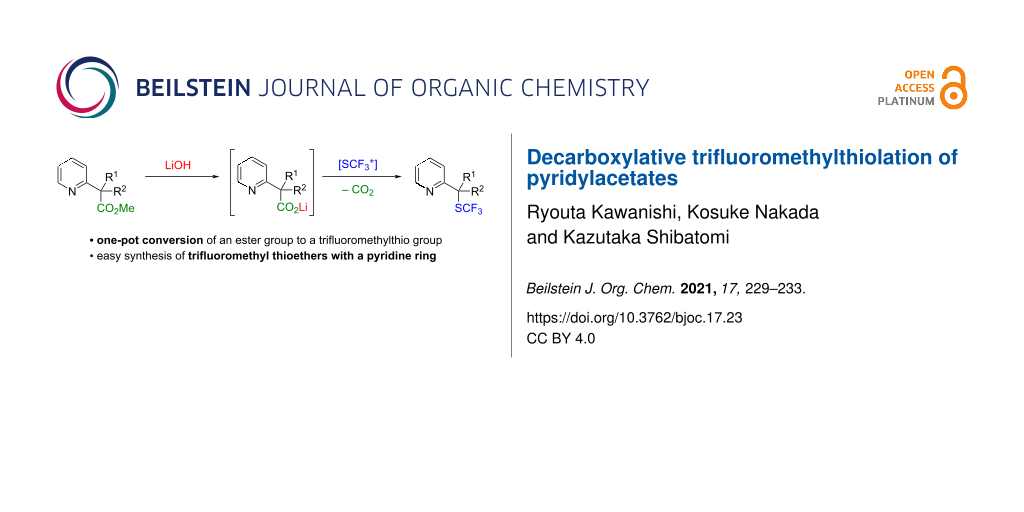
Abstract
Decarboxylative trifluoromethylthiolation of lithium pyridylacetates was achieved using N-(trifluoromethylthio)benzenesulfonimide as the electrophilic trifluoromethylthiolation reagent. The reaction afforded the corresponding trifluoromethyl thioethers in good yield. Furthermore, the preparation of lithium pyridylacetates by saponification of the corresponding methyl esters and subsequent decarboxylative trifluoromethylthiolation were performed in a one-pot fashion.

Graphical Abstract
Introduction
The pyridine ring is found in numerous biologically active compounds. Therefore, efficient methods for synthesizing substituted pyridines are in high demand in pharmaceutical and agricultural chemistry [1,2]. Because of the unique features of fluorine atoms, fluorinated functional groups have also been recognized as important substructures in the design of medicinally relevant compounds [3-6]. Introducing a trifluoromethylthio group (CF3S–), which has high lipophilicity and strong electron-withdrawing properties, into medicinal compounds can improve their pharmacokinetic properties [7-11]. Hence, the development of a synthetic method for the preparation of trifluoromethyl thioethers has recently attracted much attention [12-15].
Previously, our research group achieved decarboxylative functionalization of tertiary β-ketocarboxylic acids by exploiting their special ability to readily undergo decarboxylation [16-21]. During the course of this study, we found that lithium pyridylacetates undergo decarboxylative fluorination upon treatment with an electrophilic fluorination reagent to afford fluoromethylpyridines under catalyst-free conditions. Furthermore, we demonstrated the one-pot synthesis of fluoromethylpyridines from methyl pyridylacetates by saponification of methyl esters and subsequent decarboxylative fluorination (Scheme 1a) [21]. Herein, we describe the application of this method to decarboxylative trifluoromethylthiolation with an electrophilic trifluoromethylthiolation reagent (Scheme 1b) [22], which enables easy installation of the trifluoromethylthio group at a pyridylic carbon.
![[1860-5397-17-23-i1]](/bjoc/content/inline/1860-5397-17-23-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Electrophilic decarboxylative functionalization of 2-pyridylacetates.
Scheme 1: Electrophilic decarboxylative functionalization of 2-pyridylacetates.
Results and Discussion
First, we synthesized lithium 2-pyridylacetate 1a according to our previously reported procedure [21] and subjected it to decarboxylative trifluoromethylthiolation with N-trifluoromethylthiosuccinimide (4) in DMF at room temperature for 15 h. However, the desired product 2a was not observed (Table 1, entry 1). The use of N-trifluoromethylthiophthalimide (5) did not afford 2a either (Table 1, entry 2). Fortunately, the use of N-(trifluoromethylthio)dibenzenesulfonimide 6 [23] gave 2a in 14% yield, along with the protonated product 3a in 31% yield (Table 1, entry 3). The yield of 2a could be improved to 30% by adding MS 4 Å to the reaction mixture (Table 1, entry 4). Screening of various solvents revealed that THF was the best choice for this reaction (Table 1, entries 4–11), and the yield of 2a was dramatically improved to 89% (Table 1, entry 11). In the absence of MS 4 Å, the yield of 2a was diminished even when the reaction was carried out in THF (Table 1, entry 12).
Table 1: Screening of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-23-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | [SCF3+] | solvent | time (h) | yield of 2a (%) | yield of 3a (%) |
| 1 | 4 | DMF | 15 | 0 | 0 |
| 2 | 5 | DMF | 72 | 0 | 0 |
| 3 | 6 | DMF | 3 | 14 | 31 |
| 4a | 6 | DMF | 5 | 30 | 34 |
| 5a | 6 | DMSO | 5 | 64 | 21 |
| 6a | 6 | acetonitrile | 8 | 77 | 0 |
| 7a | 6 | toluene | 168 | 72 | 0 |
| 8a | 6 | CH2Cl2 | 72 | 54 | 0 |
| 9a | 6 | t-BuOMe | 72 | 55 | 0 |
| 10a | 6 | 1,4-dioxane | 9 | 75 | 0 |
| 11a | 6 | THF | 8 | 89 | 0 |
| 12 | 6 | THF | 8 | 63 | 26 |
| 13a,b | 6 | THF | 8 | 70 | 0 |
aThe reaction was carried out with MS 4 Å (180 mg/0.2 mmol); b1.1 equiv of 6 was used.
With the optimized reaction conditions in hand, we examined the one-pot synthesis of 2a from methyl ester 7a. Methyl 2-pyridylacetate 7a were saponified with lithium hydroxide in a MeOH/H2O system. After completion of the reaction, the solvents were evaporated under reduced pressure. Then, THF, MS 4 Å, and 6 were added to the residue, and the mixture was stirred at room temperature for 8 h. This reaction successfully afforded the desired product 2a in 85% yield over two steps (Scheme 2).
![[1860-5397-17-23-i2]](/bjoc/content/inline/1860-5397-17-23-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: One-pot procedure for the synthesis of 2a.
Scheme 2: One-pot procedure for the synthesis of 2a.
Encouraged by the aforementioned result, we applied this method to several 2-pyridylacetates (Scheme 3). Methyl 2-pyridylacetates 7b–d with arylmethyl substituents furnished the corresponding trifluoromethylthiolated products 2b−d in good yields. α,α-Dialkyl-2-pyridylacetates 7e–g also gave the desired products 2e–g in moderate yields. The method could also be applied to substrates with quinoline and isoquinoline backbones to afford the corresponding products 2h and 2i. In addition, the reaction of α-monosubstituted 2-pyridylacetate 8 was performed to yield the corresponding mono-trifluoromethylthiolated product 9 in 36% yield, along with 6% yield of disubstituted product 10 (Scheme 4). Increasing the amount of 6 did not improve the yield of products 9 and 10 significantly.
![[1860-5397-17-23-i3]](/bjoc/content/inline/1860-5397-17-23-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope. aSaponification was carried out with 2.5 equiv of LiOH, and 2.5 equiv of 6 was used for trifluoromethylthiolation. bSaponification of 7 was carried out for 39 h. cSaponification was carried out with 2.5 equiv of LiOH under reflux conditions, and 2.5 equiv of 6 was used for trifluoromethylthiolation.
Scheme 3: Substrate scope. aSaponification was carried out with 2.5 equiv of LiOH, and 2.5 equiv of 6 was use...
![[1860-5397-17-23-i4]](/bjoc/content/inline/1860-5397-17-23-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of α-monosubstituted 2-pyridylacetates.
Scheme 4: Reaction of α-monosubstituted 2-pyridylacetates.
Based on the abovementioned results and our previous study on decarboxylative fluorination [21], we propose a plausible mechanism for this reaction, as outlined in Scheme 5. An electrophilic sulfur atom of 6 approaches the nitrogen atom on the pyridine ring to promote decarboxylation via the formation of N-trifluoromethylthio-2-alkylidene-1,2-dihydropyridine intermediate I, which immediately isomerizes to afford 2 (Scheme 5). Methyl 4-pyridylacetate 11 also gave the corresponding trifluoromethylthiolated product 12 in 29% yield (Scheme 6), where the reaction was assumed to proceed via the N-trifluoromethylthio-4-alkylidene-1,4-dihydropyridine intermediate. In contrast, methyl 3-pyridylacetate 13 did not yield the trifluoromethylthiolated product at all, despite complete saponification of the methyl ester.
![[1860-5397-17-23-i5]](/bjoc/content/inline/1860-5397-17-23-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-23-i6]](/bjoc/content/inline/1860-5397-17-23-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of 3- and 4-pyridylacetates.
Scheme 6: Reaction of 3- and 4-pyridylacetates.
Conclusion
In conclusion, we demonstrated the decarboxylative trifluoromethylthiolation of lithium 2- and 4-pyridylacetates to synthesize pyridine derivatives with a trifluoromethylthio group at a tertiary carbon center adjacent to the pyridine ring. Furthermore, saponification of methyl pyridylacetates and subsequent decarboxylative trifluoromethylthiolation of the resulting lithium salts were performed in a one-pot fashion. This method can easily convert an ester group into a trifluoromethylthio group. The resulting trifluoromethyl thioethers would be useful for the preparation of various medicinally relevant compounds.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and copies of NMR spectra. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625
Return to citation in text: [1] -
Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778
Return to citation in text: [1] -
Leo, A.; Hansch, C.; Elkins, D. Chem. Rev. 1971, 71, 525–616. doi:10.1021/cr60274a001
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] -
Landelle, G.; Panossian, A.; Leroux, F. R. Curr. Top. Med. Chem. 2014, 14, 941–951. doi:10.2174/1568026614666140202210016
Return to citation in text: [1] -
Leroux, F.; Jeschke, P.; Schlosser, M. Chem. Rev. 2005, 105, 827–856. doi:10.1021/cr040075b
Return to citation in text: [1] -
Manteau, B.; Pazenok, S.; Vors, J.-P.; Leroux, F. R. J. Fluorine Chem. 2010, 131, 140–158. doi:10.1016/j.jfluchem.2009.09.009
Return to citation in text: [1] -
Toulgoat, F.; Alazet, S.; Billard, T. Eur. J. Org. Chem. 2014, 2415–2428. doi:10.1002/ejoc.201301857
Return to citation in text: [1] -
Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b
Return to citation in text: [1] -
Barata-Vallejo, S.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 7150–7182. doi:10.1039/c6ob00763e
Return to citation in text: [1] -
Barthelemy, A.-L.; Magnier, E.; Dagousset, G. Synthesis 2018, 50, 4765–4776. doi:10.1055/s-0037-1611278
Return to citation in text: [1] -
Shibatomi, K.; Kitahara, K.; Sasaki, N.; Kawasaki, Y.; Fujisawa, I.; Iwasa, S. Nat. Commun. 2017, 8, 15600. doi:10.1038/ncomms15600
Return to citation in text: [1] -
Katada, M.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Synlett 2018, 29, 2408–2411. doi:10.1055/s-0037-1611019
Return to citation in text: [1] -
Naruse, A.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Asian J. Org. Chem. 2019, 8, 691–693. doi:10.1002/ajoc.201900072
Return to citation in text: [1] -
Kawanishi, R.; Hattori, S.; Iwasa, S.; Shibatomi, K. Molecules 2019, 24, 2773. doi:10.3390/molecules24152773
Return to citation in text: [1] -
Kitahara, K.; Mizutani, H.; Iwasa, S.; Shibatomi, K. Synthesis 2019, 51, 4385–4392. doi:10.1055/s-0039-1690009
Return to citation in text: [1] -
Kawanishi, R.; Phongphane, L.; Iwasa, S.; Shibatomi, K. Chem. – Eur. J. 2019, 25, 7453–7456. doi:10.1002/chem.201900565
Return to citation in text: [1] [2] [3] [4] -
Shao, X.; Xu, C.; Lu, L.; Shen, Q. Acc. Chem. Res. 2015, 48, 1227–1236. doi:10.1021/acs.accounts.5b00047
Return to citation in text: [1] -
Liu, X.; An, R.; Zhang, X.; Luo, J.; Zhao, X. Angew. Chem., Int. Ed. 2016, 55, 5846–5850. doi:10.1002/anie.201601713
Return to citation in text: [1]
| 1. | Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625 |
| 2. | Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b |
| 16. | Shibatomi, K.; Kitahara, K.; Sasaki, N.; Kawasaki, Y.; Fujisawa, I.; Iwasa, S. Nat. Commun. 2017, 8, 15600. doi:10.1038/ncomms15600 |
| 17. | Katada, M.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Synlett 2018, 29, 2408–2411. doi:10.1055/s-0037-1611019 |
| 18. | Naruse, A.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Asian J. Org. Chem. 2019, 8, 691–693. doi:10.1002/ajoc.201900072 |
| 19. | Kawanishi, R.; Hattori, S.; Iwasa, S.; Shibatomi, K. Molecules 2019, 24, 2773. doi:10.3390/molecules24152773 |
| 20. | Kitahara, K.; Mizutani, H.; Iwasa, S.; Shibatomi, K. Synthesis 2019, 51, 4385–4392. doi:10.1055/s-0039-1690009 |
| 21. | Kawanishi, R.; Phongphane, L.; Iwasa, S.; Shibatomi, K. Chem. – Eur. J. 2019, 25, 7453–7456. doi:10.1002/chem.201900565 |
| 12. | Toulgoat, F.; Alazet, S.; Billard, T. Eur. J. Org. Chem. 2014, 2415–2428. doi:10.1002/ejoc.201301857 |
| 13. | Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b |
| 14. | Barata-Vallejo, S.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 7150–7182. doi:10.1039/c6ob00763e |
| 15. | Barthelemy, A.-L.; Magnier, E.; Dagousset, G. Synthesis 2018, 50, 4765–4776. doi:10.1055/s-0037-1611278 |
| 7. | Leo, A.; Hansch, C.; Elkins, D. Chem. Rev. 1971, 71, 525–616. doi:10.1021/cr60274a001 |
| 8. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 9. | Landelle, G.; Panossian, A.; Leroux, F. R. Curr. Top. Med. Chem. 2014, 14, 941–951. doi:10.2174/1568026614666140202210016 |
| 10. | Leroux, F.; Jeschke, P.; Schlosser, M. Chem. Rev. 2005, 105, 827–856. doi:10.1021/cr040075b |
| 11. | Manteau, B.; Pazenok, S.; Vors, J.-P.; Leroux, F. R. J. Fluorine Chem. 2010, 131, 140–158. doi:10.1016/j.jfluchem.2009.09.009 |
| 3. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 4. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 5. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 6. | Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778 |
| 23. | Liu, X.; An, R.; Zhang, X.; Luo, J.; Zhao, X. Angew. Chem., Int. Ed. 2016, 55, 5846–5850. doi:10.1002/anie.201601713 |
| 21. | Kawanishi, R.; Phongphane, L.; Iwasa, S.; Shibatomi, K. Chem. – Eur. J. 2019, 25, 7453–7456. doi:10.1002/chem.201900565 |
| 22. | Shao, X.; Xu, C.; Lu, L.; Shen, Q. Acc. Chem. Res. 2015, 48, 1227–1236. doi:10.1021/acs.accounts.5b00047 |
| 21. | Kawanishi, R.; Phongphane, L.; Iwasa, S.; Shibatomi, K. Chem. – Eur. J. 2019, 25, 7453–7456. doi:10.1002/chem.201900565 |
| 21. | Kawanishi, R.; Phongphane, L.; Iwasa, S.; Shibatomi, K. Chem. – Eur. J. 2019, 25, 7453–7456. doi:10.1002/chem.201900565 |
© 2021 Kawanishi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)