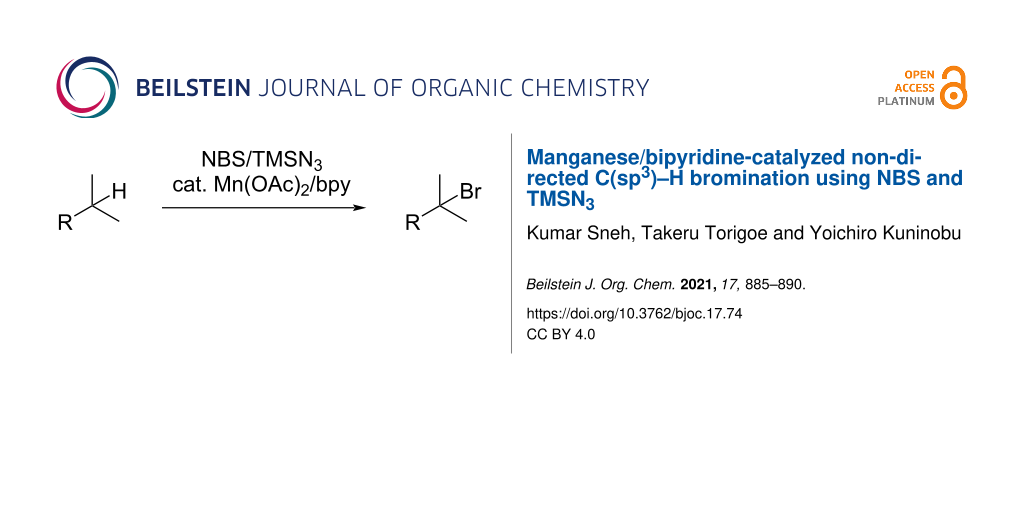
Abstract
A Mn(II)/bipyridine-catalyzed bromination reaction of unactivated aliphatic C(sp3)−H bonds has been developed using N-bromosuccinimide (NBS) as the brominating reagent. The reaction proceeded in moderate-to-good yield, even on a gram scale. The introduced bromine atom can be converted into fluorine and allyl groups.

Graphical Abstract
Introduction
Organic halides are versatile precursors for various synthetic protocols and are frequently used to introduce a variety of functionalities, such as boron-, silicon-, nitrogen-, and oxygen-based functional groups, and in C−C bond forming reactions, such as cross-coupling reactions [1-6]. The traditional method used for the preparation of alkyl bromides is the reaction of their corresponding alkyl alcohols with HBr, PBr3, or other brominating reagents [7-13].
Direct C–H halogenation is one of the most efficient methods used for the synthesis of halogenated organic molecules. This direct method involves the reaction of an alkane with Br2, CBr4, or H2O2–HBr under photolysis or at high temperatures in the absence of a catalyst (Scheme 1a) [14-16]. However, these reactions do not exhibit any selectivity due to the indiscriminate attack of bromine radicals on the C–H bonds in the substrate, which leads to the formation of a mixture of halogenated products. Electrophilic and radical C(sp3)−H halogenation at the benzylic and allylic position using N-halosuccinimide with azobisisobutyronitrile or benzoyl peroxide as a radical initiator is known as the Wohl–Ziegler bromination reaction, which requires heating, acidic/basic conditions, and/or UV irradiation (Scheme 1a) [17-20].
![[1860-5397-17-74-i1]](/bjoc/content/inline/1860-5397-17-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Several examples of C(sp3)–H halogenation.
Scheme 1: Several examples of C(sp3)–H halogenation.
There are several types of transition-metal-catalyzed C(sp3)−H halogenation reactions reported in the literature (Scheme 1b–d). Transition-metal-catalyzed 1,5-hydrogen atom transfer (1,5-HAT) is effective for promoting regioselective C(sp3)−H halogenation reactions (Scheme 1b) [21-23]. The regioselectivity is controlled by the formation of a six-membered cyclic intermediate. Directing-group-assisted C(sp3)−H halogenation reactions are efficient for promoting regioselective C(sp3)−H halogenations (Scheme 1c) [24-28]. In these reactions, the C(sp3)–H bond at the β-position of an oxazoline or amide is selectively activated using a copper or palladium catalyst.
Manganese is one of the most abundant and nontoxic transition metals found in the earth’s crust and its corresponding complexes and salts are useful in synthetic organic reactions [29-43]. Highly reactive and selective bromination reactions have been achieved using a stoichiometric amount of MnO2 [44] or a catalytic amount of Li2MnO3 [45] under fluorescent light irradiation in the presence of Br2 (Scheme 1d). Hill [46] and Groves [47-49] have reported the manganese-porphyrin-catalyzed chlorination and bromination of C(sp3)−H bonds, respectively (Scheme 1d). Groves et al. also reported the manganese-salen-catalyzed fluorination of benzylic C(sp3)−H bonds [49]. Although these methods are efficient, they have a limited substrate scope (cycloalkanes and substrates bearing a benzylic C–H group). Therefore, there remains room for the development of a simple manganese catalytic system to achieve C(sp3)−H halogenation using commercially available reagents.
Herein, we report a manganese-catalyzed C(sp3)–H bromination reaction at the methine and benzylic positions of a wide range of substrates. The manganese catalyst, brominating agent, and additives are commercially available, and the reaction can be achieved by simply mixing these reagents with the substrate.
Results and Discussion
The reaction of isoamyl alcohol derivative 1a with N-bromosuccinimide (NBS) and TMSN3 in the presence of a catalytic amount of Mn(OAc)2 and bipyridine (bpy) in 1,2-dichloroethane (DCE) at 60 °C for 18 h gave C(sp3)–H brominated product 2a in 10% yield (Table 1, entry 1). Although the yield of 2a did not increase when performing the reaction in acetonitrile (Table 1, entry 2), the yield of 2a was dramatically improved to 62% using PhCF3 as the solvent (Table 1, entry 3). Other manganese salts, such as MnBr2 and Mn(acac)2, were also effective in the reaction, giving similar yields (Table 1, entries 4 and 5). Other first-row transition metal salts, such as Fe(OAc)2 and Co(OAc)2, did not improve the yield of 2a (Table 1, entries 6 and 7). Product 2a was formed in 21 and 49% yields, respectively when the reaction was conducted in the absence of the transition metal salt and bpy ligand (Table 1, entries 8 and 9). TMSN3 was indispensable in this reaction because the C(sp3)–H bromination reaction did not occur in its absence (Table 1, entry 10). We then investigated the following experiments using the conditions described in entry 3.
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-74-i6.svg?max-width=637&scale=1.0)
|
|||
| entry | catalyst | solvent | yield (%)b |
| 1 | Mn(OAc)2 | DCE | 10 |
| 2 | Mn(OAc)2 | MeCN | 10 |
| 3 | Mn(OAc)2 | PhCF3 | 62 (53)c |
| 4 | MnBr2 | PhCF3 | 55 |
| 5 | Mn(acac)2 | PhCF3 | 54 |
| 6 | Fe(OAc)2 | PhCF3 | 42 |
| 7 | Co(OAc)2 | PhCF3 | 30 |
| 8 | – | PhCF3 | 21 |
| 9d | Mn(OAc)2 | PhCF3 | 49 |
| 10e | Mn(OAc)2 | PhCF3 | <1 |
aConditions: 1a (0.100 mmol, 1.0 equiv), NBS (0.300 mmol, 3.0 equiv), TMSN3 (0.200 mmol, 2.0 equiv), catalyst (10 mol %), bpy (10 mol %), solvent (0.50 mL). bThe 1H NMR yields were determined using 1,1,2,2-tetrachloroethane as an internal standard. cIsolated yield. dWithout bpy. eWithout TMSN3.
Under the optimized reaction conditions, we investigated the C(sp3)–H bromination reaction of several substrates (Scheme 2). The reaction proceeded regioselectively at the methine C(sp3)–H bond of isoamyl benzoate (1b) to give 2b in 64% yield. Isoamyl benzoates bearing halogen atoms, such as fluorine, chlorine, or bromine, on the phenyl ring were also suitable substrates and gave C(sp3)–H brominated products 2c–e in 49–60% yields, without any loss of the halogen substituents. Although the C(sp3)–H bromination of isobutyl benzoate 1f did not proceed at 60 °C, the corresponding C(sp3)–H brominated compound 2f was produced at higher temperature (80 °C). The C(sp3)–H bond in acetal 1g was efficiently brominated to give 2g in 79% yield. The reaction of adamantane (1h) proceeded selectively at the tertiary C(sp3)–H bond to give a mixture of mono- and dibrominated products (2h and 2h′). The selectivity of 2h and 2h′ can be controlled by varying the reaction time; mono-brominated 2h was obtained in 62% yield as the major product after 30 min of reaction and dibrominated 2h′ was afforded as the major product after 18 h. Similarly, 1,3-dimethyladamantane (1i) and methyl adamantane-1-carboxylate (1j) were successfully converted to brominated products 2i and 2j, respectively. For benzeneacetic acid methyl esters 1k, 1l and 1m, the C(sp3)–H bromination reaction proceeded selectively at the benzylic position and their corresponding brominated products (2k, 2l and 2m) were obtained in 61, 57 and 55% yield, respectively.
![[1860-5397-17-74-i2]](/bjoc/content/inline/1860-5397-17-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope. a80 °C. b45 min. c4 h. d90 °C, eGC yield of mono-brominated product 2n using mesitylene as internal standard.
Scheme 2: Substrate scope. a80 °C. b45 min. c4 h. d90 °C, eGC yield of mono-brominated product 2n using mesit...
We next investigated the regioselectivity of the reaction using substrates with two possible reaction sites. The reaction of substrate 1n bearing two methine C(sp3)–H bonds occurred selectively at the terminal position giving product 2n in 27% yield. The C(sp3)–H bromination reaction took place selectively at the methine C(sp3)–H bond when using substrate 1o, which has both methine and benzylic C(sp3)–H bonds, which gave product 2o in 30% yield.
The manganese-catalyzed C(sp3)–H bromination reaction proceeded in good yield, even on a gram scale. The reaction was performed using 2.61 g of 1a with NBS and TMSN3 in the presence of a catalytic amount of Mn(OAc)2 and bpy to give 1.98 g of 2a in 58% yield (Scheme 3).
![[1860-5397-17-74-i3]](/bjoc/content/inline/1860-5397-17-74-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The introduced bromine atom can be converted into other functional groups. The reaction of 2a with selectfluor in MeCN at 25 °C for 12 h gave fluorinated product 3 in 86% yield (Scheme 4, top) [50]. Allylated product 4 was obtained in 64% yield upon treating 2a with allyltributylstannane in the presence of a catalytic amount of AIBN (Scheme 4, bottom) [51].
![[1860-5397-17-74-i4]](/bjoc/content/inline/1860-5397-17-74-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Conversion of the C(sp3)–Br bond.
Scheme 4: Conversion of the C(sp3)–Br bond.
Table 1 shows that the C(sp3)–H bromination reaction proceeds in the absence of a transition metal salt or bpy ligand, and was accelerated by transition metal salts, especially Mn(OAc)2. In addition, the results also suggest that TMSN3 is required for the C(sp3)–H bromination reaction. The proposed reaction mechanism is shown in Scheme 5, which involves the following steps. (1) The reaction between NBS and TMSN3 generates bromine azide via the elimination of N-(trimethylsilyl)succinimide [52,53]; (2) bromine and azide radicals are then formed via homolytic cleavage of the weak Br–N3 bond in bromine azide [54,55]; (3) the bromine radical can also be generated from NBS with the formation of a succinimide radical; (4) alkyl radical intermediate A is then formed via hydrogen abstraction by the succinimidyl radical and/or azidyl radical [56,57]; (5) the Br–Mn(III) species is then formed from the Mn(II) catalyst and bromine radical; and (6) brominated product 2 formed by the reaction of intermediate A with the Br–Mn(III) species with the regeneration of the Mn(II) catalyst.
![[1860-5397-17-74-i5]](/bjoc/content/inline/1860-5397-17-74-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism of manganese-catalyzed C(sp3)–H bromination.
Scheme 5: Proposed mechanism of manganese-catalyzed C(sp3)–H bromination.
Conclusion
In summary, we have successfully developed a manganese-catalyzed bromination of unactivated aliphatic C(sp3)−H bonds. The reaction proceeded selectively at the methine and benzylic positions using simple and commercially available compounds, such as NBS, TMSN3, Mn(OAc)2, and bpy, even on a gram scale. Furthermore, the brominated products can be easily functionalized upon the introduction of other functional groups, such as fluorine and allyl groups. We hope that this C(sp3)–H bromination reaction will become a useful method to synthesize organic compounds with bromine atom(s).
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 4.8 MB | Download |
References
-
Rosseels, G.; Houben, C.; Kerckx, P. Synthesis of a metabolite of fantofarone. In Advances in Organobromine Chemistry II; Desmurs, J. R.; Gérard, B.; Goldstein, M. J., Eds.; Elsevier: Amsterdam, Netherlands, 1995; pp 152–159. doi:10.1016/s0926-9614(05)80016-4
Return to citation in text: [1] -
Cristau, H.-J.; Desmurs, J.-R. Arylation of hard heteroatomic nucleophiles using bromoarenes substrates and Cu, Ni, Pd-catalysts. In Advances in Organobromine Chemistry II; Desmurs, J. R.; Gérard, B.; Goldstein, M. J., Eds.; Elsevier: Amsterdam, Netherlands, 1995; pp 240–263. doi:10.1016/s0926-9614(05)80024-3
Return to citation in text: [1] -
Handbook of Grignard Reagents; Silverman, G. S.; Rakita, P. E., Eds.; CRC Press: Boca Raton, FL, USA, 1996. doi:10.1201/b16932
Return to citation in text: [1] -
Echavarren, A. M.; Cárdenas, D. J. Mechanistic Aspects of Metal-Catalyzed C, C- and C, X-Bond-Forming Reactions. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere, A.; Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/9783527619535
Return to citation in text: [1] -
Knochel, P.; Dohle, W.; Gommermann, N.; Kneisel, F. F.; Kopp, F.; Korn, T.; Sapountzis, I.; Vu, V. A. Angew. Chem., Int. Ed. 2003, 42, 4302–4320. doi:10.1002/anie.200300579
Return to citation in text: [1] -
Gribble, G. W. Chem. Soc. Rev. 1999, 28, 335–346. doi:10.1039/a900201d
Return to citation in text: [1] -
Kamm, O.; Marvel, C. S. Org. Synth. 1921, 1, 3–14. doi:10.15227/orgsyn.001.0003
Return to citation in text: [1] -
Wiley, G. A.; Hershkowitz, R. L.; Rein, B. M.; Chung, B. C. J. Am. Chem. Soc. 1964, 86, 964–965. doi:10.1021/ja01059a073
Return to citation in text: [1] -
Pelletier, J. D.; Poirier, D. Tetrahedron Lett. 1994, 35, 1051–1054. doi:10.1016/s0040-4039(00)79963-1
Return to citation in text: [1] -
Ferreri, C.; Costantino, C.; Chatgilialoglu, C.; Boukherroub, R.; Manuel, G. J. Organomet. Chem. 1998, 554, 135–137. doi:10.1016/s0022-328x(97)00667-0
Return to citation in text: [1] -
Iranpoor, N.; Firouzabadi, H.; Jamalian, A.; Kazemi, F. Tetrahedron 2005, 61, 5699–5704. doi:10.1016/j.tet.2005.01.115
Return to citation in text: [1] -
Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949
Return to citation in text: [1] -
Wang, G.-Z.; Shang, R.; Cheng, W.-M.; Fu, Y. J. Am. Chem. Soc. 2017, 139, 18307–18312. doi:10.1021/jacs.7b10009
Return to citation in text: [1] -
Shaw, H.; Perlmutter, H. D.; Gu, C.; Arco, S. D.; Quibuyen, T. O. J. Org. Chem. 1997, 62, 236–237. doi:10.1021/jo950371b
Return to citation in text: [1] -
Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109
Return to citation in text: [1] -
Nishina, Y.; Ohtani, B.; Kikushima, K. Beilstein J. Org. Chem. 2013, 9, 1663–1667. doi:10.3762/bjoc.9.190
Return to citation in text: [1] -
Wohl, A. Ber. Dtsch. Chem. Ges. B 1919, 52, 51–63. doi:10.1002/cber.19190520109
Return to citation in text: [1] -
Ziegler, K.; Schenck, G.; Krockow, E. W.; Siebert, A.; Wenz, A.; Weber, H. Justus Liebigs Ann. Chem. 1942, 551, 1–79. doi:10.1002/jlac.19425510102
Return to citation in text: [1] -
Djerassi, C. Chem. Rev. 1948, 43, 271–317. doi:10.1021/cr60135a004
Return to citation in text: [1] -
Wang, Y.; Li, G.-X.; Yang, G.; He, G.; Chen, G. Chem. Sci. 2016, 7, 2679–2683. doi:10.1039/c5sc04169d
Return to citation in text: [1] -
Liu, T.; Mei, T.-S.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 5871–5874. doi:10.1021/jacs.5b02065
Return to citation in text: [1] -
Liu, T.; Myers, M. C.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 306–309. doi:10.1002/anie.201608210
Return to citation in text: [1] -
Sathyamoorthi, S.; Banerjee, S.; Du Bois, J.; Burns, N. Z.; Zare, R. N. Chem. Sci. 2018, 9, 100–104. doi:10.1039/c7sc04611a
Return to citation in text: [1] -
Giri, R.; Chen, X.; Yu, J.-Q. Angew. Chem., Int. Ed. 2005, 44, 2112–2115. doi:10.1002/anie.200462884
Return to citation in text: [1] -
Giri, R.; Wasa, M.; Breazzano, S. P.; Yu, J.-Q. Org. Lett. 2006, 8, 5685–5688. doi:10.1021/ol0618858
Return to citation in text: [1] -
Rit, R. K.; Yadav, M. R.; Ghosh, K.; Shankar, M.; Sahoo, A. K. Org. Lett. 2014, 16, 5258–5261. doi:10.1021/ol502337b
Return to citation in text: [1] -
Yang, X.; Sun, Y.; Sun, T.-y.; Rao, Y. Chem. Commun. 2016, 52, 6423–6426. doi:10.1039/c6cc00234j
Return to citation in text: [1] -
Zhu, R.-Y.; Saint-Denis, T. G.; Shao, Y.; He, J.; Sieber, J. D.; Senanayake, C. H.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 5724–5727. doi:10.1021/jacs.7b02196
Return to citation in text: [1] -
Kuninobu, Y.; Takai, K. Bull. Chem. Soc. Jpn. 2012, 85, 656–671. doi:10.1246/bcsj.20120015
Return to citation in text: [1] -
Kuninobu, Y.; Sueki, S.; Kaplaneris, N.; Ackermann, L. Manganese Catalysis. In Catalysis with Earth-abundant Elements; Schneider, U.; Thomas, S., Eds.; The Royal Society of Chemistry: Cambridge. UK, 2020; pp 139–230. doi:10.1039/9781788012775-00139
Return to citation in text: [1] -
Rohit, K. R.; Radhika, S.; Saranya, S.; Anilkumar, G. Adv. Synth. Catal. 2020, 362, 1602–1650. doi:10.1002/adsc.201901389
Return to citation in text: [1] -
Wang, Y.; Liu, Q. Synlett 2020, 31, 1464–1473. doi:10.1055/s-0040-1707126
Return to citation in text: [1] -
Chandra, P.; Ghosh, T.; Choudhary, N.; Mohammad, A.; Mobin, S. M. Coord. Chem. Rev. 2020, 411, 213241. doi:10.1016/j.ccr.2020.213241
Return to citation in text: [1] -
Waiba, S.; Maji, B. ChemCatChem 2020, 12, 1891–1902. doi:10.1002/cctc.201902180
Return to citation in text: [1] -
Kuninobu, Y.; Nishina, Y.; Takeuchi, T.; Takai, K. Angew. Chem., Int. Ed. 2007, 46, 6518–6520. doi:10.1002/anie.200702256
Return to citation in text: [1] -
Kuninobu, Y.; Kikuchi, K.; Takai, K. Chem. Lett. 2008, 37, 740–741. doi:10.1246/cl.2008.740
Return to citation in text: [1] -
Kuninobu, Y.; Nishi, M.; Yudha S., S.; Takai, K. Org. Lett. 2008, 10, 3009–3011. doi:10.1021/ol800969h
Return to citation in text: [1] -
Kuninobu, Y.; Kawata, A.; Nishi, M.; Takata, H.; Takai, K. Chem. Commun. 2008, 6360–6362. doi:10.1039/b814694b
Return to citation in text: [1] -
Kuninobu, Y.; Kawata, A.; Nishi, M.; Yudha S., S.; Chen, J.; Takai, K. Chem. – Asian J. 2009, 4, 1424–1433. doi:10.1002/asia.200900137
Return to citation in text: [1] -
Kuninobu, Y.; Nishi, M.; Kawata, A.; Takata, H.; Hanatani, Y.; Yudha S., S.; Iwai, A.; Takai, K. J. Org. Chem. 2010, 75, 334–341. doi:10.1021/jo902072q
Return to citation in text: [1] -
Kuninobu, Y.; Kawata, A.; Yudha, S. S.; Takata, H.; Nishi, M.; Takai, K. Pure Appl. Chem. 2010, 82, 1491–1501. doi:10.1351/pac-con-09-09-21
Return to citation in text: [1] -
Kuninobu, Y.; Uesugi, T.; Kawata, A.; Takai, K. Angew. Chem., Int. Ed. 2011, 50, 10406–10408. doi:10.1002/anie.201104704
Return to citation in text: [1] -
Sueki, S.; Wang, Z.; Kuninobu, Y. Org. Lett. 2016, 18, 304–307. doi:10.1021/acs.orglett.5b03474
Return to citation in text: [1] -
Jiang, X.; Shen, M.; Tang, Y.; Li, C. Tetrahedron Lett. 2005, 46, 487–489. doi:10.1016/j.tetlet.2004.11.113
Return to citation in text: [1] -
Nishina, Y.; Morita, J.; Ohtani, B. RSC Adv. 2013, 3, 2158–2162. doi:10.1039/c2ra22197g
Return to citation in text: [1] -
Hill, C. L.; Smegal, J. A.; Henly, T. J. J. Org. Chem. 1983, 48, 3277–3281. doi:10.1021/jo00167a023
Return to citation in text: [1] -
Liu, W.; Groves, J. T. J. Am. Chem. Soc. 2010, 132, 12847–12849. doi:10.1021/ja105548x
Return to citation in text: [1] -
Liu, W.; Groves, J. T. Acc. Chem. Res. 2015, 48, 1727–1735. doi:10.1021/acs.accounts.5b00062
Return to citation in text: [1] -
Liu, W.; Groves, J. T. Angew. Chem., Int. Ed. 2013, 52, 6024–6027. doi:10.1002/anie.201301097
Return to citation in text: [1] [2] -
Chen, H.; Liu, Z.; Lv, Y.; Tan, X.; Shen, H.; Yu, H.-Z.; Li, C. Angew. Chem., Int. Ed. 2017, 56, 15411–15415. doi:10.1002/anie.201708197
Return to citation in text: [1] -
Keck, G. E.; Yates, J. B. J. Am. Chem. Soc. 1982, 104, 5829–5831. doi:10.1021/ja00385a066
Return to citation in text: [1] -
Van Ende, D.; Krief, A. Angew. Chem., Int. Ed. Engl. 1974, 13, 279–280. doi:10.1002/anie.197402792
Return to citation in text: [1] -
Saikia, I.; Phukan, P. C. R. Chim. 2012, 15, 688–692. doi:10.1016/j.crci.2012.05.001
Return to citation in text: [1] -
Hassner, A.; Boerwinkle, F.; Lavy, A. B. J. Am. Chem. Soc. 1970, 92, 4879–4883. doi:10.1021/ja00719a021
Return to citation in text: [1] -
Hassner, A. Acc. Chem. Res. 1971, 4, 9–16. doi:10.1021/ar50037a002
Return to citation in text: [1] -
Chalfont, G. R.; Perkins, M. J.; Horsfield, A. J. Chem. Soc. B 1970, 401–404. doi:10.1039/j29700000401
Return to citation in text: [1] -
Workentin, M. S.; Wagner, B. D.; Lusztyk, J.; Wayner, D. D. M. J. Am. Chem. Soc. 1995, 117, 119–126. doi:10.1021/ja00106a015
Return to citation in text: [1]
| 56. | Chalfont, G. R.; Perkins, M. J.; Horsfield, A. J. Chem. Soc. B 1970, 401–404. doi:10.1039/j29700000401 |
| 57. | Workentin, M. S.; Wagner, B. D.; Lusztyk, J.; Wayner, D. D. M. J. Am. Chem. Soc. 1995, 117, 119–126. doi:10.1021/ja00106a015 |
| 1. | Rosseels, G.; Houben, C.; Kerckx, P. Synthesis of a metabolite of fantofarone. In Advances in Organobromine Chemistry II; Desmurs, J. R.; Gérard, B.; Goldstein, M. J., Eds.; Elsevier: Amsterdam, Netherlands, 1995; pp 152–159. doi:10.1016/s0926-9614(05)80016-4 |
| 2. | Cristau, H.-J.; Desmurs, J.-R. Arylation of hard heteroatomic nucleophiles using bromoarenes substrates and Cu, Ni, Pd-catalysts. In Advances in Organobromine Chemistry II; Desmurs, J. R.; Gérard, B.; Goldstein, M. J., Eds.; Elsevier: Amsterdam, Netherlands, 1995; pp 240–263. doi:10.1016/s0926-9614(05)80024-3 |
| 3. | Handbook of Grignard Reagents; Silverman, G. S.; Rakita, P. E., Eds.; CRC Press: Boca Raton, FL, USA, 1996. doi:10.1201/b16932 |
| 4. | Echavarren, A. M.; Cárdenas, D. J. Mechanistic Aspects of Metal-Catalyzed C, C- and C, X-Bond-Forming Reactions. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere, A.; Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/9783527619535 |
| 5. | Knochel, P.; Dohle, W.; Gommermann, N.; Kneisel, F. F.; Kopp, F.; Korn, T.; Sapountzis, I.; Vu, V. A. Angew. Chem., Int. Ed. 2003, 42, 4302–4320. doi:10.1002/anie.200300579 |
| 6. | Gribble, G. W. Chem. Soc. Rev. 1999, 28, 335–346. doi:10.1039/a900201d |
| 21. | Liu, T.; Mei, T.-S.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 5871–5874. doi:10.1021/jacs.5b02065 |
| 22. | Liu, T.; Myers, M. C.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 306–309. doi:10.1002/anie.201608210 |
| 23. | Sathyamoorthi, S.; Banerjee, S.; Du Bois, J.; Burns, N. Z.; Zare, R. N. Chem. Sci. 2018, 9, 100–104. doi:10.1039/c7sc04611a |
| 52. | Van Ende, D.; Krief, A. Angew. Chem., Int. Ed. Engl. 1974, 13, 279–280. doi:10.1002/anie.197402792 |
| 53. | Saikia, I.; Phukan, P. C. R. Chim. 2012, 15, 688–692. doi:10.1016/j.crci.2012.05.001 |
| 17. | Wohl, A. Ber. Dtsch. Chem. Ges. B 1919, 52, 51–63. doi:10.1002/cber.19190520109 |
| 18. | Ziegler, K.; Schenck, G.; Krockow, E. W.; Siebert, A.; Wenz, A.; Weber, H. Justus Liebigs Ann. Chem. 1942, 551, 1–79. doi:10.1002/jlac.19425510102 |
| 19. | Djerassi, C. Chem. Rev. 1948, 43, 271–317. doi:10.1021/cr60135a004 |
| 20. | Wang, Y.; Li, G.-X.; Yang, G.; He, G.; Chen, G. Chem. Sci. 2016, 7, 2679–2683. doi:10.1039/c5sc04169d |
| 54. | Hassner, A.; Boerwinkle, F.; Lavy, A. B. J. Am. Chem. Soc. 1970, 92, 4879–4883. doi:10.1021/ja00719a021 |
| 55. | Hassner, A. Acc. Chem. Res. 1971, 4, 9–16. doi:10.1021/ar50037a002 |
| 14. | Shaw, H.; Perlmutter, H. D.; Gu, C.; Arco, S. D.; Quibuyen, T. O. J. Org. Chem. 1997, 62, 236–237. doi:10.1021/jo950371b |
| 15. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109 |
| 16. | Nishina, Y.; Ohtani, B.; Kikushima, K. Beilstein J. Org. Chem. 2013, 9, 1663–1667. doi:10.3762/bjoc.9.190 |
| 50. | Chen, H.; Liu, Z.; Lv, Y.; Tan, X.; Shen, H.; Yu, H.-Z.; Li, C. Angew. Chem., Int. Ed. 2017, 56, 15411–15415. doi:10.1002/anie.201708197 |
| 7. | Kamm, O.; Marvel, C. S. Org. Synth. 1921, 1, 3–14. doi:10.15227/orgsyn.001.0003 |
| 8. | Wiley, G. A.; Hershkowitz, R. L.; Rein, B. M.; Chung, B. C. J. Am. Chem. Soc. 1964, 86, 964–965. doi:10.1021/ja01059a073 |
| 9. | Pelletier, J. D.; Poirier, D. Tetrahedron Lett. 1994, 35, 1051–1054. doi:10.1016/s0040-4039(00)79963-1 |
| 10. | Ferreri, C.; Costantino, C.; Chatgilialoglu, C.; Boukherroub, R.; Manuel, G. J. Organomet. Chem. 1998, 554, 135–137. doi:10.1016/s0022-328x(97)00667-0 |
| 11. | Iranpoor, N.; Firouzabadi, H.; Jamalian, A.; Kazemi, F. Tetrahedron 2005, 61, 5699–5704. doi:10.1016/j.tet.2005.01.115 |
| 12. | Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949 |
| 13. | Wang, G.-Z.; Shang, R.; Cheng, W.-M.; Fu, Y. J. Am. Chem. Soc. 2017, 139, 18307–18312. doi:10.1021/jacs.7b10009 |
| 51. | Keck, G. E.; Yates, J. B. J. Am. Chem. Soc. 1982, 104, 5829–5831. doi:10.1021/ja00385a066 |
| 45. | Nishina, Y.; Morita, J.; Ohtani, B. RSC Adv. 2013, 3, 2158–2162. doi:10.1039/c2ra22197g |
| 47. | Liu, W.; Groves, J. T. J. Am. Chem. Soc. 2010, 132, 12847–12849. doi:10.1021/ja105548x |
| 48. | Liu, W.; Groves, J. T. Acc. Chem. Res. 2015, 48, 1727–1735. doi:10.1021/acs.accounts.5b00062 |
| 49. | Liu, W.; Groves, J. T. Angew. Chem., Int. Ed. 2013, 52, 6024–6027. doi:10.1002/anie.201301097 |
| 44. | Jiang, X.; Shen, M.; Tang, Y.; Li, C. Tetrahedron Lett. 2005, 46, 487–489. doi:10.1016/j.tetlet.2004.11.113 |
| 49. | Liu, W.; Groves, J. T. Angew. Chem., Int. Ed. 2013, 52, 6024–6027. doi:10.1002/anie.201301097 |
| 29. | Kuninobu, Y.; Takai, K. Bull. Chem. Soc. Jpn. 2012, 85, 656–671. doi:10.1246/bcsj.20120015 |
| 30. | Kuninobu, Y.; Sueki, S.; Kaplaneris, N.; Ackermann, L. Manganese Catalysis. In Catalysis with Earth-abundant Elements; Schneider, U.; Thomas, S., Eds.; The Royal Society of Chemistry: Cambridge. UK, 2020; pp 139–230. doi:10.1039/9781788012775-00139 |
| 31. | Rohit, K. R.; Radhika, S.; Saranya, S.; Anilkumar, G. Adv. Synth. Catal. 2020, 362, 1602–1650. doi:10.1002/adsc.201901389 |
| 32. | Wang, Y.; Liu, Q. Synlett 2020, 31, 1464–1473. doi:10.1055/s-0040-1707126 |
| 33. | Chandra, P.; Ghosh, T.; Choudhary, N.; Mohammad, A.; Mobin, S. M. Coord. Chem. Rev. 2020, 411, 213241. doi:10.1016/j.ccr.2020.213241 |
| 34. | Waiba, S.; Maji, B. ChemCatChem 2020, 12, 1891–1902. doi:10.1002/cctc.201902180 |
| 35. | Kuninobu, Y.; Nishina, Y.; Takeuchi, T.; Takai, K. Angew. Chem., Int. Ed. 2007, 46, 6518–6520. doi:10.1002/anie.200702256 |
| 36. | Kuninobu, Y.; Kikuchi, K.; Takai, K. Chem. Lett. 2008, 37, 740–741. doi:10.1246/cl.2008.740 |
| 37. | Kuninobu, Y.; Nishi, M.; Yudha S., S.; Takai, K. Org. Lett. 2008, 10, 3009–3011. doi:10.1021/ol800969h |
| 38. | Kuninobu, Y.; Kawata, A.; Nishi, M.; Takata, H.; Takai, K. Chem. Commun. 2008, 6360–6362. doi:10.1039/b814694b |
| 39. | Kuninobu, Y.; Kawata, A.; Nishi, M.; Yudha S., S.; Chen, J.; Takai, K. Chem. – Asian J. 2009, 4, 1424–1433. doi:10.1002/asia.200900137 |
| 40. | Kuninobu, Y.; Nishi, M.; Kawata, A.; Takata, H.; Hanatani, Y.; Yudha S., S.; Iwai, A.; Takai, K. J. Org. Chem. 2010, 75, 334–341. doi:10.1021/jo902072q |
| 41. | Kuninobu, Y.; Kawata, A.; Yudha, S. S.; Takata, H.; Nishi, M.; Takai, K. Pure Appl. Chem. 2010, 82, 1491–1501. doi:10.1351/pac-con-09-09-21 |
| 42. | Kuninobu, Y.; Uesugi, T.; Kawata, A.; Takai, K. Angew. Chem., Int. Ed. 2011, 50, 10406–10408. doi:10.1002/anie.201104704 |
| 43. | Sueki, S.; Wang, Z.; Kuninobu, Y. Org. Lett. 2016, 18, 304–307. doi:10.1021/acs.orglett.5b03474 |
| 24. | Giri, R.; Chen, X.; Yu, J.-Q. Angew. Chem., Int. Ed. 2005, 44, 2112–2115. doi:10.1002/anie.200462884 |
| 25. | Giri, R.; Wasa, M.; Breazzano, S. P.; Yu, J.-Q. Org. Lett. 2006, 8, 5685–5688. doi:10.1021/ol0618858 |
| 26. | Rit, R. K.; Yadav, M. R.; Ghosh, K.; Shankar, M.; Sahoo, A. K. Org. Lett. 2014, 16, 5258–5261. doi:10.1021/ol502337b |
| 27. | Yang, X.; Sun, Y.; Sun, T.-y.; Rao, Y. Chem. Commun. 2016, 52, 6423–6426. doi:10.1039/c6cc00234j |
| 28. | Zhu, R.-Y.; Saint-Denis, T. G.; Shao, Y.; He, J.; Sieber, J. D.; Senanayake, C. H.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 5724–5727. doi:10.1021/jacs.7b02196 |
| 46. | Hill, C. L.; Smegal, J. A.; Henly, T. J. J. Org. Chem. 1983, 48, 3277–3281. doi:10.1021/jo00167a023 |
© 2021 Sneh et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)