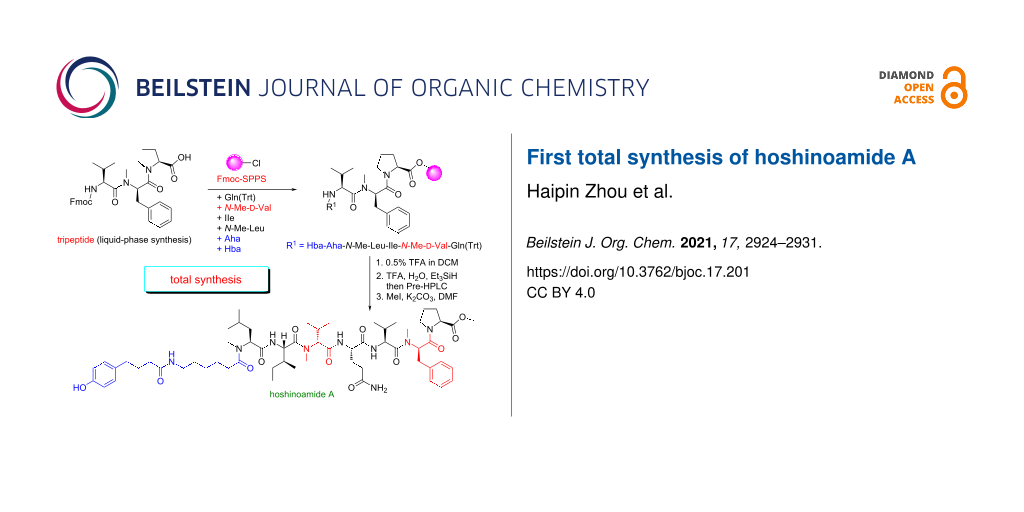
Abstract
Hoshinoamides A, B and C, linear lipopeptides, were isolated from the marine cyanobacterium Caldora penicillata, with potent antiplasmodial activity against chloroquine-sensitive Plasmodium falciparum. Herein, we describe the first total synthesis of hoshinoamide A by the combination of liquid-phase and solid-phase peptide synthesis. Liquid-phase synthesis is to improve the coupling yield of ʟ-Val3 and N-Me-ᴅ-Phe2. Connecting other amino acids efficiency and convergence is achieved by solid-state synthesis. Our synthetic strategy could synthesize the target peptide in high yield with good purity

Graphical Abstract
Introduction
Malaria is an insect-borne infectious disease caused by parasites of the genus Plasmodium, which seriously threatens human life and health [1]. Half of the world's population is at the risk of malaria, causing 200 million new infections and killing hundreds of thousands of people each year [2]. Current medicines for malaria include quinolone [3,4], folic acid antagonist [5,6] and artemisinin derivatives [7]. The emergence of drug resistance makes the efficacy of these drugs decline year by year, forcing scientists to constantly search for new antimalarial drugs [8-10].
In recent years, Iwasaki and co-workers have reported three novel linear lipopeptide natural products, hoshinoamides A, B [11] and C [12], from a microbial metabolite of marine cyanobacterium Caldora penicillata (Figure 1). Hoshinoamides A and B showed potent activities against chloroquine-sensitive Plasmodium falciparum 3D7 with IC50 values of 0.52 and 1.0 μM, respectively. Hoshinoamide C inhibited the growth of the malaria parasites (IC50 = 0.96 μM) and African sleeping sickness (IC50 = 2.9 μM). Hoshinoamide C was firstly synthesized from Boc-protected amino acids in liquid phase [12]. Both hoshinoamides A and B are highly methylated polypeptides containing three N-methyl amino acids: N-Me-ʟ-Leu7, N-Me-ᴅ-Val5 and N-Me-ᴅ/ʟ-Phe2. Hoshinoamide C includes two N-methyl amino acids: N-Me-ᴅ-Phe2 and N-Me-ᴅ-IIe5. The C-terminal Pro is functionalized as methyl ester, while the N-terminal polypeptide is linked to the long alkyl chain amino acid Aha8/Ana8/Ama7 and p-hydroxybenzoic acid Hba9/Hba8. Hoshinoamides have a relatively simple structure and therefore make an attractive target for further medicinal chemistry studies. To enable these new SAR studies, we need to develop an efficient synthetic method to provide sufficient materials firstly. Hoshinoamide A shows a better antimalarial activity and less cytotoxicity compared to hoshinoamide B. Herein, we report the initial progress on the total synthesis of hoshinomaide A.
![[1860-5397-17-201-1]](/bjoc/content/figures/1860-5397-17-201-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Structures of hoshinoamides A and B. b) Structure of hoshinoamide C.
Figure 1: a) Structures of hoshinoamides A and B. b) Structure of hoshinoamide C.
The key challenges for the total synthesis of hoshinoamide A are the coupling of highly methylated amino acids and the purification of hydrophobic peptides.
Results and Discussion
As shown in Scheme 1, we initially tested Fmoc solid-phase peptide synthesis (SPPS) [13] to get 2-chlorotrityl resin-bound Pro1-(N-Me)-Phe2 dipeptide 2 under the conditions of HCTU and DIPEA. Unfortunately, the N-Me coupling proceeded in low yield (<10%).
![[1860-5397-17-201-i1]](/bjoc/content/inline/1860-5397-17-201-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of resin-bound tripeptide 3 by SPPS. DIPEA: N,N-diisopropylethylamine; HCTU: O-(6-chloro-1-hydrocibenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphat.
Scheme 1: Synthesis of resin-bound tripeptide 3 by SPPS. DIPEA: N,N-diisopropylethylamine; HCTU: O-(6-chloro-...
In order to improve the coupling yield of the hindered peptide, we tried the condensation of Val3 with the dipeptide in solution. Firstly, Pro-OBn 5 was coupled with Fmoc-N-Me-ᴅ-Phe-OH by the treatment of HATU and DIPEA [14], giving dipeptide 6 in 83% yield (Scheme 2).
![[1860-5397-17-201-i2]](/bjoc/content/inline/1860-5397-17-201-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of dipeptide 6. HATU: 2-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate.
Scheme 2: Synthesis of dipeptide 6. HATU: 2-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorop...
In principle, the coupling of 6 and Fmoc-Val-OH under suitable conditions would deliver tripeptide 7. Keeping this in mind, we next screened a series of coupling reagents [15]. As shown in Table 1, the coupling reagents have a significant effect on the efficiency of the reactions. At the very beginning, we selected HCTU and DIPEA as the coupling reagent (Table 1, entry 1). The desired tripeptide 7 was obtained in 36% yield. Similar conditions were selected by Iwasaki’s group during the total synthesis of hoshinoamide C. To our delight, when HATU was used, the overall yield of tripeptide 7 was rised to more than 70% (Table 1, entries 2 and 3). Tripeptide 7 was produced in 78% isolated yield when HATU/DIPEA as the coupling reagent was used (Table 1, entry 2). Only trace product could be detected with EEDQ (Table 1, entry 4). Further screening did not give better results (Table 1, entries 5 and 6). The purity of tripeptide 7 was determined by HPLC and no racemization was observed, ensuring the smooth progress of the total synthesis of hoshinoamide A.
Table 1: Hindered peptide coupling: conditions and yields.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-201-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Coupling reagenta | Yieldb (%) | Racemizationc |
| 1 | HCTU, DIPEA | 36 | NO |
| 2 | HATU, DIPEA | 78d | NO |
| 3 | HATU, HOATe, DIPEA | 75 | NO |
| 4 | EEDQf | trace | NO |
| 5 | DICg, HOAT | 41 | NO |
| 6 | DIC, Oxymah | 24 | NO |
a0.1 mmol/L of dipeptide 6 and Fmoc-Val in DMF, 1.5 equiv coupling reagent, rt, 3 h. bYield was determined by 1H NMR data analysis. cRacemization was determined by HPLC. dIsolated yield. eHOAT: 3H-[1,2,3]-triazolo[4,5-b]pyridin-3-ol; fEEDQ: 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline; gDIC: N,N'-diisopropylcarbodiimide; hOxyma: 17-(acetyloxy)-3-methoxy-20-oxopregna-3,5-diene-6-carboxaldehyde.
With the tripeptide 7 in hand, we went on to construct the peptide scaffold (Scheme 3). When tripeptide 7 was subjected to Pd-catalyzed hydrogenation conditions [16], the benzyl group was selectively cleaved to generate 8. Treatment of 2-chlorotrityl chloride (CTC) resin with 8 (4 equiv) and DIPEA successfully produced the resin-bound tripeptide 3 in good yield [17]. It should be noted that unreacted 8 can be largely recovered by a quick silica gel chromatography. The N terminus of 3 was then sequentially extended with properly protected ʟ-Gln4, N-Me-ᴅ-Val5, ʟ-Ile6, N-Me- ʟ-Leu7, Ana8, and Hba9 units to give 9-mer peptide 9 using the standard SPPS procedure [13]. When ʟ-Ile6 and Ana8 were coupled, the reaction time was prolonged to 3 h in order to increase the yield. The peptide chain was then cleaved from resin by the solution of 0.5% TFA in DCM. The Trt group was removed with aqueous solution of TFA to give 9-mer peptide 10 in good yield. The carboxylic acid of 9-mer peptide 10 was converted to the methyl ester with MeI and K2CO3 in DMF [18], delivering the final natural product hoshinoamide A in 2% yield (10 mg). The spectroscopic data of synthetic hoshinoamide A were in excellent agreement with the data previously reported for the natural product.
![[1860-5397-17-201-i3]](/bjoc/content/inline/1860-5397-17-201-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have completed the first total synthesis of hoshinoamide A. By combining the liquid and solid-phase peptide synthetic strategy, hoshinoamide A was synthesized in high efficiency. After systematic screening of the coupling reagents in liquid phase, the key intermediate tripeptide 7 was obtained in high yield. The solid-phase synthesis improves the entire efficiency of the synthetic route. This strategy could be applied to the stereoselective synthesis of hoshinoamide A and other highly methylated polypeptide analogues. Our strategy was also helpful to further study its antimalarial activity. Structure-and-activity and functional studies with the fluorescent-labeled analogs are currently under investigation in our lab.
Experimental
General experimental procedures. 1H NMR spectra were obtained using a Bruker AVANCE AV 400 spectrometer at a frequency of 400 MHz, respectively in CDCl3, CD3OD or D2O. Chemical shifts are reported in parts per million (ppm) and coupling constants in Hertz (Hz). The residual solvent peaks were used as internal standards. 1H NMR data are reported as follows: chemical shift values (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant and relative integral. 13C NMR spectra were obtained using a Bruker AVANCE AV 400 at 100 MHz in CDCl3, CD3OD or D2O. 13C NMR data are reported as chemical shift values (ppm).
LC–MS was performed on a Thermo Scientific MSQ instrument with the spectrometer operating in positive mode. Separations on the LC–MS system were performed on two methods using a thermo accucore C18 (2.6 µm, 100 × 2.1 mm) column. Method A: Linear gradient of 10–90% CH3CN/H2O and 0.1% TFA over 40 min were applied at a flow rate of 1.0 mL/min and detection at 220 nm. Method B: Linear gradient of 10–90–90–10% CH3CN/H2O and 0.1% TFA over 10 min (10–90 vol % MeCN over 6 min, 90–90 vol % over 3 min, 90–10 vol % MeCN over 1 min) were applied at a flow rate of 1.0 mL/min and detection at 220 nm. Preparative reverse-phase HPLC was performed by using Thermo Scientific Ultimate 3000 equipped with a Thermo Hypersil Gold (5 µm, 150 × 21.2 mm) column adpoting the following buffer systems: A: 0.1% TFA in water. B: 0.1% TFA in MeCN using a 10–90–90–10 vol % MeCN gradient (10–90 vol % MeCN over 30 min, 90–90 vol % over 10 min, 90–10 vol % MeCN over 10 min) at a flow rate of 8 mL/min.
Standard SPPS (solid-phase peptide synthesis) method:
- General procedures for coupling on resin: The loaded resin was shaken for 2 h at room temperature with a solution of the desired Fmoc-AA-OH (4 equiv), HATU/coupling reagent (4 equiv) and DIPEA (8 equiv) in DMF (20 mL). The coupling mixture was filtered and the resin was washed with CH2Cl2 (10 mL × 5) and CH3OH (10 mL × 5).
- General procedures for deprotection of Fmoc: The loaded resin was treated with a solution of 20 vol % piperidine in DMF (20 mL) for 30 min and then filtered. The resin was washed with CH2Cl2 (20 mL × 5) and CH3OH (20 mL × 5).
- General procedures for cleavage the peptide from the resin: 0.5% TFA in DCM (20 mL) were added on the resin and the mixture was shaken for 2 h before filtered. The resin was washed with CH2Cl2 (20 mL × 5) and CH3OH (20 mL × 5).
Fmoc-Pro-OH (674 mg, 2 mmol) was then dissolved in a mixture of DCM (10 mL) and DMF (10 mL). DIPEA (1.7 mL, 10 mmol) and 2-CTC resin (1 g) were added to this mixture and the reaction was stirred at room temperature for 2 h. The resin was filtered and washed with MeOH (3 × 20 mL) and DCM (3 × 20 mL). The unreacted resin was capped with MeOH in a mixture of MeOH/DIPEA/DCM (1:2:7, 10 mL) for 3 h. The resin-bound peptide was added to a mixture of 20% piperidine in DMF (20 mL), and the mixture was shaken for 30 minutes. Then the mixture was filtered, and the resin was washed with MeOH (3 × 20 mL) and DCM (3 × 20 mL). Fmoc-N-Me-ᴅ-Phe-OH (1000 mg, 2.5 mmol), HCTU (1.0 g, 2.5 mmol) and DIPEA (871 µL, 5.0 mmol) in DMF were added on the resin and the reactor was shaken for 1 h at room temperature. Then the mixture was filtered, the resin was washed with MeOH (3 × 20 mL) and DCM (3 × 20 mL) to afford the resin-bound dipeptide. The resin-bound dipeptide was added to a mixture of 20% piperidine in DMF (20 mL), and the mixture was shaken for 30 minutes. Then the mixture was filtered, and the resin was washed with MeOH (3 × 20 mL) and DCM (3 × 20 mL). Fmoc-Val-OH (848 mg, 2.5 mmol), HATU (1.0 g, 2.5 mmol) and DIPEA (871 µL, 5.0 mmol) in DMF were added on the resin and the reactor was shaken for 1 h at room temperature. The resulting tripeptide was analyzed on a Thermo Scientific MSQ instrument, and few product was observed.
Fmoc-N-Me-ᴅ-Phe-Pro-OBn (6). Pro-OBn.HCl (2.41 g, 10 mmol), Fmoc-N-Me-ᴅ-Phe-OH (4.01 g, 10 mmol) and DIPEA (5.2 mL, 30 mmol) were dissolved in 50 mL anhydrous DCM. HATU (5.7 g, 15 mmol) was added to the solution and the mixture was stirred at room temperature for 6 h. The reaction mixture was then washed with 1.0 M HCl (20 mL), aqueous NaHCO3 (20 mL) and brine (20 mL). The organic phase was dried with anhydrous Na2SO4 and concentrated in vacuo. The crude residue was purified by flash column chromatography (n-hexanes/EA 2:1) to afford dipeptide 6 (4.9 g, 83%). 1H NMR (400 MHz, methanol-d4) δ 7.39–7.27 (m, 19H), 5.20–5.12 (m, 4H), 4.85 (s, 6H), 4.44 (m, 4H), 3.50 (m, 2H), 3.31–3.23 (m, 4H), 3.07 (dd, J = 12.8, 10.3 Hz, 2H), 2.57 (s, 6H), 2.46 (dt, J = 9.9, 7.0 Hz, 2H), 2.03–1.94 (m, 2H), 1.89– 1.71 (m, 4H), 1.48 (m, 2H); 13C NMR (101 MHz, methanol-d4) δ 171.21, 165.90, 135.71, 133.51, 129.37, 128.70, 128.24, 128.07, 127.93, 127.74, 66.74, 60.75, 59.32, 36.55, 30.88, 28.45, 23.94; HRMS–ESI (m/z): [M + H]+ calcd. for C22H26N2O3, 588.2671; found, 588.2673.
Fmoc-Val-N-Me-ᴅ-Phe-Val-Pro-OBn (7). To a stirred solution of dipeptide 6 (118 mg, 0.20 mmol) was added 20% Et2NH in CH3CN (5 mL) at rt for 0.5 h. The Et2NH and CH3CN were evaporated in vacuo and the residue was triturated with ether (10 mL), the crude amine was washed with ether (quintic, 10 mL each time) and dried in vacuo. The crude amine and Fmoc-Val-OH (71 mg, 0.20 mmol) were dissolved in 10 mL anhydrous DMF. The coupling reagent was added to the solution and the mixture was stirred at room temperature for 3 h. This mixture was poured onto water (10 mL) and extracted with CH2Cl2 (3 × 10 mL), then washed with 1.0 M HCl (10 mL), aqueous NaHCO3 (10 mL) and brine (10 mL). The organic phase was dried with anhydrous Na2SO4 and concentrated in vacuo. The crude residue was purified by flash column chromatography (n-hexanes/EA 2:1) to afford tripeptide 7. 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.7 Hz, 2H), 7.57 (t, J = 7.2 Hz, 2H), 7.40–7.17 (m, 14H), 5.72 (dd, J = 8.9, 6.5 Hz, 1H), 5.46 (d, J = 9.5 Hz, 1H), 5.23–5.20 (m, 1H), 5.06 (d, J = 12.2 Hz, 1H), 4.50–4.18 (m, 5H), 3.48–3.43 (m, 1H), 3.28 (dt, J = 11.3, 5.8 Hz, 2H), 3.10 (s, 1H), 2.95–2.86 (m, 3H), 2.21–2.14 (m, 2H), 1.78–1.62 (m, 5H), 1.28 (s, 2H), 0.76 (m, 3H), 0.47 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 171.70, 171.60, 168.26, 156.39, 143.88, 141.30, 137.01, 129.54, 128.88, 128.69, 128.60, 128.50, 128.39, 128.31, 128.20, 127.72, 127.08, 126.65, 125.17, 125.08, 119.99, 67.04, 66.84, 59.43, 55.87, 55.59, 47.17, 46.92, 35.00, 30.72, 30.47, 28.78, 25.25, 19.82, 16.33; HRMS–ESI (m/z): [M + H]+ calcd. for C42H45N3O6, 688.3381; found, 688.3384. Comparison of the effects of different coupling reagents on the reaction yield is shown in Table 1.
Fmoc-Val-N-Me-ᴅ-Phe-Val-Pro-OH (8). Tripeptide 7 (2.3 g, 3.3 mmol) was dissolved in 30 mL of MeOH/HCOOH (v/v 9:1) and hydrogenized with Pd(OH)2 (500 mg) under H2 atmosphere for 10 hours to remove the benzyl groups. The reaction mixture was filtered through a pad of celite and the filtrate was concentrated in vacuo to give a brown oil which was purified by flash chromatography (n-hexanes/EA 2:1), affording tripeptide 8 (1.87 g, 95%) as a white foam. 1H NMR (400 MHz, methanol-d4) δ 7.59 (d, J = 8 Hz, 2H), 7.45 (d, J = 8 Hz, 2H), 7.21–6.96 (m, 10H), 5.52 (m, 1H), 4.81 (s, 2H), 4.20–3.98 (m, 4H), 3.29–3.10 (m, 2H), 3.04–2.91 (m, 4H), 2.80–2.67 (m, 2H), 2.62 (s, 3H), 2.01–1.83 (m, 2H), 1.53 (m, 4H), 1.13–1.09 (m, 2H), 0.52–0.45 (m, 4H); 13C NMR (101 MHz, methanol-d4) δ 175.49, 175.06, 174.95, 173.81, 173.14, 171.16, 170.16, 167.42, 164.85, 158.64, 158.58, 158.53, 145.38, 145.35, 145.20, 145.13, 145.09, 142.88, 142.55, 138.50, 138.41, 131.03, 130.75, 130.66, 130.27, 129.77, 129.64, 129.57, 129.42, 128.86, 128.83, 128.55, 128.28, 128.21, 127.77, 127.66, 126.35, 126.29, 126.21, 125.76, 121.02, 70.63, 68.11, 68.04, 66.85, 60.78, 60.68, 59.15, 58.19, 57.76, 57.67, 57.41, 57.06, 56.41, 56.08, 38.96, 37.58, 37.01, 36.60, 35.79, 35.61, 33.11, 32.20, 31.89, 31.73, 31.53, 31.40, 30.88, 30.83, 30.66, 30.53, 30.39, 29.98, 29.64, 28.18, 26.97, 26.10, 23.80, 23.34, 19.88, 19.48, 18.17, 18.07, 17.64, 14.58; HRMS–ESI (m/z): [M + H]+ calcd. for C35H39N3O6, 598.2912; found, 598.2915.
Solid-phase synthesis of hoshinoamide A from tripeptide 8
Tripeptide 8 (1.20 g, 2 mmol) was dissolved in a mixture of DCM (10 mL) and DMF (10 mL). DIPEA (1.7 mL, 10 mmol) and 2-CTC resin (1 g) were added to this mixture and the reaction was stirred at room temperature for 3 h. The resin was filtered and washed with MeOH (3 × 20 mL) and DCM (3 × 20 mL). Tripeptide 8 was recovered (1.6 mmol). The unreacted resin was capped with MeOH in a mixture of MeOH/DIPEA/DCM (1:2:7, 10 mL) for 5 h. The Fmoc protecting group was removed following the general procedure and the remaining amino acids were successively coupled using the standard SPPS method. 0.5% TFA in DCM (20 mL) was added on the resin and the mixture was shaken for 2 h to cleave the peptide from the resin. The mixture was filtered and the filtrate was concentrated in vacuo to give a white foam. The peptide was re-dissolved in a mixture of TFA/Et3SiH/H2O (10 mL, 50:50:50 v/v/v). The reaction mixture was stirred for 3 h, and then concentrated in vacuo. The crude peptide was precipitated using cold Et2O and centrifuged at 7000 rpm to give a white solid. This solid was further purified by RP-HPLC through protocols described in the general method section. Fractions were collected, concentrated and lyophilized to give nanopeptide 10 as a white solid. Some nanopeptide 10 (23 mg, 0.02 mmol) was dissolved in dry DMF (5 mL). The remaining nanopeptide 10 was recovered. K2CO3 (3.1 mg, 0.022 mmol) and MeI (3.13 mg, 0.022 mmol) was added to this solution. The reaction mixture was stirred for 3 h. This mixture was poured onto water (5 mL) and extracted with CH2Cl2 (3 × 5 mL). then washed with 1.0 M HCl (10 mL), aqueous NaHCO3 (10 mL) and brine (10 mL). The organic phase was dried with anhydrous Na2SO4 and concentrated in vacuo to give a brown oil. This oil was further purified by RP-HPLC using protocols described in the general method section. Fractions were collected, concentrated and lyophilized to give hoshinoamide A as a white solid (10 mg, 2% yield). The 1H NMR and 13C NMR spectra of the synthetic product were fully consistent with the data of isolated samples reported in the literature [11]. See Supporting Information File 1, Tables S2 and S3 for details. 1H NMR (400 MHz, methanol-d4) δ 7.30–7.11 (m, 6H), 7.02–6.94 (m, 2H), 6.72–6.65 (m, 2H), 5.73 (dd, J = 9.2, 6.4 Hz, 1H), 4.82–4.73 (m, 1H), 4.64–4.53 (m, 2H), 4.43–4.25 (m, 2H), 3.69 (s, 2H), 3.50 (dt, J = 11.2, 6.0 Hz, 1H), 3.41–3.31 (m, 1H), 3.20–3.06 (m, 9H), 2.99–2.92 (m, 3H), 2.91–2.86 (m, 1H), 2.52 (t, J = 7.6 Hz, 2H), 2.47–2.38 (m, 2H), 2.29–2.22 (m, 3H), 2.18 (q, J = 7.7 Hz, 3H), 2.05–1.99 (m, 1H), 1.99–1.71 (m, 9H), 1.68–1.46 (m, 6H), 1.46–1.34 (m, 4H), 1.11–0.97 (m, 3H), 0.97–0.81 (m, 16H), 0.68–0.55 (m, 6H). 13C NMR (101 MHz, methanol-d4) δ 177.43, 176.52, 176.00, 174.55, 173.86, 173.15, 173.09, 172.87, 171.89, 170.47, 156.60, 138.40, 133.75, 130.69, 130.40, 129.45, 127.70, 116.18, 64.21, 60.84, 57.23, 55.69, 55.64, 55.52, 54.32, 52.69, 40.23, 38.43, 37.82, 36.62, 35.76, 35.51, 34.59, 32.62, 31.80, 31.59, 31.56, 30.22, 29.98, 29.20, 27.72, 26.21, 26.17, 26.06, 25.95, 25.90, 25.11, 23.80, 22.17, 20.39, 19.90, 19.49, 18.13, 16.50, 11.60; HRMS–ESI (m/z): [M + H]+ calcd. for C61H95N9O12, 1146.7173; found, 1146.7173.
Supporting Information
| Supporting Information File 1: NMR (1H NMR and 13C NMR) spectra of compounds 2−8, and comparison of the spectral data of natural and synthetic hoshinoamide A. | ||
| Format: PDF | Size: 1.6 MB | Download |
Acknowledgements
The authors also sincerely thank Mr. Timothy Barnes (The Department of Chemistry and Biochemistry of Auburn University) for the help of preparing the manuscript.
Funding
This research was supported by the Natural Science key Project of Anhui Education Department (Grant No. KJ2019A0635); Provincial University Students’ Innovation and Entrepreneurship Program (Grant No. 2019CXXL077); Hubei Province Key Laboratory of Occupational Hazard Identification and Control, Wuhan University of Science and Technology (Grant No. OHIC2020Y06).
References
-
Cowman, A. F.; Healer, J.; Marapana, D.; Marsh, K. Cell 2016, 167, 610–624. doi:10.1016/j.cell.2016.07.055
Return to citation in text: [1] -
World malaria report 2020. World Health Organization, 2020; https://www.who.int/publications/i/item/9789240015791.
Return to citation in text: [1] -
Singh, S. K.; Singh, S. Int. J. Pharm. Sci. Rev. Res. 2014, 25, 295–302.
Return to citation in text: [1] -
Achan, J.; Talisuna, A. O.; Erhart, A.; Yeka, A.; Tibenderana, J. K.; Baliraine, F. N.; Rosenthal, P. J.; D'Alessandro, U. Malar. J. 2011, 10, 144. doi:10.1186/1475-2875-10-144
Return to citation in text: [1] -
Kumar, S.; Bhardwaj, T. R.; Prasad, D. N.; Singh, R. K. Biomed. Pharmacother. 2018, 104, 8–27. doi:10.1016/j.biopha.2018.05.009
Return to citation in text: [1] -
Golenser, J.; Waknine, J. H.; Krugliak, M.; Hunt, N. H.; Grau, G. E. Int. J. Parasitol. 2006, 36, 1427–1441. doi:10.1016/j.ijpara.2006.07.011
Return to citation in text: [1] -
Hsu, E. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 505–508. doi:10.1016/j.trstmh.2005.09.020
Return to citation in text: [1] -
Devine, S. M.; Challis, M. P.; Kigotho, J. K.; Siddiqui, G.; De Paoli, A.; MacRaild, C. A.; Avery, V. M.; Creek, D. J.; Norton, R. S.; Scammells, P. J. Eur. J. Med. Chem. 2021, 221, 113518. doi:10.1016/j.ejmech.2021.113518
Return to citation in text: [1] -
Shibeshi, M. A.; Kifle, Z. D.; Atnafie, S. A. Infect. Drug Resist. 2020, 13, 4047–4060. doi:10.2147/idr.s279433
Return to citation in text: [1] -
Ojha, P. K.; Kumar, V.; Roy, J.; Roy, K. Expert Opin. Drug Discovery 2021, 16, 659–695. doi:10.1080/17460441.2021.1866535
Return to citation in text: [1] -
Iwasaki, A.; Tadenuma, T.; Sumimoto, S.; Shiota, I.; Matsubara, T.; Saito-Nakano, Y.; Nozaki, T.; Sato, T.; Suenaga, K. J. Nat. Prod. 2018, 81, 2545–2552. doi:10.1021/acs.jnatprod.8b00643
Return to citation in text: [1] [2] -
Iwasaki, A.; Ohtomo, K.; Kurisawa, N.; Shiota, I.; Rahmawati, Y.; Jeelani, G.; Nozaki, T.; Suenaga, K. J. Nat. Prod. 2021, 84, 126–135. doi:10.1021/acs.jnatprod.0c01209
Return to citation in text: [1] [2] -
Zong, Y.; Sun, X.; Gao, H.; Meyer, K. J.; Lewis, K.; Rao, Y. J. Med. Chem. 2018, 61, 3409–3421. doi:10.1021/acs.jmedchem.7b01241
Return to citation in text: [1] [2] -
Bu, X.; Xie, G.; Law, C. W.; Guo, Z. Tetrahedron Lett. 2002, 43, 2419–2422. doi:10.1016/s0040-4039(02)00237-x
Return to citation in text: [1] -
Dunetz, J. R.; Magano, J.; Weisenburger, G. A. Org. Process Res. Dev. 2016, 20, 140–177. doi:10.1021/op500305s
Return to citation in text: [1] -
Li, Y.; Manickam, G.; Ghoshal, A.; Subramaniam, P. Synth. Commun. 2006, 36, 925–928. doi:10.1080/00397910500466199
Return to citation in text: [1] -
Liu, L.; Wu, S.; Wang, Q.; Zhang, M.; Wang, B.; He, G.; Chen, G. Org. Chem. Front. 2018, 5, 1431–1435. doi:10.1039/c8qo00145f
Return to citation in text: [1] -
Guo, W.; Li, J.; Fan, N.; Wu, W.; Zhou, P.; Xia, C. Synth. Commun. 2005, 35, 145–152. doi:10.1081/scc-200046532
Return to citation in text: [1]
| 11. | Iwasaki, A.; Tadenuma, T.; Sumimoto, S.; Shiota, I.; Matsubara, T.; Saito-Nakano, Y.; Nozaki, T.; Sato, T.; Suenaga, K. J. Nat. Prod. 2018, 81, 2545–2552. doi:10.1021/acs.jnatprod.8b00643 |
| 1. | Cowman, A. F.; Healer, J.; Marapana, D.; Marsh, K. Cell 2016, 167, 610–624. doi:10.1016/j.cell.2016.07.055 |
| 7. | Hsu, E. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 505–508. doi:10.1016/j.trstmh.2005.09.020 |
| 13. | Zong, Y.; Sun, X.; Gao, H.; Meyer, K. J.; Lewis, K.; Rao, Y. J. Med. Chem. 2018, 61, 3409–3421. doi:10.1021/acs.jmedchem.7b01241 |
| 5. | Kumar, S.; Bhardwaj, T. R.; Prasad, D. N.; Singh, R. K. Biomed. Pharmacother. 2018, 104, 8–27. doi:10.1016/j.biopha.2018.05.009 |
| 6. | Golenser, J.; Waknine, J. H.; Krugliak, M.; Hunt, N. H.; Grau, G. E. Int. J. Parasitol. 2006, 36, 1427–1441. doi:10.1016/j.ijpara.2006.07.011 |
| 18. | Guo, W.; Li, J.; Fan, N.; Wu, W.; Zhou, P.; Xia, C. Synth. Commun. 2005, 35, 145–152. doi:10.1081/scc-200046532 |
| 3. | Singh, S. K.; Singh, S. Int. J. Pharm. Sci. Rev. Res. 2014, 25, 295–302. |
| 4. | Achan, J.; Talisuna, A. O.; Erhart, A.; Yeka, A.; Tibenderana, J. K.; Baliraine, F. N.; Rosenthal, P. J.; D'Alessandro, U. Malar. J. 2011, 10, 144. doi:10.1186/1475-2875-10-144 |
| 16. | Li, Y.; Manickam, G.; Ghoshal, A.; Subramaniam, P. Synth. Commun. 2006, 36, 925–928. doi:10.1080/00397910500466199 |
| 2. | World malaria report 2020. World Health Organization, 2020; https://www.who.int/publications/i/item/9789240015791. |
| 17. | Liu, L.; Wu, S.; Wang, Q.; Zhang, M.; Wang, B.; He, G.; Chen, G. Org. Chem. Front. 2018, 5, 1431–1435. doi:10.1039/c8qo00145f |
| 12. | Iwasaki, A.; Ohtomo, K.; Kurisawa, N.; Shiota, I.; Rahmawati, Y.; Jeelani, G.; Nozaki, T.; Suenaga, K. J. Nat. Prod. 2021, 84, 126–135. doi:10.1021/acs.jnatprod.0c01209 |
| 14. | Bu, X.; Xie, G.; Law, C. W.; Guo, Z. Tetrahedron Lett. 2002, 43, 2419–2422. doi:10.1016/s0040-4039(02)00237-x |
| 12. | Iwasaki, A.; Ohtomo, K.; Kurisawa, N.; Shiota, I.; Rahmawati, Y.; Jeelani, G.; Nozaki, T.; Suenaga, K. J. Nat. Prod. 2021, 84, 126–135. doi:10.1021/acs.jnatprod.0c01209 |
| 15. | Dunetz, J. R.; Magano, J.; Weisenburger, G. A. Org. Process Res. Dev. 2016, 20, 140–177. doi:10.1021/op500305s |
| 11. | Iwasaki, A.; Tadenuma, T.; Sumimoto, S.; Shiota, I.; Matsubara, T.; Saito-Nakano, Y.; Nozaki, T.; Sato, T.; Suenaga, K. J. Nat. Prod. 2018, 81, 2545–2552. doi:10.1021/acs.jnatprod.8b00643 |
| 8. | Devine, S. M.; Challis, M. P.; Kigotho, J. K.; Siddiqui, G.; De Paoli, A.; MacRaild, C. A.; Avery, V. M.; Creek, D. J.; Norton, R. S.; Scammells, P. J. Eur. J. Med. Chem. 2021, 221, 113518. doi:10.1016/j.ejmech.2021.113518 |
| 9. | Shibeshi, M. A.; Kifle, Z. D.; Atnafie, S. A. Infect. Drug Resist. 2020, 13, 4047–4060. doi:10.2147/idr.s279433 |
| 10. | Ojha, P. K.; Kumar, V.; Roy, J.; Roy, K. Expert Opin. Drug Discovery 2021, 16, 659–695. doi:10.1080/17460441.2021.1866535 |
| 13. | Zong, Y.; Sun, X.; Gao, H.; Meyer, K. J.; Lewis, K.; Rao, Y. J. Med. Chem. 2018, 61, 3409–3421. doi:10.1021/acs.jmedchem.7b01241 |
© 2021 Zhou et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.