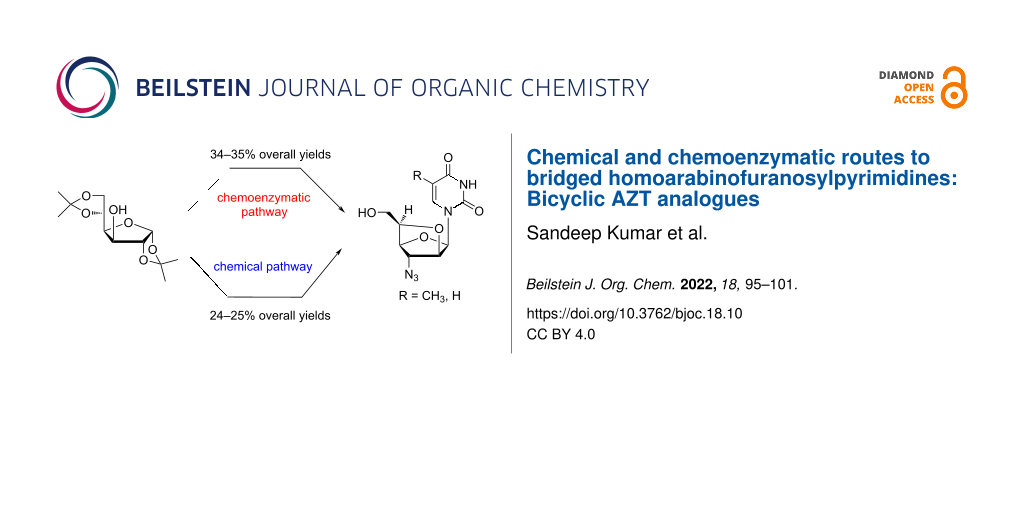
Abstract
Conformationally restricted diastereomeric homoarabinofuranosylpyrimidines (AZT analogue), i.e., (5′R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosylthymine and -uracil had been synthesized starting from diacetone ᴅ-glucofuranose following chemoenzymatic and chemical routes in 34–35% and 24–25% overall yields, respectively. The quantitative and diastereoselective acetylation of primary hydroxy over two secondary hydroxy groups present in the key nucleoside precursor was mediated with Lipozyme® TL IM in 2-methyltetrahydrofuran following a chemoenzymatic pathway. Whereas, the protection of the primary hydroxy over the lone secondary hydroxy group in the key azido sugar precursor was achieved using bulky tert-butyldiphenylsilyl chloride (TBDPS-Cl) in pyridine in 92% yield following a chemical synthetic pathway. The chemoenzymatic method was found to be superior over the chemical method in respect of the number of synthetic steps and overall yield of the final product.

Graphical Abstract
Introduction
In the last few decades, modification of nucleoside/nucleotide analogues has been a field of keen interest to researchers due to their therapeutic properties for treatment of cancer, viral and microbial infections [1-9]. The very first cytotoxic chemotherapeutic agents used for the treatment of cancer were nucleoside analogues and nucleobases [10]. Azidothymidine (1, AZT) was the first approved drug for the treatment of human immunodeficiency virus (HIV) [11,12]. Subsequently, a large number of sugar modified nucleosides, such as ddC (zalcitabine) [13,14], ddI (didanosine) [15], d4T (stavudine) [16,17], 3TC (lamivudine) [18,19] and AZT analogues were synthesized and evaluated towards inhibition of HIV reverse transcriptase (HIV RT). However, the toxic side effects associated with these molecules and development of drug-resistant viruses eventually brought about the need for newly designed and improved anti-HIV drugs with respect to their improved pharmacological properties [20]. With an idea to keep the presence of azide functionality and to introduce structural rigidity, Marquez et al. [21] developed a methodology for the synthesis of azido-carbobicyclic nucleosides 2 and 3, where compound 2 showed activity similar to that of AZT while compound 3 was devoid of any type of activity (Figure 1). Nielsen et al. [22] synthesized the conformationally restricted nucleoside 4, which was devoid of any anti-HIV activity. Further, Zhang et al. [23] synthesized the tricyclic azido-isonucleoside analogue 5 and Imanishi et al. [24] synthesized the azido-nucleoside 6 with a conformationally restricted sugar moiety. Recently, we have developed a greener and efficient chemoenzymatic synthetic methodology for the synthesis of conformationally restricted bicyclic azido-nucleosides 6 [25] and 7 [26], and bicyclic homonucleosides 8 [27]. Herein, we report an efficient chemo-enzymatic and chemical route to synthesize two novel conformationally restricted azido-homoarabino nucleosides 9a,b in excellent overall yields (Figure 1). To the best of our knowledge, this is the first report of a synthesis of azido-bicyclic β-ᴅ-homoarabinofuranosyl nucleosides, i.e., bicyclic homonucleoside analogues of AZT.
![[1860-5397-18-10-1]](/bjoc/content/figures/1860-5397-18-10-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of AZT and representative related bicyclic nucleosides.
Figure 1: Structure of AZT and representative related bicyclic nucleosides.
Results and Discussion
It was envisioned to synthesize targeted bridged homoarabinofuranosyl nucleosides starting from diacetone-ᴅ-glucofuranose following chemoenzymatic and chemical pathways (Scheme 1).
![[1860-5397-18-10-i1]](/bjoc/content/inline/1860-5397-18-10-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic routes for the synthesis of (5′R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosylpyrimidines following a chemoenzymatic pathway (a) and a chemical pathway (b).
Scheme 1: Retrosynthetic routes for the synthesis of (5′R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabin...
Thus, in the chemoenzymatic approach, the synthesis of targeted bicyclic homonucleosides started with the conversion of diacetone ᴅ-glucofuranose 10 into 3-azido-3-deoxydiacetone-ᴅ-allofuranoside 11 following a literature procedure in 53% yield [28]. Azidofuranoside 11 on acetolysis with acetic acid/acetic anhydride/sulfuric acid (100:10:0.1) at room temperature afforded an anomeric mixture of 1,2,5,6-tetra-O-acetyl-3-azido-3-deoxy-α,β-ᴅ-allofuranose (12a,b), which on Vorbrüggen base coupling [29] with thymine and uracil in the presence of N,O-bis(trimethylsilyl)acetamide (BSA) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) afforded nucleosides 13a,b. Further, deacetylation of the triacetylated azidoallofuranosyl nucleosides 13a,b with K2CO3 in methanol/water led to the formation of 3′-azido-3′-deoxy-β-ᴅ-allofuranosyl nucleosides 14a,b in 85% overall yield starting from compound 11, respectively (Scheme 2).
![[1860-5397-18-10-i2]](/bjoc/content/inline/1860-5397-18-10-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 3′-azido-3′-deoxy-β-ᴅ-allofuranosylthymine (14a) and -uracil (14b).
Scheme 2: Synthesis of 3′-azido-3′-deoxy-β-ᴅ-allofuranosylthymine (14a) and -uracil (14b).
The use of lipase as biocatalyst was employed for the selective acetylation of the primary hydroxy group present in trihydroxy nucleosides 14a,b. This led to the screening of two different lipases, viz Candida antarctica lipase-B (CAL-B) immobilized on polyacrylate (Lewatit), commonly known as Novozyme® 435 and Thermomyces lanuginosus lipase immobilized on silica, commonly known as Lipozyme® TL IM in different organic solvents, such as THF, acetonitrile, toluene, acetone, DIPE and 2-Me-THF. Vinyl acetate was used as acetyl donor at temperatures ranging from 25–45 °C in an incubator shaker at 200 rpm. Lipozyme® TL IM (10 wt % of the substrate) in 2-Me-THF at 40 °C was found to be the most suitable solvent for exclusive acetylation of the primary hydroxy group present in compounds 14a,b in quantitative yields (Scheme 3). Further, the reusability of the recovered Lipozyme® TL IM was also checked and it was found that the lipase could be used up to seven cycles of selective acetylation of 3′-azido-3′-deoxy-β-ᴅ-allofuranosyl nucleosides 14a,b without noticeable loss of regioselectivity and efficiency. Thus, trihydroxy nucleosides 14a,b were treated in 2-Me-THF with Lipozyme® TL IM (10 wt % of the substrate nucleoside) and vinyl acetate at 40 °C and 200 rpm in an incubator shaker to afford the monoacetylated nucleosides 15a,b in quantitative yields in 2 h (Scheme 3).
![[1860-5397-18-10-i3]](/bjoc/content/inline/1860-5397-18-10-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Biocatalytic acetylation of the primary hydroxy group of nucleosides 14a,b.
Scheme 3: Biocatalytic acetylation of the primary hydroxy group of nucleosides 14a,b.
The monoacetylated nucleosides 15a,b were mesylated using mesyl chloride in pyridine to afford dimesylated nucleosides 16a,b in 93 and 94% yields, respectively. The reaction of nucleosides 16a,b with NaOH in dioxane/water (1:1) underwent a cascade reaction pathway to form 9a,b in 82 and 84% yields, respectively (Scheme 4). The overall yields for the synthesis of nucleosides 9a,b starting from diacetone ᴅ-glucofuranose were found to be 33.9 and 35.1%, respectively.
![[1860-5397-18-10-i4]](/bjoc/content/inline/1860-5397-18-10-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of (5'R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosyl nucleosides 9a,b.
Scheme 4: Synthesis of (5'R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosyl nucleosides 9a,b.
In our earlier research work, bridged homolyxofuranosylpyrimidines were synthesized following a chemoenzymatic methodology where biocatalyst Novozyme 435 was used for regioselective acetylation of the primary hydroxy group present in 3′-O-benzyl-β-ᴅ-glucofuranosylpyrimidines [27]. In this article, regioselective acylation at the primary hydroxy group of 3′-azido-3′-deoxy-β-ᴅ-allofuranosylpyrimidines was carried out with the different biocatalyst Lipozyme TL IM using the same acylating agent, i.e., vinyl acetate but in a different solvent, i.e., 2-Me-THF instead of THF and at a different temperature, i.e., 40 °C instead of 45 oC as mentioned in the previous reaction conditions. It is noteworthy to mention that both of these biocatalytic reactions afforded the desired products in quantitative yield.
At first, trihydroxy nucleoside 14b was protected with TBDPS to carry out the synthesis of the target nucleoside monomer 9b through a chemical pathway. However, the poor yield of this reaction compelled us to abandon the route as high solubility of the TBDPS-protected dihydroxy nucleoside in aqueous phase withstood its extraction in the organic phase during work up procedure. So, the classical chemical route to synthesize nucleosides 9a,b started with the conversion of diacetone ᴅ-glucofuranose 10 to 3′-azido-3′-deoxy-β-ᴅ-allofuranoside 11, which on acidification with 80% acetic acid gave furanoside diol 17. The regioselective protection of the primary hydroxy group of diol 17 using TBDPS-Cl in pyridine at room temperature afforded TBDPS-protected furanoside 18, which on acetolysis using AcOH/Ac2O/H2SO4 (100:10:0.1) afforded the anomeric mixture of coupling sugar 19a,b in 80% yield. The Vorbrüggen coupling [29] of anomeric triacetylated sugars 19a,b with thymine and uracil in the presence of BSA and TMSOTf in acetonitrile afforded nucleosides 20a,b, respectively in high yields. The deacetylation of these nucleosides was carried out using K2CO3 in a solvent mixture of methanol and water (9:1) to afford nucleoside 21a,b, which in turn on mesylation with mesyl chloride in pyridine afforded dimesylated nucleosides 22a,b. Nucleosides 22a,b on treatment with aqueous sodium hydroxide in a solvent mixture of dioxane and water (1:1) afforded 2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosyl nucleosides 9a,b in 84 and 83% yields, respectively (Scheme 5). The overall yields of the synthesis of nucleosides 9a and 9b starting from diacetone ᴅ-glucofuranose following the classical chemical pathway were found to be 24.2 and 24.4%, respectively.
![[1860-5397-18-10-i5]](/bjoc/content/inline/1860-5397-18-10-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Chemical synthesis of nucleosides 9a,b starting from diacetone ᴅ-glucofuranose.
Scheme 5: Chemical synthesis of nucleosides 9a,b starting from diacetone ᴅ-glucofuranose.
The stereochemistry at the C5′ carbon in bicyclic homonucleosides 9a,b was established on the basis of 1H,1H-COSY and NOESY NMR spectral data analysis of nucleoside 9a (Figure 2). Herein, cross peaks were observed in the NOESY spectrum of compound 9a due to the spatial interaction of the C6′ methylene protons resonating at δ 3.41 with the C3′ proton resonating at δ 4.73 and the C5′ proton resonating at δ 4.15 with the C6 proton of the nucleobase resonating at δ 7.67 (see Supporting Information File 1). Based on this analysis of the spatial arrangement of the groups attached at the C5′ chiral centre, the stereochemistry of this carbon centre was assigned to be (R) for both of these nucleosides.
![[1860-5397-18-10-2]](/bjoc/content/figures/1860-5397-18-10-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure elucidation of (5′R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosylthymine (9a).
Figure 2: Structure elucidation of (5′R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosylthymine (...
The study of NOESY NMR spectral data of compound 9a enabled us to assign the stereochemistry at the C5′ chiral centre in the compound as (R). Following this trend, we assigned the same stereochemistry for the C5′ centre of nucleoside 9b. Interestingly, we noticed that the stereochemistry of the C5′ centre was unaltered in the dimesylated allofuranosyl nucleosides 16a,b or 22a,b and the corresponding bridged homoarabinofuranosylpyrimidine nucleosides 9a,b, whereas we expected an inversion of stereochemistry as it was noticed in earlier literature [30,31]. Depending on this observation, we postulated a mechanistic pathway for this cyclization process, where two consecutive inversions of configuration at the C5′ centre enabled the retention of stereochemistry. Thus, formation of 5,6-epoxide moieties was common in case of hexose carbohydrates, where the C6-OH group attacked the C5 centre and substituted the mesyl group present at C5 to form 5,6-epoxide moieties [32-34]. Herein, we elucidated the mechanistic pathway for conversion of 16a,b into 9a,b through the formation of an intermediate epoxide II, which has (S) stereochemistry at the C5′ due to first inversion of configuration by attack from the C6′-OH group (Scheme 6). The second inversion at the C5′ centre occurred when the C2′-OH attacked the same chiral centre and opened the epoxide ring by SN2 reaction and inverted the configuration of the centre into (R). In the chemical route for synthesis of bridged nucleosides 9a,b, an unusual removal of TBDPS was observed when nucleosides 22a,b were treated with 2 M NaOH in dioxane and water (1:1). A literature search showed that the TBDPS protecting group could be removed under basic conditions [35,36]. However, removal of the TBDPS group from the primary hydroxy group of a nucleoside under basic conditions of 2 M NaOH in dioxane and water (1:1) was the first of its kind in the literature. The progress of the reaction (from nucleoside 22b to nucleoside 9b) was investigated by mass spectrometry (MS) and a sample of the reaction mixture was collected during the reaction. The mass spectral analysis showed molecular ion peaks ([M + H]+) corresponding to the intermediates I′, I′′, II′ and III′ (Scheme 6), which illustrated the removal of TBDPS from the nucleoside and explained the retention of configuration at the C5′ centre. So, we assembled the identified intermediates in a most possible sequence to explain the progress of the reaction (Scheme 6). The sequence of the intermediates also described that epoxide formation occurred after removal of TBDPS and then attack from C2′-OH happened at the C5′ centre, i.e., two consecutive SN2 reactions at the C5′ centre, which retained the configuration of that chiral centre as (R) in the 2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosyl-nucleosides 9a,b.
![[1860-5397-18-10-i6]](/bjoc/content/inline/1860-5397-18-10-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: a) Plausible mechanism for the synthesis of (5′R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabinofuranosyl nucleosides 9a,b from 16a,b and b) intermediates detected on the basis of mass spectrometry during the conversion of 22b into 9b.
Scheme 6: a) Plausible mechanism for the synthesis of (5′R)-3′-azido-3′-deoxy-2′-O,5′-C-bridged-β-ᴅ-homoarabi...
The structures of all the synthesized compounds 9a,b, 11, 12a,b, 13a,b, 14a,b, 15a,b, 16a,b, 17, 18, 19, 20a,b, 21a,b, and 22a,b were unambiguously established on the basis of their spectral (IR, 1H, 13C NMR, 1H,1H-COSY NMR, 1H,13C-HETCOR NMR, NOESY NMR and HRMS) data analysis. The structures of known compounds 11 and 17 were further confirmed by comparison of their physical and spectral data with those reported in literature [28].
Conclusion
The efficiency of Lipozyme® TL IM was discovered for exclusive acetylation of the primary hydroxy group in 3′-azido-3′-deoxy-β-ᴅ-allofuranosylthymine and -uracil over the two secondary hydroxy groups present in the molecule. The enzymatically prepared monoacetylated 3′-azido-3′-deoxy-β-ᴅ-allofuranosylthymine and -uracil were conveniently converted into novel AZT bicyclic analogues, 3′-azido-3′-deoxy-2′-O,5′-C-bridged-homoarabinofuranosylthymine and -uracil in excellent yields. These nucleosides were also accessed through the chemical methodology in good yields. The biological activity, particularly antiviral activity study of these compounds will be carried out and reported separately.
Supporting Information
| Supporting Information File 1: Experimental part and NMR spectra. | ||
| Format: PDF | Size: 3.6 MB | Download |
Funding
We are grateful to the Institute of Imminence, University of Delhi for providing financial support to strengthen research and development. We are thankful to the DST-FIST program, CIF-USIC and Department of Chemistry, University of Delhi for providing the NMR spectral and HRMS recording facilities. Sandeep Kumar thanks CSIR, New Delhi for an award of a SPM Research Fellowship [File No. 09/045(0269)/2018-EMR-1]. Sumit Kumar thanks CSIR, New Delhi for an award of a Junior Research Fellowship.
References
-
Sharma, V. K.; Watts, J. K. Future Med. Chem. 2015, 7, 2221–2242. doi:10.4155/fmc.15.144
Return to citation in text: [1] -
Sharma, V. K.; Sharma, R. K.; Singh, S. K. Med. Chem. Commun. 2014, 5, 1454–1471. doi:10.1039/c4md00184b
Return to citation in text: [1] -
Jahnz-Wechmann, Z.; Framski, G. R.; Januszczyk, P. A.; Boryski, J. Front. Chem. (Lausanne, Switz.) 2016, 4, 19. doi:10.3389/fchem.2016.00019
Return to citation in text: [1] -
Shelton, J.; Lu, X.; Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2016, 116, 14379–14455. doi:10.1021/acs.chemrev.6b00209
Return to citation in text: [1] -
Mahmoud, S.; Hasabelnaby, S.; Hammad, S.; Sakr, T. M. J. Adv. Pharm. Res. 2018, 2, 73–88. doi:10.21608/aprh.2018.5829
Return to citation in text: [1] -
Seley-Radtke, K. L.; Yates, M. K. Antiviral Res. 2018, 154, 66–86. doi:10.1016/j.antiviral.2018.04.004
Return to citation in text: [1] -
Sugimoto, I.; Shuto, S.; Mori, S.; Shigeta, S.; Matsuda, A. Bioorg. Med. Chem. Lett. 1999, 9, 385–388. doi:10.1016/s0960-894x(99)00010-4
Return to citation in text: [1] -
Waga, T.; Ohrui, H.; Meguro, H. Nucleosides Nucleotides 1996, 15, 287–304. doi:10.1080/07328319608002385
Return to citation in text: [1] -
Thomson, J. M.; Lamont, I. L. Front. Microbiol. 2019, 10, 952. doi:10.3389/fmicb.2019.00952
Return to citation in text: [1] -
Galmarini, C. M.; Mackey, J. R.; Dumontet, C. Lancet Oncol. 2002, 3, 415–424. doi:10.1016/s1470-2045(02)00788-x
Return to citation in text: [1] -
https://www.drugs.com/monograph/zidovudine.html.
Return to citation in text: [1] -
Mitsuya, H.; Weinhold, K. J.; Furman, P. A.; St Clair, M. H.; Lehrman, S. N.; Gallo, R. C.; Bolognesi, D.; Barry, D. W.; Broder, S. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 7096–7100. doi:10.1073/pnas.82.20.7096
Return to citation in text: [1] -
Collier, A. C.; Coombs, R. W.; Schoenfeld, D. A.; Bassett, R. L.; Timpone, J.; Baruch, A.; Jones, M.; Facey, K.; Whitacre, C.; McAuliffe, V. J.; Friedman, H. M.; Merigan, T. C.; Reichman, R. C.; Hooper, C.; Corey, L. N. Engl. J. Med. 1996, 334, 1011–1018. doi:10.1056/nejm199604183341602
Return to citation in text: [1] -
Whittington, R.; Brogden, R. N. Drugs 1992, 44, 656–683. doi:10.2165/00003495-199244040-00009
Return to citation in text: [1] -
Perry, C. M.; Noble, S. Drugs 1999, 58, 1099–1135. doi:10.2165/00003495-199958060-00009
Return to citation in text: [1] -
Hurst, M.; Noble, S. Drugs 1999, 58, 919–949. doi:10.2165/00003495-199958050-00012
Return to citation in text: [1] -
Rana, K. Z.; Dudley, M. N. Clin. Pharmacokinet. 1997, 33, 276–284. doi:10.2165/00003088-199733040-00003
Return to citation in text: [1] -
Jarvis, B.; Faulds, D. Drugs 1999, 58, 101–141. doi:10.2165/00003495-199958010-00015
Return to citation in text: [1] -
Maag, H.; Rydzewski, R. M.; McRoberts, M. J.; Crawford-Ruth, D.; Verheyden, J. P. H.; Prisbe, E. J. J. Med. Chem. 1992, 35, 1440–1451. doi:10.1021/jm00086a013
Return to citation in text: [1] -
Bryld, T.; Sørensen, M. H.; Nielsen, P.; Koch, T.; Nielsen, C.; Wengel, J. J. Chem. Soc., Perkin Trans. 1 2002, 1655–1662. doi:10.1039/b203484k
Return to citation in text: [1] -
Marquez, V. E.; Ezzitouni, A.; Russ, P.; Siddiqui, M. A.; Ford, H., Jr.; Feldman, R. J.; Mitsuya, H.; George, C.; Barchi, J. J., Jr. J. Am. Chem. Soc. 1998, 120, 2780–2789. doi:10.1021/ja973535+
Return to citation in text: [1] -
Sørensen, M. H.; Nielsen, C.; Nielsen, P. J. Org. Chem. 2001, 66, 4878–4886. doi:10.1021/jo010299j
Return to citation in text: [1] -
Li, Z.-S.; Qiao, R.-P.; Yang, Z.-J.; Zhang, L.-R.; Zhang, L.-H. Tetrahedron: Asymmetry 2006, 17, 1056–1061. doi:10.1016/j.tetasy.2006.03.034
Return to citation in text: [1] -
Obika, S.; Andoh, J.-i.; Sugimoto, T.; Miyashita, K.; Imanishi, T. Tetrahedron Lett. 1999, 40, 6465–6468. doi:10.1016/s0040-4039(99)01324-6
Return to citation in text: [1] -
Kumar, M.; Sharma, V. K.; Olsen, C. E.; Prasad, A. K. RSC Adv. 2014, 4, 37231–37235. doi:10.1039/c4ra06805j
Return to citation in text: [1] -
Rana, N.; Kumar, M.; Singh, A.; Maity, J.; Shukla, P.; Prasad, A. K. Nucleosides, Nucleotides Nucleic Acids 2018, 37, 217–231. doi:10.1080/15257770.2018.1460476
Return to citation in text: [1] -
Kumar, S.; Singla, H.; Maity, J.; Mangla, P.; Prasad, A. K. Carbohydr. Res. 2020, 492, 108013. doi:10.1016/j.carres.2020.108013
Return to citation in text: [1] [2] -
Nagy, A.; Csordás, B.; Zsoldos-Mády, V.; Pintér, I.; Farkas, V.; Perczel, A. Amino Acids 2017, 49, 223–240. doi:10.1007/s00726-016-2346-5
Return to citation in text: [1] [2] -
Niedballa, U.; Vorbrüggen, H. Angew. Chem., Int. Ed. Engl. 1970, 9, 461–462. doi:10.1002/anie.197004612
Return to citation in text: [1] [2] -
Mangla, P.; Maity, J.; Rungta, P.; Verma, V.; Sanghvi, Y. S.; Prasad, A. K. ChemistrySelect 2019, 4, 3241–3246. doi:10.1002/slct.201900809
Return to citation in text: [1] -
Hojland, T.; Babu, B. R.; Wengel, J. Nucleic Acids Symp. Ser. 2008, 52, 271–272. doi:10.1093/nass/nrn137
Return to citation in text: [1] -
Takahashi, S.; Kuzuhara, H.; Nakajima, M. Tetrahedron 2001, 57, 6915–6926. doi:10.1016/s0040-4020(01)00642-1
Return to citation in text: [1] -
Sun, Z.-H.; Wang, B. J. Org. Chem. 2008, 73, 2462–2465. doi:10.1021/jo7026596
Return to citation in text: [1] -
Hung, S.-C.; Lu, X.-A.; Lee, J.-C.; Chang, M. D.-T.; Fang, S.-l.; Fan, T.-c.; Zulueta, M. M. L.; Zhong, Y.-Q. Org. Biomol. Chem. 2012, 10, 760–772. doi:10.1039/c1ob06415k
Return to citation in text: [1] -
Loh, T.-P.; Feng, L.-C. Tetrahedron Lett. 2001, 42, 3223–3226. doi:10.1016/s0040-4039(01)00401-4
Return to citation in text: [1] -
Schuster, H.; Martinez, R.; Bruss, H.; Antonchick, A. P.; Kaiser, M.; Schürmann, M.; Waldmann, H. Chem. Commun. 2011, 47, 6545–6547. doi:10.1039/c1cc11388g
Return to citation in text: [1]
| 29. | Niedballa, U.; Vorbrüggen, H. Angew. Chem., Int. Ed. Engl. 1970, 9, 461–462. doi:10.1002/anie.197004612 |
| 29. | Niedballa, U.; Vorbrüggen, H. Angew. Chem., Int. Ed. Engl. 1970, 9, 461–462. doi:10.1002/anie.197004612 |
| 27. | Kumar, S.; Singla, H.; Maity, J.; Mangla, P.; Prasad, A. K. Carbohydr. Res. 2020, 492, 108013. doi:10.1016/j.carres.2020.108013 |
| 1. | Sharma, V. K.; Watts, J. K. Future Med. Chem. 2015, 7, 2221–2242. doi:10.4155/fmc.15.144 |
| 2. | Sharma, V. K.; Sharma, R. K.; Singh, S. K. Med. Chem. Commun. 2014, 5, 1454–1471. doi:10.1039/c4md00184b |
| 3. | Jahnz-Wechmann, Z.; Framski, G. R.; Januszczyk, P. A.; Boryski, J. Front. Chem. (Lausanne, Switz.) 2016, 4, 19. doi:10.3389/fchem.2016.00019 |
| 4. | Shelton, J.; Lu, X.; Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2016, 116, 14379–14455. doi:10.1021/acs.chemrev.6b00209 |
| 5. | Mahmoud, S.; Hasabelnaby, S.; Hammad, S.; Sakr, T. M. J. Adv. Pharm. Res. 2018, 2, 73–88. doi:10.21608/aprh.2018.5829 |
| 6. | Seley-Radtke, K. L.; Yates, M. K. Antiviral Res. 2018, 154, 66–86. doi:10.1016/j.antiviral.2018.04.004 |
| 7. | Sugimoto, I.; Shuto, S.; Mori, S.; Shigeta, S.; Matsuda, A. Bioorg. Med. Chem. Lett. 1999, 9, 385–388. doi:10.1016/s0960-894x(99)00010-4 |
| 8. | Waga, T.; Ohrui, H.; Meguro, H. Nucleosides Nucleotides 1996, 15, 287–304. doi:10.1080/07328319608002385 |
| 9. | Thomson, J. M.; Lamont, I. L. Front. Microbiol. 2019, 10, 952. doi:10.3389/fmicb.2019.00952 |
| 15. | Perry, C. M.; Noble, S. Drugs 1999, 58, 1099–1135. doi:10.2165/00003495-199958060-00009 |
| 27. | Kumar, S.; Singla, H.; Maity, J.; Mangla, P.; Prasad, A. K. Carbohydr. Res. 2020, 492, 108013. doi:10.1016/j.carres.2020.108013 |
| 13. | Collier, A. C.; Coombs, R. W.; Schoenfeld, D. A.; Bassett, R. L.; Timpone, J.; Baruch, A.; Jones, M.; Facey, K.; Whitacre, C.; McAuliffe, V. J.; Friedman, H. M.; Merigan, T. C.; Reichman, R. C.; Hooper, C.; Corey, L. N. Engl. J. Med. 1996, 334, 1011–1018. doi:10.1056/nejm199604183341602 |
| 14. | Whittington, R.; Brogden, R. N. Drugs 1992, 44, 656–683. doi:10.2165/00003495-199244040-00009 |
| 28. | Nagy, A.; Csordás, B.; Zsoldos-Mády, V.; Pintér, I.; Farkas, V.; Perczel, A. Amino Acids 2017, 49, 223–240. doi:10.1007/s00726-016-2346-5 |
| 11. | https://www.drugs.com/monograph/zidovudine.html. |
| 12. | Mitsuya, H.; Weinhold, K. J.; Furman, P. A.; St Clair, M. H.; Lehrman, S. N.; Gallo, R. C.; Bolognesi, D.; Barry, D. W.; Broder, S. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 7096–7100. doi:10.1073/pnas.82.20.7096 |
| 25. | Kumar, M.; Sharma, V. K.; Olsen, C. E.; Prasad, A. K. RSC Adv. 2014, 4, 37231–37235. doi:10.1039/c4ra06805j |
| 10. | Galmarini, C. M.; Mackey, J. R.; Dumontet, C. Lancet Oncol. 2002, 3, 415–424. doi:10.1016/s1470-2045(02)00788-x |
| 26. | Rana, N.; Kumar, M.; Singh, A.; Maity, J.; Shukla, P.; Prasad, A. K. Nucleosides, Nucleotides Nucleic Acids 2018, 37, 217–231. doi:10.1080/15257770.2018.1460476 |
| 21. | Marquez, V. E.; Ezzitouni, A.; Russ, P.; Siddiqui, M. A.; Ford, H., Jr.; Feldman, R. J.; Mitsuya, H.; George, C.; Barchi, J. J., Jr. J. Am. Chem. Soc. 1998, 120, 2780–2789. doi:10.1021/ja973535+ |
| 23. | Li, Z.-S.; Qiao, R.-P.; Yang, Z.-J.; Zhang, L.-R.; Zhang, L.-H. Tetrahedron: Asymmetry 2006, 17, 1056–1061. doi:10.1016/j.tetasy.2006.03.034 |
| 35. | Loh, T.-P.; Feng, L.-C. Tetrahedron Lett. 2001, 42, 3223–3226. doi:10.1016/s0040-4039(01)00401-4 |
| 36. | Schuster, H.; Martinez, R.; Bruss, H.; Antonchick, A. P.; Kaiser, M.; Schürmann, M.; Waldmann, H. Chem. Commun. 2011, 47, 6545–6547. doi:10.1039/c1cc11388g |
| 20. | Bryld, T.; Sørensen, M. H.; Nielsen, P.; Koch, T.; Nielsen, C.; Wengel, J. J. Chem. Soc., Perkin Trans. 1 2002, 1655–1662. doi:10.1039/b203484k |
| 24. | Obika, S.; Andoh, J.-i.; Sugimoto, T.; Miyashita, K.; Imanishi, T. Tetrahedron Lett. 1999, 40, 6465–6468. doi:10.1016/s0040-4039(99)01324-6 |
| 28. | Nagy, A.; Csordás, B.; Zsoldos-Mády, V.; Pintér, I.; Farkas, V.; Perczel, A. Amino Acids 2017, 49, 223–240. doi:10.1007/s00726-016-2346-5 |
| 18. | Jarvis, B.; Faulds, D. Drugs 1999, 58, 101–141. doi:10.2165/00003495-199958010-00015 |
| 19. | Maag, H.; Rydzewski, R. M.; McRoberts, M. J.; Crawford-Ruth, D.; Verheyden, J. P. H.; Prisbe, E. J. J. Med. Chem. 1992, 35, 1440–1451. doi:10.1021/jm00086a013 |
| 30. | Mangla, P.; Maity, J.; Rungta, P.; Verma, V.; Sanghvi, Y. S.; Prasad, A. K. ChemistrySelect 2019, 4, 3241–3246. doi:10.1002/slct.201900809 |
| 31. | Hojland, T.; Babu, B. R.; Wengel, J. Nucleic Acids Symp. Ser. 2008, 52, 271–272. doi:10.1093/nass/nrn137 |
| 16. | Hurst, M.; Noble, S. Drugs 1999, 58, 919–949. doi:10.2165/00003495-199958050-00012 |
| 17. | Rana, K. Z.; Dudley, M. N. Clin. Pharmacokinet. 1997, 33, 276–284. doi:10.2165/00003088-199733040-00003 |
| 22. | Sørensen, M. H.; Nielsen, C.; Nielsen, P. J. Org. Chem. 2001, 66, 4878–4886. doi:10.1021/jo010299j |
| 32. | Takahashi, S.; Kuzuhara, H.; Nakajima, M. Tetrahedron 2001, 57, 6915–6926. doi:10.1016/s0040-4020(01)00642-1 |
| 33. | Sun, Z.-H.; Wang, B. J. Org. Chem. 2008, 73, 2462–2465. doi:10.1021/jo7026596 |
| 34. | Hung, S.-C.; Lu, X.-A.; Lee, J.-C.; Chang, M. D.-T.; Fang, S.-l.; Fan, T.-c.; Zulueta, M. M. L.; Zhong, Y.-Q. Org. Biomol. Chem. 2012, 10, 760–772. doi:10.1039/c1ob06415k |
© 2022 Kumar et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.