coordinated with alkenes, which then acted as a primary photocatalyst. After light irradiation, the excited alkene–copper–amido species offered a benzyl radical and the organocopper via SET with hydrogen atom abstraction from CH3CN. Subsequently, the benzyl radical was captured by the organocopper to generate the hydroamination products (Scheme 10). In 2020, the same group [60] reported the copper-catalyzed asymmetric dual carbofunctionalization of alkenes with alkynes and alkyl halides (Scheme 11). The alkynyl copper-ligand served as the photoactive species and delivered a single electron to the alkyl halide to produce the alkyl radical, which then reacted with the alkene and alkyne to generate the coupling products (Scheme 10).
![[1860-5397-17-169-i10]](/bjoc/content/inline/1860-5397-17-169-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-169-i11]](/bjoc/content/inline/1860-5397-17-169-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Cross-coupling reaction of alkenes, alkyl halides with nucleophiles.
Scheme 11: Cross-coupling reaction of alkenes, alkyl halides with nucleophiles.
From 2018 to 2020, Xiao and Yu et al. [61,62] disclosed a series of copper-catalyzed cyanoalkylation reactions among alkenes, oxime esters, and boronic acids or alkynes. Mechanistic studies implied that the CuI complex gets photoexcited via a SET process to generate a cyanoalkyl radical from the oxime esters. The resulting cyanoalkyl radical then adds to the alkene to form a new alkyl radical. This radical is captured by a high-valent CuIII complex, which undergoes a reductive elimination to give the target product (Scheme 12).
![[1860-5397-17-169-i12]](/bjoc/content/inline/1860-5397-17-169-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Cross-coupling of alkenes with oxime esters.
Scheme 12: Cross-coupling of alkenes with oxime esters.
In 2018, Reiser and co-worker [63] established a CuII-catalyzed oxo-azidation of vinyl arenes (Scheme 13). In this transformation, the azide anion is oxidized to its radical, and this process requires a high reduction potential that cannot be achieved by iridium and ruthenium-based catalysts. In contrast, [Cu(dap)2]Cl and [Cu(dap)Cl2] were found suitable for the oxidation. Based on a mechanistic study, CuII serves as the catalytically active species that undergoes homolytic cleavage to form a CuI species and an azide radical. The latter adds to the alkene to form an alkyl radical, which is then trapped by oxygen to form the desired product. The homolytic cleavage of the active species represents a new platform for copper-based photocatalysis (see section 3.3). In 2019, Yu et al. [64] developed a similar copper-catalyzed azidation of activated alkenes with 1-azido-1,2-benziodoxole as the azide radical precursor (Scheme 14).
![[1860-5397-17-169-i13]](/bjoc/content/inline/1860-5397-17-169-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Oxo-azidation of vinyl arenes.
Scheme 13: Oxo-azidation of vinyl arenes.
![[1860-5397-17-169-i14]](/bjoc/content/inline/1860-5397-17-169-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Azidation/difunctionalization of vinyl arenes.
Scheme 14: Azidation/difunctionalization of vinyl arenes.
3.3 Functionalization of alkynes
A series of photoinduced copper-catalyzed coupling reactions of terminal alkynes was published by Hwang and co-worker. In 2012, they [65] achieved a photoinitiated Sonogashira reaction using aryl halides (bromides and iodides) 20 and aryl- or alkylacetylenes 19. In control experiments, the replacement of the copper salt with palladium salts or the substitution of thermal heating for LED irradiation resulted in a low yield of the cross-coupling products 22 and 23, indicating the necessity of the copper salt and LED irradiation. Mild conditions, such as Pd-free and high reaction yields at room temperature, make this method a very promising, scalable green process that can be used as an alternative to the conventional Sonogashira cross-coupling reactions. In 2018, Lalic and co-workers [66] extended this approach to alkyl halides and reported the photoinduced copper-catalyzed Sonogashira coupling of alkynes and alkyl iodide 21. The proposed mechanism is shown in Scheme 15. Under blue visible light, the haloalkane is reduced by the copper acetylide to form the alkyl radical intermediate R2•. If aryl halide is the haloalkane, the copper acetylide is attacked by the aryl halide to form transition-state intermediate A. The copper acetylide is transformed into a high-valent CuIII complex, which subsequently undergoes reductive elimination or dissociation of the transition-state intermediate in the case of aryl halide to generate the target product (Scheme 15).
![[1860-5397-17-169-i15]](/bjoc/content/inline/1860-5397-17-169-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Photoinitiated copper-catalyzed Sonogashira reaction.
Scheme 15: Photoinitiated copper-catalyzed Sonogashira reaction.
From 2015 to 2020, Hwang and co-workers [67-71] investigated the copper-catalyzed oxidative coupling of alkynes, nucleophiles (e.g., phenols 25 and 28; amines 24, 27, 29, and 32; 2-hydrazinylpyridine 26; alkyne 33; and alcohol 30), and oxidants (benzoquinone or O2). Based on the literature and mechanistic experiments [72,73], the reaction is initiated by the photoirradiation of in situ-generated CuI phenylacetylide. From the excited state of CuI phenylacetylide an electron is transferred to the oxidants (benzoquinone or O2) via a SET, thereby forming a CuII phenylacetylide species and a radical anion. The resulting CuII phenylacetylide species is involved in the bond-forming reaction [74]. As a notable exception, in 2016, Hwang’s group [75] reported the novel synthesis of unsymmetrical 1,3-conjugated diynes 31 from terminal alkynes under LED irradiation. The reaction mechanism involved a bipolar heterodimeric copper phenylacetylide species that showed similar photophysical properties (Scheme 16).
![[1860-5397-17-169-i16]](/bjoc/content/inline/1860-5397-17-169-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Alkyne functionalization reactions.
Scheme 16: Alkyne functionalization reactions.
In 2019, Vlla’s group [76] explored the copper-catalyzed alkynylation of dihydroquinoxalin-2-ones 34 with terminal alkynes under irradiation. 4-Benzyl-3,4-dihydroquinoxalin-2(1H)-one 35 was subjected to an oxidation process with a CuII salt to generate a nitrogen radical cation I and a CuI species. This process regenerated CuII in the presence of molecular oxygen. The deprotonation of the nitrogen radical cation produces an α–amino radical II, which was further oxidized to the iminium ion III to which the copper alkynylide added forming the desired product (Scheme 17).
![[1860-5397-17-169-i17]](/bjoc/content/inline/1860-5397-17-169-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Alkynylation of dihydroquinoxalin-2-ones with terminal alkynes.
Scheme 17: Alkynylation of dihydroquinoxalin-2-ones with terminal alkynes.
In 2020, Zhang’s group [77] described the photoinduced copper-catalyzed decarboxylative alkynylation of redox-active esters with terminal alkynes. N-Hydroxy-tetrachlorophthalimide (TCNHPI, 36) derived from carboxylic acids was identified as the ideal radical precursor. Under irradiation, the CuI acetylide–ligand species A generated in situ was irradiated to form the activated state A*, which transferred a single electron to TCNHPI to form an alkyl radical and tetrachlorophthalimide anion and concurrently generated the oxidative CuI acetylide species B. The intermediate B was subsequently trapped by the alkyl radical and underwent reductive elimination to deliver the desired product. Liu’s group [78] further applied this protocol to the asymmetric decarboxylative alkynylation of N-hydroxy 2,3-naphthalimide-derived ester 37 with terminal alkynes. Remarkably, the N-hydroxy 2,3-naphthalimide-derived ester acted as an ideal radical precursor and accepted a single electron from the excited state CuI-acetylide complex. The copper catalyst plays a dual role, namely, as a photoredox catalyst and a cross-coupling catalyst. NHP-type esters inhibited the homodimerization of the alkyl radical and terminal alkyne (Scheme 18).
![[1860-5397-17-169-i18]](/bjoc/content/inline/1860-5397-17-169-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Decarboxylative alkynylation of redox-active esters.
Scheme 18: Decarboxylative alkynylation of redox-active esters.
Under visible-light irradiation, disulfides are easy transformed to thiyl radicals via the homolytic cleavage of the S–S bond [79]. In 2020, Anandhan and co-workers [80] explored the C(sp)–S coupling of terminal alkynes with 2-aminothiophenol dimer 38 as a radical precursor. Under photoexcitation the CuI acetylide A undergoes a SET process to form the CuII phenylacetylide species B and a superoxide radical anion. In parallel, under irradiation the homolytic S–S-bond cleavage in 2-aminothiophenol dimer 38 forms thiol radicals 40. The nucleophilic addition of the amino group in radical 40 to the CuII acetylide B generates the CuIII acetylide species C, which coordinates with the thiol radical to give the CuII-aminothiophenol complex D. Finally, the intermediate CuI-aminothiophenol complex E reacted with HCl and O2 to generate the C(sp)-S coupling product 39 (Scheme 19).
![[1860-5397-17-169-i19]](/bjoc/content/inline/1860-5397-17-169-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Aerobic oxidative C(sp)–S coupling reaction.
Scheme 19: Aerobic oxidative C(sp)–S coupling reaction.
3.4 Functionalization of organic halides
As demonstrated by the different photoredox mechanisms of Cu complexes, the CuI complexes have strong reduction ability and can promote electron transfer to organic substrates. Thus, the high reduction potentials of CuI complexes are also applied to the functionalization of organic halides. The seminal work by Fu and Peters demonstrated that with the help of light, copper–nucleophile complexes undergo excitation, and the resulting complex engages with organic halides in a SET process to generate alkyl or aryl radicals. Next, a C–X (X = O, N, S, C) bond is formed between the nucleophile and the alkyl or aryl radicals. From 2013 to 2019, the authors disclosed a series of nitrogen, sulfur, oxygen, and carbon nucleophiles for photoinduced, copper-catalyzed cross-couplings with organic halides. The copper–nucleophile complexes that were generated in situ as photoredox catalysts transferred electrons to organic halides, thereby achieving the cross-coupling. The detailed mechanistic studies are shown in section 3.2.
In 2013, Fu and co-workers [81] reported the photoinduced copper-catalyzed alkylation of carbazoles 41 with alkyl halides 42 and completed the corresponding mechanistic study [41,42] in 2017. This metal-catalyzed, photoinduced, and asymmetric radical transformation requires two catalysts, namely, (i) a metal catalyst that promotes electron transfer and (ii) a separate chiral catalyst that facilitates the highly stereoselective bond formation. In 2016, Fu [82] discovered the asymmetric cross-coupling of racemic tertiary alkyl halides 43 with carbazoles or indoles 44 in the CuI/chiral phosphine system. Under irradiation conditions, excitation of the copper–nucleophile complex A results in the excited state species B that engages in the electron transfer with the alkyl halide to generate a copperII–nucleophile complex C and an alkyl radical. The formation of the R–Nu bond might occur through an in-cage pathway involving complex C (Scheme 20).
![[1860-5397-17-169-i20]](/bjoc/content/inline/1860-5397-17-169-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Copper-catalyzed alkylation of carbazoles with alkyl halides.
Scheme 20: Copper-catalyzed alkylation of carbazoles with alkyl halides.
In addition to carbazoles, the authors further described the C–N coupling of organic halides 45 with amides [83] and aliphatic amines [84] 46. The results of the mechanistic studies showed that a copper/tridentate carbazolide-bisphosphine ligand complex serves as a new photoredox catalyst engaged in the electron transfer to the electrophile. Under photoexcitation, the excited photoredox catalyst F reduces the alkyl halide, producing an alkyl radical and a copperII intermediate G, which oxidizes a copperI–nucleophile complex A to the corresponding copperII–nucleophile complex C. Complex C then couples with the alkyl radical to generate the product Nu–R in an out-of-cage process (Scheme 21).
![[1860-5397-17-169-i21]](/bjoc/content/inline/1860-5397-17-169-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: C–N coupling of organic halides with amides and aliphatic amines.
Scheme 21: C–N coupling of organic halides with amides and aliphatic amines.
The same group was interested in extending this protocol from C–N bond formation reactions to C–O [85], C–S [86], and C–C [87,88] bond formations. Recently, a photoredox catalysis was applied to these types of cross-coupling reactions, with key contributions from the groups of Ackermann [87], Evano [55], Zhang [89], Nguyen [90], and Bissember [91]. In 2013, Peters’ group [86] established the copper-catalyzed C–S cross-coupling between thiols and aryl halides. The mechanistic studies revealed that the reaction runs with the inexpensive precatalyst (CuI) and no ligand co-additive is necessary. In 2014, the same group [85] reported the copper-catalyzed C–O cross-coupling reaction. Results of mechanistic studies indicated that in this case a CuI–phenoxide complex is a competent intermediate in the photoinduced C–O bond formation. In 2020, Nguyen and co-worker [90] reported copper-catalyzed C–O cross-coupling of glycosyl bromides with aliphatic alcohols. In 2015, Ackermann’s group [88] disclosed the visible light-induced copper-catalyzed arylation of azoles. In this case, the mechanistic studies revealed that amino acid ligands accelerated the cross-coupling (Scheme 22).
![[1860-5397-17-169-i22]](/bjoc/content/inline/1860-5397-17-169-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Copper-catalyzed C–X (N, S, O) bond formation reactions.
Scheme 22: Copper-catalyzed C–X (N, S, O) bond formation reactions.
In 2020, a visible-light-induced copper-catalyzed arylation of C(sp2)–H bonds of azoles was developed by Zhang [89]. A 2,2’-bipyridine copper coordination compound served as the photoredox catalyst and accomplished the azole C–H arylations. Under irradiation with blue LED, the photoexcited state [LnCuI-benzoxazole]* (C) engages in a double electron-transfer process with aryl iodides to generate intermediate D, which then undergoes reductive elimination to generate the desired products (Scheme 23).
![[1860-5397-17-169-i23]](/bjoc/content/inline/1860-5397-17-169-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Arylation of C(sp2)–H bonds of azoles.
Scheme 23: Arylation of C(sp2)–H bonds of azoles.
In 2017, Evano’s group [55] established a photoinduced, copper-catalyzed C–C cross-coupling of aryl halides, and heteroarenes. The cyclization of N-allyl-o-iodoanilines was further studied in intramolecular processes. [(DPEphos)(bcp)Cu]PF6, as a photocatalyst, was applied in these transformations. In 2018, the Bissember group [91] reported the photoinduced and copper-catalyzed dual α-amino-C–H/C–F functionalization reaction (Scheme 24).
![[1860-5397-17-169-i24]](/bjoc/content/inline/1860-5397-17-169-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: C–C cross-coupling of aryl halides and heteroarenes.
Scheme 24: C–C cross-coupling of aryl halides and heteroarenes.
3.5 Alkyl C–H functionalization reactions
Benzylic or α-amino C–H groups and even the stable C(sp3)–H group were functionalized through the corresponding benzylic radical, α-amino radical, or alkyl radical. In 2016, Greaney and co-workers [92] investigated the direct C–H azidation with benzylic C–H compounds 47 and the Zhdankin reagent. After investigating a range of reaction parameters, copper salts and visible light were found to be necessary for the transformation. The reaction is highly selective for the benzylic position. In the same year, Bissember’s group [93] reported a copper-photocatalyzed α-amino C–H functionalization. In this work, N,N-dialkylanilines or N-aryltetrahydroisoquinolines 48 reacted with N-substituted maleimide 49 via annulation to provide a range of tetrahydroquinolines or tetrahydroisoquinolines 50, respectively, with good yield. The mechanistic investigation revealed that an α-amino radical undergoes radical addition with the N-substituted maleimide (Scheme 25).
![[1860-5397-17-169-i25]](/bjoc/content/inline/1860-5397-17-169-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Benzylic or α-amino C–H functionalization.
Scheme 25: Benzylic or α-amino C–H functionalization.
In 2017, Wu and co-workers [94] reported the α-amino C−H functionalization of aromatic amines 51 with nucleophiles, including arynes or aromatic olefins 52, indoles, acyclic β-ketoester 53, and β-diketone 54 (Scheme 26). Mechanistic observations revealed a new avenue for copper-based photocatalysis. The transformation was initiated by a SET process from the amine to the CuII ion to generate the visible-light-driven species I [CuI-NH•+]. Under visible-light irradiation, the intermediate [CuI-NH•+] was oxidized to the imine 60 by O2. Next, the imine 60 transferred a single electron to the CuII ion, thereby providing intermediate II [CuI-N•+]. [CuI-N+•] was equal to CuI and N+• of the imine, which is activated for the nucleophilic addition. With the help of O2, CuI regenerates CuII to complete the catalytic cycle (Scheme 27). A series of quinolones 56, indolo[3,2-c]quinolines 57, β-amino acids 58, and 1,4-dihydropyridine derivatives 59 were obtained through this route in moderate yields (Scheme 26). Besides nucleophiles, aromatic amines also reacted with redox-active radical precursors, such as NHPI [95] and N-alkoxyphthalimides 55 [96].
![[1860-5397-17-169-i26]](/bjoc/content/inline/1860-5397-17-169-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: α-Amino C–H functionalization of aromatic amines.
Scheme 26: α-Amino C–H functionalization of aromatic amines.
![[1860-5397-17-169-i27]](/bjoc/content/inline/1860-5397-17-169-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: C–H functionalization of aromatic amines.
Scheme 27: C–H functionalization of aromatic amines.
The functionalization of stable alkanes C(sp3)–H is generally difficult. In 2020, the König group [97] explored the photoinduced copperII catalyzed N–H alkylation of a broad range of nitrogen-containing compounds 61 with unactivated alkanes 62. A tert-butoxy radical abstracted a hydrogen atom from the alkane via the photolysis of DTBP producing an alkyl radical, which reacted with nitrogen-containing compounds to give the target products 63. The catalytic cycle involves a photoinduced copperII peroxide system with an in situ-generated CuII–N complex as the key catalytic species. In 2020, Anandhan’s group [98] developed photoinduced copper-catalyzed α-C(sp3)–H cyclization of aliphatic alcohols with o-aminobenzamide. However, the aliphatic alcohols were limited to methanol and ethanol. In this transformation, α-C(sp3)–H of MeOH/EtOH undergoes a hydrogen atom transfer (HAT) process to synthesize quinazolinones involving ligand-CuII superoxo complexes A. Under light irradiation, complex A produces the excited-state ligand-CuII superoxo complex A*, which undergoes coordination with the aliphatic alcohol to form complex B. The latter initiates the oxidation reaction and transfers a hydrogen atom from α-C(sp3)–H of the alcohol to generate the CuII hydroperoxo complex C and the corresponding aldehyde. Complex C can undergo a reductive elimination to recover 64a. The liberated aminobenzamide 64a and the aldehyde undergo a condensation reaction to produce quinazolinone 66′, followed by oxidation with molecular oxygen to produce the desired quinazolinone 66 (Scheme 28).
![[1860-5397-17-169-i28]](/bjoc/content/inline/1860-5397-17-169-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: α-Amino-C–H and alkyl C–H functionalization reactions.
Scheme 28: α-Amino-C–H and alkyl C–H functionalization reactions.
3.6 Other copper-photocatalyzed reactions
With the advances in photocatalyzed reactions, radical precursors have received considerable research attention as practical and mild functional reagents. Extensive studies have been reported. The Fu [99] and Wang [100] groups reported that NHPI esters (67, 68) have been used as alkyl radical precursors in decarboxylative coupling reactions. These reactions feature a wide substrate scope. Primary, secondary, and tertiary alkyl carboxylic acids exhibit good yield, such as decarboxylative coupling reactions between N-heteroarenes 69 and redox-active esters 68. In 2018, Gong and co-workers [43] used benzyltrifluoroborates 71 as a benzylic radical source for the visible-light-induced alkylation of imines 70. In the catalytic system, chiral ligands initiated benzylic radical formation and governed the subsequent stereoselective transformations. In addition, Fimognari’s group [101] utilized copper photoredox catalysts to achieve the N-desulfonylation of benzenesulfonyl-protected N-heterocycles 72 (Scheme 29).
![[1860-5397-17-169-i29]](/bjoc/content/inline/1860-5397-17-169-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Other copper-photocatalyzed reactions.
Scheme 29: Other copper-photocatalyzed reactions.
In 2019, Xiao’s group [102] observed that under visible light or copper catalysis, cycloketone oxime esters 73 formed cyclic iminyl radicals, which then formed cyanoalkyl radicals through a selective β-C–C bond scission. This protocol was further applied to the aminocarbonylation of cycloketone oxime esters with CO gas and amines 74. Cycloketone oxime esters are reduced by the photoexcited [LnCuI–NHR]* complex C or the ground-state LnCuI–NHR species B to generate a cyclic iminyl radical 73a-A, which oxidizes the LnCuII–NHR complex D (Scheme 30, path a or b). Subsequently, radical 73a-A undergoes a β-C–C bond scission to provide the cyanoalkyl radical 73a-B, which is trapped by complex D and converted to the high-valent CuIII complex E. Next, CO inserts into complex E to generate intermediates F or G, which undergo elimination to furnish the final product 75. In 2020, Chen and co-worker [103] further explored the potential of this method and accomplished photoinduced the copper-catalyzed C(sp3)–O cross-coupling using oxime esters and phenols 76 (Scheme 30).
![[1860-5397-17-169-i30]](/bjoc/content/inline/1860-5397-17-169-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Cross-coupling of oxime esters with phenols or amines.
Scheme 30: Cross-coupling of oxime esters with phenols or amines.
In 2020, Loh and co-workers [104] reported the copper-catalyzed highly site-selective alkylation of heteroarene N-oxides in the presence of hypervalent iodineIII carboxylates. As an alkylating agent, the hypervalent iodineIII carboxylates were reduced by active copperI complexes and produced an alkyl radical, which was then captured by a copperIII active species. Finally, after reductive elimination, the target products were obtained (Scheme 31).
![[1860-5397-17-169-i31]](/bjoc/content/inline/1860-5397-17-169-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Alkylation of heteroarene N-oxides.
Scheme 31: Alkylation of heteroarene N-oxides.
Conclusion
This review highlighted the special features and applications of photoinduced copper-catalyzed reactions. Copper photoredox catalysts are powerful photocatalysts used for cross-coupling reactions. Their function is based on the strong reducing power of copper complexes and the ability of copper complexes to coordinate substrates or trap reactive intermediates. The applications of photoinduced copper-catalyzed reactions include alkene/alkyne functionalization, organic halide functionalization, and alkyl C–H functionalization. This review introduced the photoinduced copper-catalyzed stereoselective reactions within these broad reaction categories. Copper salts coordinate with diverse chiral ligands to provide a chiral environment for asymmetric control. Despite the remarkable achievements in this field, copper-based catalytic asymmetric reactions still remain a challenging task because of the difficulty of stereocontrol of the highly reactive radical intermediates. This review discussed the fundamental mechanisms underlying copper-based photocatalysis, including CuI/CuII-mediated and copper substrate-mediated catalytic cycles, which are important in metallo-photoredox mechanisms. The excited-state properties of Cu-based photosensitizers can be efficiently tuned by ligand modification. Although remarkable efforts have been made to elucidate and modify Cu complexes as photoredox catalysts for organic synthesis, the design of these complexes has not received much attention. If new complexes with improved redox and photophysical performances are designed, then Cu-based complexes could replace ruthenium- or iridium-based photocatalysts in the future.
References
-
Deisenhofer, J.; Epp, O.; Miki, K.; Huber, R.; Michel, H. Nature 1985, 318, 618–624. doi:10.1038/318618a0
Return to citation in text: [1] -
Baranoff, E.; Collin, J. P.; Flamigni, L.; Sauvage, J. P. Chem. Soc. Rev. 2004, 33, 147–155. doi:10.1039/b308983e
Return to citation in text: [1] -
Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. doi:10.1002/anie.201709766
Return to citation in text: [1] -
Meyer, T. J. Acc. Chem. Res. 1989, 22, 163–170. doi:10.1021/ar00161a001
Return to citation in text: [1] -
Balzani, V.; Credi, A.; Venturi, M. ChemSusChem 2008, 1, 26–58. doi:10.1002/cssc.200700087
Return to citation in text: [1] -
Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A. Chem. Rev. 2007, 107, 2725–2756. doi:10.1002/chin.200737237
Return to citation in text: [1] -
Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056
Return to citation in text: [1] -
Teplý, F. Collect. Czech. Chem. Commun. 2011, 76, 859–917. doi:10.1135/cccc2011078
Return to citation in text: [1] -
Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] -
Ravelli, D.; Fagnoni, M. ChemCatChem 2012, 4, 169–171. doi:10.1002/cctc.201100363
Return to citation in text: [1] -
Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449
Return to citation in text: [1] -
Musacchio, A. J.; Nguyen, L. Q.; Beard, G. H.; Knowles, R. R. J. Am. Chem. Soc. 2014, 136, 12217–12220. doi:10.1021/ja5056774
Return to citation in text: [1] -
Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057
Return to citation in text: [1] -
Hari, D. P.; Knig, B. Chem. Commun. 2014, 50, 6688–6699. doi:10.1039/c4cc00751d
Return to citation in text: [1] -
Fukuzumi, S.; Ohkubo, K. Org. Biomol. Chem. 2014, 12, 6059–6071. doi:10.1039/c4ob00843j
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Angerani, S.; Winssinger, N. Chem. – Eur. J. 2019, 25, 6661–6672. doi:10.1002/chem.201806024
Return to citation in text: [1] -
You, Y.-M.; Nam, W. Chem. Soc. Rev. 2012, 41, 7061–7084. doi:10.1039/c2cs35171d
Return to citation in text: [1] -
Marin, V.; Holder, E.; Hoogenboom, R.; Schubert, U. S. Chem. Soc. Rev. 2007, 36, 618–635. doi:10.1039/b610016c
Return to citation in text: [1] -
Paria, S.; Reiser, O. ChemCatChem 2014, 6, 2477–2483. doi:10.1002/cctc.201402237
Return to citation in text: [1] [2] [3] [4] -
Armaroli, N. Chem. Soc. Rev. 2001, 30, 113–124. doi:10.1039/b000703j
Return to citation in text: [1] [2] [3] [4] [5] -
Lang, X.-J.; Zhao, J.-C.; Chen, X.-D. Chem. Soc. Rev. 2016, 45, 3026–3038. doi:10.1039/c5cs00659g
Return to citation in text: [1] -
Nicholls, T. P.; Bissember, A. C. Tetrahedron Lett. 2019, 60, 150883–150893. doi:10.1016/j.tetlet.2019.06.042
Return to citation in text: [1] -
Wang, C.-S.; Dixneuf, P. H.; Soulé, J.-F. Chem. Rev. 2018, 118, 7532–7585. doi:10.1021/acs.chemrev.8b00077
Return to citation in text: [1] -
Akita, M.; Koike, T. J. Synth. Org. Chem., Jpn. 2016, 74, 1036–1046. doi:10.5059/yukigoseikyokaishi.74.1036
Return to citation in text: [1] -
Hopkinson, M. N.; Sahoo, B.; Li, J.-L.; Glorius, F. Chem. – Eur. J. 2014, 20, 3874–3886. doi:10.1002/chem.201304823
Return to citation in text: [1] -
Reiser, O. Acc. Chem. Res. 2016, 49, 1990–1996. doi:10.1021/acs.accounts.6b00296
Return to citation in text: [1] [2] [3] -
Hernandez-Perez, A. C.; Collins, S. K. Acc. Chem. Res. 2016, 49, 1557–1565. doi:10.1021/acs.accounts.6b00250
Return to citation in text: [1] -
Wang, B.; Shelar, D. P.; Han, X.-Z.; Li, T.-T.; Guan, X.-G.; Lu, W.; Liu, K.; Chen, Y.; Fu, W.-F.; he, C.-M. Chem. – Eur. J. 2014, 20, 1–8. doi:10.1002/chem.201405356
Return to citation in text: [1] [2] [3] -
Cuttell, D. G.; Kuang, S.-M.; Fanwick, P. E.; McMillin, D. R.; Walton, R. A. J. Am. Chem. Soc. 2002, 124, 6–7. doi:10.1021/ja012247h
Return to citation in text: [1] [2] -
Lavie-Cambot, A.; Cantuel, M.; Leydet, Y.; Jonusauskas, G.; Bassani, D. M.; McClenaghan, N. D. Chem. Rev. 2008, 252, 2572–2584. doi:10.1016/j.ccr.2008.03.013
Return to citation in text: [1] -
Khnayzer, R. S.; McCusker, C. E.; Olaiya, B. S.; Castellano, F. N. J. Am. Chem. Soc. 2013, 135, 14068–14070. doi:10.1021/ja407816f
Return to citation in text: [1] -
Czerwieniec, R.; Kowalski, K.; Yersin, H. Dalton Trans. 2013, 42, 9826–9831. doi:10.1039/c3dt51006a
Return to citation in text: [1] -
Smith, C. S.; Branham, C. W.; Marquardt, B. J.; Mann, K. R. J. Am. Chem. Soc. 2010, 132, 14079–14085. doi:10.1021/ja103112m
Return to citation in text: [1] -
Knorn, M.; Rawner, T.; Czerwieniec, R.; Reiser, O. ACS Catal. 2015, 5, 5186–5193. doi:10.1021/acscatal.5b01071
Return to citation in text: [1] -
Hossain, A.; Bhattacharyya, A.; Reiser, O. Science 2019, 364, eaav9713. doi:10.1126/science.aav9713
Return to citation in text: [1] [2] -
Mitani, M.; Kato, I.; Koyama, K. J. Am. Chem. Soc. 1983, 105, 6719–6721. doi:10.1021/ja00360a033
Return to citation in text: [1] [2] -
Abderrazak, Y.; Bhattacharyya, A.; Reiser, O. Angew. Chem., Int. Ed. 2021, 60, 21100–21115. doi:10.1002/anie.202100270
Return to citation in text: [1] [2] -
Xiong, Y.; Sun, Y.-W.; Zhang, G.-Z. Org. Lett. 2018, 20, 6250–6254. doi:10.1021/acs.orglett.8b02735
Return to citation in text: [1] -
Ahn, J. M.; Ratani, T. S.; Hannoun, K. I.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2017, 139, 12716–12723. doi:10.1021/jacs.7b07052
Return to citation in text: [1] [2] [3] -
Creutz, S. E.; Lotito, K. J.; Fu, G. C.; Peters, J. C. Science 2012, 338, 647–651. doi:10.1126/science.1226458
Return to citation in text: [1] [2] -
Li, Y.-Y.; Zhou, K.-X.; Wen, Z.-R.; Cao, S.; Shen, X.; Lei, M.; Gong, L. J. Am. Chem. Soc. 2018, 140, 15850–15858. doi:10.1021/jacs.8b09251
Return to citation in text: [1] [2] -
Pirtsch, M.; Paria, S.; Matsuno, T.; Isobe, H.; Reiser, O. Chem. – Eur. J. 2012, 18, 7336–7340. doi:10.1002/chem.201200967
Return to citation in text: [1] -
Baralle, A.; Fensterbank, L.; Goddard, J.-P.; Ollivier, C. Chem. – Eur. J. 2013, 19, 10809–10813. doi:10.1002/chem.201301449
Return to citation in text: [1] -
Lei, W.-L.; Wang, T.; Feng, K.-W.; Wu, L.-Z.; Liu, Q. ACS Catal. 2017, 7, 7941–7945. doi:10.1021/acscatal.7b02818
Return to citation in text: [1] -
Bagal, D. B.; Kachkovskyi, G.; Knorn, M.; Rawner, T.; Bhanage, B. M.; Reiser, O. Angew. Chem., Int. Ed. 2015, 54, 6999–7002. doi:10.1002/anie.201501880
Return to citation in text: [1] -
Rawner, T.; Knorn, M.; Lutsker, E.; Hossain, A.; Reiser, O. J. Org. Chem. 2016, 81, 7139–7147. doi:10.1021/acs.joc.6b01001
Return to citation in text: [1] -
Hossain, A.; Engl, S.; Lutsker, E.; Reiser, O. ACS Catal. 2019, 9, 1103–1109. doi:10.1021/acscatal.8b04188
Return to citation in text: [1] -
Engl, S.; Reiser, O. Eur. J. Org. Chem. 2020, 1523–1533. doi:10.1002/ejoc.201900839
Return to citation in text: [1] -
Alkan-Zambada, M.; Hu, X. J. Org. Chem. 2019, 84, 4525–4533. doi:10.1021/acs.joc.9b00238
Return to citation in text: [1] -
Rawner, T.; Lutsker, E.; Kaiser, C. A.; Reiser, O. ACS Catal. 2018, 8, 3950–3956. doi:10.1021/acscatal.8b00847
Return to citation in text: [1] -
Engl, S.; Reiser, O. ACS Catal. 2020, 10, 9899–9906. doi:10.1021/acscatal.0c02984
Return to citation in text: [1] -
Guo, Q.; Wang, M.; Wang, Y.; Xu, Z.; Wang, R. Chem. Commun. 2017, 53, 12317–12320. doi:10.1039/c7cc07128k
Return to citation in text: [1] -
Michelet, B.; Deldaele, C.; Kajouj, S.; Moucheron, C.; Evano, G. Org. Lett. 2017, 19, 3576–3579. doi:10.1021/acs.orglett.7b01518
Return to citation in text: [1] [2] [3] -
Guo, Q.; Wang, M.; Peng, Q.; Huo, Y.; Liu, Q.; Wang, R.; Xu, Z. ACS Catal. 2019, 9, 4470–4476. doi:10.1021/acscatal.9b00209
Return to citation in text: [1] -
He, J.; Chen, C.; Fu, G. C.; Peters, J. C. ACS Catal. 2018, 8, 11741–11748. doi:10.1021/acscatal.8b04094
Return to citation in text: [1] -
Xiong, Y.; Ma, X.; Zhang, G. Org. Lett. 2019, 21, 1699–1703. doi:10.1021/acs.orglett.9b00252
Return to citation in text: [1] -
Xiong, Y.; Zhang, G. Org. Lett. 2019, 21, 7873–7877. doi:10.1021/acs.orglett.9b02863
Return to citation in text: [1] -
Zhang, Y.; Sun, Y.; Chen, B.; Xu, M.; Li, C.; Zhang, D.; Zhang, G. Org. Lett. 2020, 22, 1490–1494. doi:10.1021/acs.orglett.0c00071
Return to citation in text: [1] -
Chen, J.; He, B.-Q.; Wang, P.-Z.; Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.-R.; Xiao, W.-J. Org. Lett. 2019, 21, 4359–4364. doi:10.1021/acs.orglett.9b01529
Return to citation in text: [1] -
Lou, J.; Ma, J.; Xu, B.-H.; Zhou, Y.-G.; Yu, Z. Org. Lett. 2020, 22, 5202–5206. doi:10.1021/acs.orglett.0c01645
Return to citation in text: [1] -
Hossain, A.; Vidyasagar, A.; Eichinger, C.; Lankes, C.; Phan, J.; Rehbein, J.; Reiser, O. Angew. Chem., Int. Ed. 2018, 57, 8288–8292. doi:10.1002/anie.201801678
Return to citation in text: [1] -
Wu, D.; Cui, S.-S.; Lin, Y.; Li, L.; Yu, W. J. Org. Chem. 2019, 84, 10978–10989. doi:10.1021/acs.joc.9b01569
Return to citation in text: [1] -
Sagadevan, A.; Hwang, K. C. Adv. Synth. Catal. 2012, 354, 3421–3427. doi:10.1002/adsc.201200683
Return to citation in text: [1] -
Hazra, A.; Lee, M. T.; Chiu, J. F.; Lalic, G. Angew. Chem., Int. Ed. 2018, 57, 5492–5496. doi:10.1002/anie.201801085
Return to citation in text: [1] -
Sagadevan, A.; Charpe, V. P.; Ragupathi, A.; Hwang, K. C. J. Am. Chem. Soc. 2017, 139, 2896–2899. doi:10.1021/jacs.6b13113
Return to citation in text: [1] -
Sagadevan, A.; Ragupathi, A.; Lin, C.-C.; Hwu, J. R.; Hwang, K. C. Green Chem. 2015, 17, 1113–1119. doi:10.1039/c4gc01623h
Return to citation in text: [1] -
Ragupathi, A.; Sagadevan, A.; Lin, C.-C.; Hwu, J.-R.; Hwang, K. C. Chem. Commun. 2016, 52, 11756–11759. doi:10.1039/c6cc05506k
Return to citation in text: [1] -
Ragupathi, A.; Sagadevan, A.; Charpe, V. P.; Lin, C.-C.; Hwu, J.-R.; Hwang, K. C. Chem. Commun. 2019, 55, 5151–5154. doi:10.1039/c9cc01801h
Return to citation in text: [1] -
Sagadevan, A.; Pampana, V. K. K.; Hwang, K. C. Angew. Chem., Int. Ed. 2019, 58, 3838–3842. doi:10.1002/anie.201813315
Return to citation in text: [1] -
Sagadevan, A.; Ragupathi, A.; Hwang, K. C. Angew. Chem. 2015, 127, 14102–14107. doi:10.1002/ange.201506579
Return to citation in text: [1] -
Charpe, V. P.; Sagadevan, A.; Hwang, K. C. Green Chem. 2020, 22, 4426–4432. doi:10.1039/d0gc00975j
Return to citation in text: [1] -
Xiao, P.; Li, C.-X.; Fang, W.-H.; Cui, G.; Thiel, W. J. Am. Chem. Soc. 2018, 140, 15099–15113. doi:10.1021/jacs.8b10387
Return to citation in text: [1] -
Sagadevan, A.; Lyu, P.-C.; Hwang, K. C. Green Chem. 2016, 18, 4526–4530. doi:10.1039/c6gc01463a
Return to citation in text: [1] -
Rostoll-Berenguer, J.; Blay, G.; Pedro, J. R.; Vila, C. Synthesis 2020, 52, 544–552. doi:10.1055/s-0039-1690244
Return to citation in text: [1] -
Zhang, Y.; Zhang, D. Org. Biomol. Chem. 2020, 18, 4479–4483. doi:10.1039/d0ob00835d
Return to citation in text: [1] -
Xia, H.-D.; Li, Z.-L.; Gu, Q.-S.; Dong, X.-Y.; Fang, J.-H.; Du, X.-Y.; Wang, L.-L.; Liu, X.-Y. Angew. Chem., Int. Ed. 2020, 59, 16926–16932. doi:10.1002/anie.202006317
Return to citation in text: [1] -
Chatterjee, A.; König, B.; Natarajan, P. ChemPhotoChem 2020, 4, 291–293. doi:10.1002/cptc.201900266
Return to citation in text: [1] -
Reddy, M. B.; Anandhan, R. Chem. Commun. 2020, 56, 3781–3784. doi:10.1039/d0cc00815j
Return to citation in text: [1] -
Bissember, A. C.; Lundgren, R. J.; Creutz, S. E.; Peters, J. C.; Fu, G. C. Angew. Chem. 2013, 125, 5233–5237. doi:10.1002/ange.201301202
Return to citation in text: [1] -
Kainz, Q. M.; Matier, C. D.; Bartoszewicz, A.; Zultanski, S. L.; Peters, J. C.; Fu, G. C. Science 2016, 351, 681–684. doi:10.1126/science.aad8313
Return to citation in text: [1] -
Ahn, J. M.; Peters, J. C.; Fu, G. C. J. Am. Chem. Soc. 2017, 139, 18101–18106. doi:10.1021/jacs.7b10907
Return to citation in text: [1] -
Matier, C. D.; Schwaben, J.; Peters, J. C.; Fu, G. C. J. Am. Chem. Soc. 2017, 139, 17707–17710. doi:10.1021/jacs.7b09582
Return to citation in text: [1] -
Tan, Y.; Muñoz-Molina, J. M.; Fu, G. C.; Peters, J. C. Chem. Sci. 2014, 5, 2831–2835. doi:10.1039/c4sc00368c
Return to citation in text: [1] [2] -
Uyeda, C.; Tan, Y.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2013, 135, 9548–9552. doi:10.1021/ja404050f
Return to citation in text: [1] [2] -
Ratani, T. S.; Bachman, S.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2015, 137, 13902–13907. doi:10.1021/jacs.5b08452
Return to citation in text: [1] [2] -
Yang, F.; Koeller, J.; Ackermann, L. Angew. Chem., Int. Ed. 2016, 55, 4759–4762. doi:10.1002/anie.201512027
Return to citation in text: [1] [2] -
Ma, X.; Zhang, G. Chin. J. Chem. 2020, 38, 1299–1303. doi:10.1002/cjoc.201900527
Return to citation in text: [1] [2] -
Yu, F.; Dickson, J. L.; Loka, R. S.; Xu, H.; Schaugaard, R. N.; Schlegel, H. B.; Luo, L.; Nguyen, H. M. ACS Catal. 2020, 10, 5990–6001. doi:10.1021/acscatal.0c01470
Return to citation in text: [1] [2] -
Nicholls, T. P.; Robertson, J. C.; Gardiner, M. G.; Bissember, A. C. Chem. Commun. 2018, 54, 4589–4592. doi:10.1039/c8cc02244e
Return to citation in text: [1] [2] -
Rabet, P. T. G.; Fumagalli, G.; Boyd, S.; Greaney, M. F. Org. Lett. 2016, 18, 1646–1649. doi:10.1021/acs.orglett.6b00512
Return to citation in text: [1] -
Nicholls, T. P.; Constable, G. E.; Robertson, J. C.; Gardiner, M. G.; Bissember, A. C. ACS Catal. 2016, 6, 451–457. doi:10.1021/acscatal.5b02014
Return to citation in text: [1] -
Meng, Q.-Y.; Gao, X.-W.; Lei, T.; Liu, Z.; Zhan, F.; Li, Z.-J.; Zhong, J.-J.; Xiao, H.; Feng, K.; Chen, B.; Tao, Y.; Tung, C.-H.; Wu, L.-Z. Sci. Adv. 2017, 3, e1700666. doi:10.1126/sciadv.1700666
Return to citation in text: [1] -
Wang, C.; Guo, M.; Qi, R.; Shang, Q.; Liu, Q.; Wang, S.; Zhao, L.; Wang, R.; Xu, Z. Angew. Chem., Int. Ed. 2018, 57, 15841–15846. doi:10.1002/anie.201809400
Return to citation in text: [1] -
Wang, C.; Yu, Y.; Liu, W.-L.; Duan, W.-L. Org. Lett. 2019, 21, 9147–9152. doi:10.1021/acs.orglett.9b03524
Return to citation in text: [1] -
Zheng, Y.-W.; Narobe, R.; Donabauer, K.; Yakubov, S.; König, B. ACS Catal. 2020, 10, 8582–8589. doi:10.1021/acscatal.0c01924
Return to citation in text: [1] -
Reddy, M. B.; Prasanth, K.; Anandhan, R. Org. Biomol. Chem. 2020, 18, 9601–9605. doi:10.1039/d0ob02234a
Return to citation in text: [1] -
Zhao, W.; Wurz, R. P.; Peters, J. C.; Fu, G. C. J. Am. Chem. Soc. 2017, 139, 12153–12156. doi:10.1021/jacs.7b07546
Return to citation in text: [1] -
Lyu, X.-L.; Huang, S.-S.; Song, H.-J.; Liu, Y.-X.; Wang, Q.-M. Org. Lett. 2019, 21, 5728–5732. doi:10.1021/acs.orglett.9b02105
Return to citation in text: [1] -
Hunter, C. J.; Boyd, M. J.; May, G. D.; Fimognari, R. J. Org. Chem. 2020, 85, 8732–8739. doi:10.1021/acs.joc.0c00983
Return to citation in text: [1] -
Lu, B.; Cheng, Y.; Chen, L.-Y.; Chen, J.-R.; Xiao, W.-J. ACS Catal. 2019, 9, 8159–8164. doi:10.1021/acscatal.9b02830
Return to citation in text: [1] -
Yu, X.-Y.; Chen, J.; Chen, H.-W.; Xiao, W.-J.; Chen, J.-R. Org. Lett. 2020, 22, 2333–2338. doi:10.1021/acs.orglett.0c00532
Return to citation in text: [1] -
Liu, D.-Y.; Liu, X.; Gao, Y.; Wang, C.-Q.; Tian, J.-S.; Loh, T.-P. Org. Lett. 2020, 22, 8978–8983. doi:10.1021/acs.orglett.0c03382
Return to citation in text: [1]
| 46. | Lei, W.-L.; Wang, T.; Feng, K.-W.; Wu, L.-Z.; Liu, Q. ACS Catal. 2017, 7, 7941–7945. doi:10.1021/acscatal.7b02818 |
| 47. | Bagal, D. B.; Kachkovskyi, G.; Knorn, M.; Rawner, T.; Bhanage, B. M.; Reiser, O. Angew. Chem., Int. Ed. 2015, 54, 6999–7002. doi:10.1002/anie.201501880 |
| 88. | Yang, F.; Koeller, J.; Ackermann, L. Angew. Chem., Int. Ed. 2016, 55, 4759–4762. doi:10.1002/anie.201512027 |
| 48. | Rawner, T.; Knorn, M.; Lutsker, E.; Hossain, A.; Reiser, O. J. Org. Chem. 2016, 81, 7139–7147. doi:10.1021/acs.joc.6b01001 |
| 89. | Ma, X.; Zhang, G. Chin. J. Chem. 2020, 38, 1299–1303. doi:10.1002/cjoc.201900527 |
| 85. | Tan, Y.; Muñoz-Molina, J. M.; Fu, G. C.; Peters, J. C. Chem. Sci. 2014, 5, 2831–2835. doi:10.1039/c4sc00368c |
| 90. | Yu, F.; Dickson, J. L.; Loka, R. S.; Xu, H.; Schaugaard, R. N.; Schlegel, H. B.; Luo, L.; Nguyen, H. M. ACS Catal. 2020, 10, 5990–6001. doi:10.1021/acscatal.0c01470 |
| 86. | Uyeda, C.; Tan, Y.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2013, 135, 9548–9552. doi:10.1021/ja404050f |
| 56. | Guo, Q.; Wang, M.; Peng, Q.; Huo, Y.; Liu, Q.; Wang, R.; Xu, Z. ACS Catal. 2019, 9, 4470–4476. doi:10.1021/acscatal.9b00209 |
| 57. | He, J.; Chen, C.; Fu, G. C.; Peters, J. C. ACS Catal. 2018, 8, 11741–11748. doi:10.1021/acscatal.8b04094 |
| 54. | Guo, Q.; Wang, M.; Wang, Y.; Xu, Z.; Wang, R. Chem. Commun. 2017, 53, 12317–12320. doi:10.1039/c7cc07128k |
| 94. | Meng, Q.-Y.; Gao, X.-W.; Lei, T.; Liu, Z.; Zhan, F.; Li, Z.-J.; Zhong, J.-J.; Xiao, H.; Feng, K.; Chen, B.; Tao, Y.; Tung, C.-H.; Wu, L.-Z. Sci. Adv. 2017, 3, e1700666. doi:10.1126/sciadv.1700666 |
| 55. | Michelet, B.; Deldaele, C.; Kajouj, S.; Moucheron, C.; Evano, G. Org. Lett. 2017, 19, 3576–3579. doi:10.1021/acs.orglett.7b01518 |
| 51. | Alkan-Zambada, M.; Hu, X. J. Org. Chem. 2019, 84, 4525–4533. doi:10.1021/acs.joc.9b00238 |
| 92. | Rabet, P. T. G.; Fumagalli, G.; Boyd, S.; Greaney, M. F. Org. Lett. 2016, 18, 1646–1649. doi:10.1021/acs.orglett.6b00512 |
| 52. | Rawner, T.; Lutsker, E.; Kaiser, C. A.; Reiser, O. ACS Catal. 2018, 8, 3950–3956. doi:10.1021/acscatal.8b00847 |
| 53. | Engl, S.; Reiser, O. ACS Catal. 2020, 10, 9899–9906. doi:10.1021/acscatal.0c02984 |
| 93. | Nicholls, T. P.; Constable, G. E.; Robertson, J. C.; Gardiner, M. G.; Bissember, A. C. ACS Catal. 2016, 6, 451–457. doi:10.1021/acscatal.5b02014 |
| 49. | Hossain, A.; Engl, S.; Lutsker, E.; Reiser, O. ACS Catal. 2019, 9, 1103–1109. doi:10.1021/acscatal.8b04188 |
| 55. | Michelet, B.; Deldaele, C.; Kajouj, S.; Moucheron, C.; Evano, G. Org. Lett. 2017, 19, 3576–3579. doi:10.1021/acs.orglett.7b01518 |
| 28. | Reiser, O. Acc. Chem. Res. 2016, 49, 1990–1996. doi:10.1021/acs.accounts.6b00296 |
| 50. | Engl, S.; Reiser, O. Eur. J. Org. Chem. 2020, 1523–1533. doi:10.1002/ejoc.201900839 |
| 91. | Nicholls, T. P.; Robertson, J. C.; Gardiner, M. G.; Bissember, A. C. Chem. Commun. 2018, 54, 4589–4592. doi:10.1039/c8cc02244e |
| 58. | Xiong, Y.; Ma, X.; Zhang, G. Org. Lett. 2019, 21, 1699–1703. doi:10.1021/acs.orglett.9b00252 |
| 41. | Ahn, J. M.; Ratani, T. S.; Hannoun, K. I.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2017, 139, 12716–12723. doi:10.1021/jacs.7b07052 |
| 99. | Zhao, W.; Wurz, R. P.; Peters, J. C.; Fu, G. C. J. Am. Chem. Soc. 2017, 139, 12153–12156. doi:10.1021/jacs.7b07546 |
| 59. | Xiong, Y.; Zhang, G. Org. Lett. 2019, 21, 7873–7877. doi:10.1021/acs.orglett.9b02863 |
| 100. | Lyu, X.-L.; Huang, S.-S.; Song, H.-J.; Liu, Y.-X.; Wang, Q.-M. Org. Lett. 2019, 21, 5728–5732. doi:10.1021/acs.orglett.9b02105 |
| 97. | Zheng, Y.-W.; Narobe, R.; Donabauer, K.; Yakubov, S.; König, B. ACS Catal. 2020, 10, 8582–8589. doi:10.1021/acscatal.0c01924 |
| 98. | Reddy, M. B.; Prasanth, K.; Anandhan, R. Org. Biomol. Chem. 2020, 18, 9601–9605. doi:10.1039/d0ob02234a |
| 95. | Wang, C.; Guo, M.; Qi, R.; Shang, Q.; Liu, Q.; Wang, S.; Zhao, L.; Wang, R.; Xu, Z. Angew. Chem., Int. Ed. 2018, 57, 15841–15846. doi:10.1002/anie.201809400 |
| 96. | Wang, C.; Yu, Y.; Liu, W.-L.; Duan, W.-L. Org. Lett. 2019, 21, 9147–9152. doi:10.1021/acs.orglett.9b03524 |
| 67. | Sagadevan, A.; Charpe, V. P.; Ragupathi, A.; Hwang, K. C. J. Am. Chem. Soc. 2017, 139, 2896–2899. doi:10.1021/jacs.6b13113 |
| 68. | Sagadevan, A.; Ragupathi, A.; Lin, C.-C.; Hwu, J. R.; Hwang, K. C. Green Chem. 2015, 17, 1113–1119. doi:10.1039/c4gc01623h |
| 69. | Ragupathi, A.; Sagadevan, A.; Lin, C.-C.; Hwu, J.-R.; Hwang, K. C. Chem. Commun. 2016, 52, 11756–11759. doi:10.1039/c6cc05506k |
| 70. | Ragupathi, A.; Sagadevan, A.; Charpe, V. P.; Lin, C.-C.; Hwu, J.-R.; Hwang, K. C. Chem. Commun. 2019, 55, 5151–5154. doi:10.1039/c9cc01801h |
| 71. | Sagadevan, A.; Pampana, V. K. K.; Hwang, K. C. Angew. Chem., Int. Ed. 2019, 58, 3838–3842. doi:10.1002/anie.201813315 |
| 72. | Sagadevan, A.; Ragupathi, A.; Hwang, K. C. Angew. Chem. 2015, 127, 14102–14107. doi:10.1002/ange.201506579 |
| 73. | Charpe, V. P.; Sagadevan, A.; Hwang, K. C. Green Chem. 2020, 22, 4426–4432. doi:10.1039/d0gc00975j |
| 65. | Sagadevan, A.; Hwang, K. C. Adv. Synth. Catal. 2012, 354, 3421–3427. doi:10.1002/adsc.201200683 |
| 66. | Hazra, A.; Lee, M. T.; Chiu, J. F.; Lalic, G. Angew. Chem., Int. Ed. 2018, 57, 5492–5496. doi:10.1002/anie.201801085 |
| 63. | Hossain, A.; Vidyasagar, A.; Eichinger, C.; Lankes, C.; Phan, J.; Rehbein, J.; Reiser, O. Angew. Chem., Int. Ed. 2018, 57, 8288–8292. doi:10.1002/anie.201801678 |
| 102. | Lu, B.; Cheng, Y.; Chen, L.-Y.; Chen, J.-R.; Xiao, W.-J. ACS Catal. 2019, 9, 8159–8164. doi:10.1021/acscatal.9b02830 |
| 64. | Wu, D.; Cui, S.-S.; Lin, Y.; Li, L.; Yu, W. J. Org. Chem. 2019, 84, 10978–10989. doi:10.1021/acs.joc.9b01569 |
| 103. | Yu, X.-Y.; Chen, J.; Chen, H.-W.; Xiao, W.-J.; Chen, J.-R. Org. Lett. 2020, 22, 2333–2338. doi:10.1021/acs.orglett.0c00532 |
| 60. | Zhang, Y.; Sun, Y.; Chen, B.; Xu, M.; Li, C.; Zhang, D.; Zhang, G. Org. Lett. 2020, 22, 1490–1494. doi:10.1021/acs.orglett.0c00071 |
| 43. | Li, Y.-Y.; Zhou, K.-X.; Wen, Z.-R.; Cao, S.; Shen, X.; Lei, M.; Gong, L. J. Am. Chem. Soc. 2018, 140, 15850–15858. doi:10.1021/jacs.8b09251 |
| 61. | Chen, J.; He, B.-Q.; Wang, P.-Z.; Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.-R.; Xiao, W.-J. Org. Lett. 2019, 21, 4359–4364. doi:10.1021/acs.orglett.9b01529 |
| 62. | Lou, J.; Ma, J.; Xu, B.-H.; Zhou, Y.-G.; Yu, Z. Org. Lett. 2020, 22, 5202–5206. doi:10.1021/acs.orglett.0c01645 |
| 101. | Hunter, C. J.; Boyd, M. J.; May, G. D.; Fimognari, R. J. Org. Chem. 2020, 85, 8732–8739. doi:10.1021/acs.joc.0c00983 |
| 75. | Sagadevan, A.; Lyu, P.-C.; Hwang, K. C. Green Chem. 2016, 18, 4526–4530. doi:10.1039/c6gc01463a |
| 76. | Rostoll-Berenguer, J.; Blay, G.; Pedro, J. R.; Vila, C. Synthesis 2020, 52, 544–552. doi:10.1055/s-0039-1690244 |
| 74. | Xiao, P.; Li, C.-X.; Fang, W.-H.; Cui, G.; Thiel, W. J. Am. Chem. Soc. 2018, 140, 15099–15113. doi:10.1021/jacs.8b10387 |
| 1. | Deisenhofer, J.; Epp, O.; Miki, K.; Huber, R.; Michel, H. Nature 1985, 318, 618–624. doi:10.1038/318618a0 |
| 104. | Liu, D.-Y.; Liu, X.; Gao, Y.; Wang, C.-Q.; Tian, J.-S.; Loh, T.-P. Org. Lett. 2020, 22, 8978–8983. doi:10.1021/acs.orglett.0c03382 |
| 13. | Musacchio, A. J.; Nguyen, L. Q.; Beard, G. H.; Knowles, R. R. J. Am. Chem. Soc. 2014, 136, 12217–12220. doi:10.1021/ja5056774 |
| 32. | Lavie-Cambot, A.; Cantuel, M.; Leydet, Y.; Jonusauskas, G.; Bassani, D. M.; McClenaghan, N. D. Chem. Rev. 2008, 252, 2572–2584. doi:10.1016/j.ccr.2008.03.013 |
| 33. | Khnayzer, R. S.; McCusker, C. E.; Olaiya, B. S.; Castellano, F. N. J. Am. Chem. Soc. 2013, 135, 14068–14070. doi:10.1021/ja407816f |
| 82. | Kainz, Q. M.; Matier, C. D.; Bartoszewicz, A.; Zultanski, S. L.; Peters, J. C.; Fu, G. C. Science 2016, 351, 681–684. doi:10.1126/science.aad8313 |
| 10. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 11. | Ravelli, D.; Fagnoni, M. ChemCatChem 2012, 4, 169–171. doi:10.1002/cctc.201100363 |
| 12. | Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449 |
| 30. | Wang, B.; Shelar, D. P.; Han, X.-Z.; Li, T.-T.; Guan, X.-G.; Lu, W.; Liu, K.; Chen, Y.; Fu, W.-F.; he, C.-M. Chem. – Eur. J. 2014, 20, 1–8. doi:10.1002/chem.201405356 |
| 34. | Czerwieniec, R.; Kowalski, K.; Yersin, H. Dalton Trans. 2013, 42, 9826–9831. doi:10.1039/c3dt51006a |
| 35. | Smith, C. S.; Branham, C. W.; Marquardt, B. J.; Mann, K. R. J. Am. Chem. Soc. 2010, 132, 14079–14085. doi:10.1021/ja103112m |
| 6. | Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A. Chem. Rev. 2007, 107, 2725–2756. doi:10.1002/chin.200737237 |
| 7. | Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056 |
| 8. | Teplý, F. Collect. Czech. Chem. Commun. 2011, 76, 859–917. doi:10.1135/cccc2011078 |
| 9. | Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223 |
| 28. | Reiser, O. Acc. Chem. Res. 2016, 49, 1990–1996. doi:10.1021/acs.accounts.6b00296 |
| 30. | Wang, B.; Shelar, D. P.; Han, X.-Z.; Li, T.-T.; Guan, X.-G.; Lu, W.; Liu, K.; Chen, Y.; Fu, W.-F.; he, C.-M. Chem. – Eur. J. 2014, 20, 1–8. doi:10.1002/chem.201405356 |
| 81. | Bissember, A. C.; Lundgren, R. J.; Creutz, S. E.; Peters, J. C.; Fu, G. C. Angew. Chem. 2013, 125, 5233–5237. doi:10.1002/ange.201301202 |
| 2. | Baranoff, E.; Collin, J. P.; Flamigni, L.; Sauvage, J. P. Chem. Soc. Rev. 2004, 33, 147–155. doi:10.1039/b308983e |
| 3. | Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. doi:10.1002/anie.201709766 |
| 4. | Meyer, T. J. Acc. Chem. Res. 1989, 22, 163–170. doi:10.1021/ar00161a001 |
| 5. | Balzani, V.; Credi, A.; Venturi, M. ChemSusChem 2008, 1, 26–58. doi:10.1002/cssc.200700087 |
| 22. | Armaroli, N. Chem. Soc. Rev. 2001, 30, 113–124. doi:10.1039/b000703j |
| 31. | Cuttell, D. G.; Kuang, S.-M.; Fanwick, P. E.; McMillin, D. R.; Walton, R. A. J. Am. Chem. Soc. 2002, 124, 6–7. doi:10.1021/ja012247h |
| 41. | Ahn, J. M.; Ratani, T. S.; Hannoun, K. I.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2017, 139, 12716–12723. doi:10.1021/jacs.7b07052 |
| 42. | Creutz, S. E.; Lotito, K. J.; Fu, G. C.; Peters, J. C. Science 2012, 338, 647–651. doi:10.1126/science.1226458 |
| 21. | Paria, S.; Reiser, O. ChemCatChem 2014, 6, 2477–2483. doi:10.1002/cctc.201402237 |
| 22. | Armaroli, N. Chem. Soc. Rev. 2001, 30, 113–124. doi:10.1039/b000703j |
| 23. | Lang, X.-J.; Zhao, J.-C.; Chen, X.-D. Chem. Soc. Rev. 2016, 45, 3026–3038. doi:10.1039/c5cs00659g |
| 24. | Nicholls, T. P.; Bissember, A. C. Tetrahedron Lett. 2019, 60, 150883–150893. doi:10.1016/j.tetlet.2019.06.042 |
| 22. | Armaroli, N. Chem. Soc. Rev. 2001, 30, 113–124. doi:10.1039/b000703j |
| 28. | Reiser, O. Acc. Chem. Res. 2016, 49, 1990–1996. doi:10.1021/acs.accounts.6b00296 |
| 79. | Chatterjee, A.; König, B.; Natarajan, P. ChemPhotoChem 2020, 4, 291–293. doi:10.1002/cptc.201900266 |
| 21. | Paria, S.; Reiser, O. ChemCatChem 2014, 6, 2477–2483. doi:10.1002/cctc.201402237 |
| 22. | Armaroli, N. Chem. Soc. Rev. 2001, 30, 113–124. doi:10.1039/b000703j |
| 29. | Hernandez-Perez, A. C.; Collins, S. K. Acc. Chem. Res. 2016, 49, 1557–1565. doi:10.1021/acs.accounts.6b00250 |
| 80. | Reddy, M. B.; Anandhan, R. Chem. Commun. 2020, 56, 3781–3784. doi:10.1039/d0cc00815j |
| 17. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 18. | Angerani, S.; Winssinger, N. Chem. – Eur. J. 2019, 25, 6661–6672. doi:10.1002/chem.201806024 |
| 19. | You, Y.-M.; Nam, W. Chem. Soc. Rev. 2012, 41, 7061–7084. doi:10.1039/c2cs35171d |
| 20. | Marin, V.; Holder, E.; Hoogenboom, R.; Schubert, U. S. Chem. Soc. Rev. 2007, 36, 618–635. doi:10.1039/b610016c |
| 77. | Zhang, Y.; Zhang, D. Org. Biomol. Chem. 2020, 18, 4479–4483. doi:10.1039/d0ob00835d |
| 14. | Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057 |
| 15. | Hari, D. P.; Knig, B. Chem. Commun. 2014, 50, 6688–6699. doi:10.1039/c4cc00751d |
| 16. | Fukuzumi, S.; Ohkubo, K. Org. Biomol. Chem. 2014, 12, 6059–6071. doi:10.1039/c4ob00843j |
| 25. | Wang, C.-S.; Dixneuf, P. H.; Soulé, J.-F. Chem. Rev. 2018, 118, 7532–7585. doi:10.1021/acs.chemrev.8b00077 |
| 26. | Akita, M.; Koike, T. J. Synth. Org. Chem., Jpn. 2016, 74, 1036–1046. doi:10.5059/yukigoseikyokaishi.74.1036 |
| 27. | Hopkinson, M. N.; Sahoo, B.; Li, J.-L.; Glorius, F. Chem. – Eur. J. 2014, 20, 3874–3886. doi:10.1002/chem.201304823 |
| 78. | Xia, H.-D.; Li, Z.-L.; Gu, Q.-S.; Dong, X.-Y.; Fang, J.-H.; Du, X.-Y.; Wang, L.-L.; Liu, X.-Y. Angew. Chem., Int. Ed. 2020, 59, 16926–16932. doi:10.1002/anie.202006317 |
| 21. | Paria, S.; Reiser, O. ChemCatChem 2014, 6, 2477–2483. doi:10.1002/cctc.201402237 |
| 22. | Armaroli, N. Chem. Soc. Rev. 2001, 30, 113–124. doi:10.1039/b000703j |
| 31. | Cuttell, D. G.; Kuang, S.-M.; Fanwick, P. E.; McMillin, D. R.; Walton, R. A. J. Am. Chem. Soc. 2002, 124, 6–7. doi:10.1021/ja012247h |
| 36. | Knorn, M.; Rawner, T.; Czerwieniec, R.; Reiser, O. ACS Catal. 2015, 5, 5186–5193. doi:10.1021/acscatal.5b01071 |
| 21. | Paria, S.; Reiser, O. ChemCatChem 2014, 6, 2477–2483. doi:10.1002/cctc.201402237 |
| 30. | Wang, B.; Shelar, D. P.; Han, X.-Z.; Li, T.-T.; Guan, X.-G.; Lu, W.; Liu, K.; Chen, Y.; Fu, W.-F.; he, C.-M. Chem. – Eur. J. 2014, 20, 1–8. doi:10.1002/chem.201405356 |
| 85. | Tan, Y.; Muñoz-Molina, J. M.; Fu, G. C.; Peters, J. C. Chem. Sci. 2014, 5, 2831–2835. doi:10.1039/c4sc00368c |
| 86. | Uyeda, C.; Tan, Y.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2013, 135, 9548–9552. doi:10.1021/ja404050f |
| 83. | Ahn, J. M.; Peters, J. C.; Fu, G. C. J. Am. Chem. Soc. 2017, 139, 18101–18106. doi:10.1021/jacs.7b10907 |
| 84. | Matier, C. D.; Schwaben, J.; Peters, J. C.; Fu, G. C. J. Am. Chem. Soc. 2017, 139, 17707–17710. doi:10.1021/jacs.7b09582 |
| 44. | Pirtsch, M.; Paria, S.; Matsuno, T.; Isobe, H.; Reiser, O. Chem. – Eur. J. 2012, 18, 7336–7340. doi:10.1002/chem.201200967 |
| 45. | Baralle, A.; Fensterbank, L.; Goddard, J.-P.; Ollivier, C. Chem. – Eur. J. 2013, 19, 10809–10813. doi:10.1002/chem.201301449 |
| 41. | Ahn, J. M.; Ratani, T. S.; Hannoun, K. I.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2017, 139, 12716–12723. doi:10.1021/jacs.7b07052 |
| 42. | Creutz, S. E.; Lotito, K. J.; Fu, G. C.; Peters, J. C. Science 2012, 338, 647–651. doi:10.1126/science.1226458 |
| 90. | Yu, F.; Dickson, J. L.; Loka, R. S.; Xu, H.; Schaugaard, R. N.; Schlegel, H. B.; Luo, L.; Nguyen, H. M. ACS Catal. 2020, 10, 5990–6001. doi:10.1021/acscatal.0c01470 |
| 39. | Abderrazak, Y.; Bhattacharyya, A.; Reiser, O. Angew. Chem., Int. Ed. 2021, 60, 21100–21115. doi:10.1002/anie.202100270 |
| 43. | Li, Y.-Y.; Zhou, K.-X.; Wen, Z.-R.; Cao, S.; Shen, X.; Lei, M.; Gong, L. J. Am. Chem. Soc. 2018, 140, 15850–15858. doi:10.1021/jacs.8b09251 |
| 91. | Nicholls, T. P.; Robertson, J. C.; Gardiner, M. G.; Bissember, A. C. Chem. Commun. 2018, 54, 4589–4592. doi:10.1039/c8cc02244e |
| 39. | Abderrazak, Y.; Bhattacharyya, A.; Reiser, O. Angew. Chem., Int. Ed. 2021, 60, 21100–21115. doi:10.1002/anie.202100270 |
| 55. | Michelet, B.; Deldaele, C.; Kajouj, S.; Moucheron, C.; Evano, G. Org. Lett. 2017, 19, 3576–3579. doi:10.1021/acs.orglett.7b01518 |
| 37. | Hossain, A.; Bhattacharyya, A.; Reiser, O. Science 2019, 364, eaav9713. doi:10.1126/science.aav9713 |
| 38. | Mitani, M.; Kato, I.; Koyama, K. J. Am. Chem. Soc. 1983, 105, 6719–6721. doi:10.1021/ja00360a033 |
| 40. | Xiong, Y.; Sun, Y.-W.; Zhang, G.-Z. Org. Lett. 2018, 20, 6250–6254. doi:10.1021/acs.orglett.8b02735 |
| 89. | Ma, X.; Zhang, G. Chin. J. Chem. 2020, 38, 1299–1303. doi:10.1002/cjoc.201900527 |
| 37. | Hossain, A.; Bhattacharyya, A.; Reiser, O. Science 2019, 364, eaav9713. doi:10.1126/science.aav9713 |
| 87. | Ratani, T. S.; Bachman, S.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2015, 137, 13902–13907. doi:10.1021/jacs.5b08452 |
| 88. | Yang, F.; Koeller, J.; Ackermann, L. Angew. Chem., Int. Ed. 2016, 55, 4759–4762. doi:10.1002/anie.201512027 |
| 38. | Mitani, M.; Kato, I.; Koyama, K. J. Am. Chem. Soc. 1983, 105, 6719–6721. doi:10.1021/ja00360a033 |
| 87. | Ratani, T. S.; Bachman, S.; Fu, G. C.; Peters, J. C. J. Am. Chem. Soc. 2015, 137, 13902–13907. doi:10.1021/jacs.5b08452 |
© 2021 Zhang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)