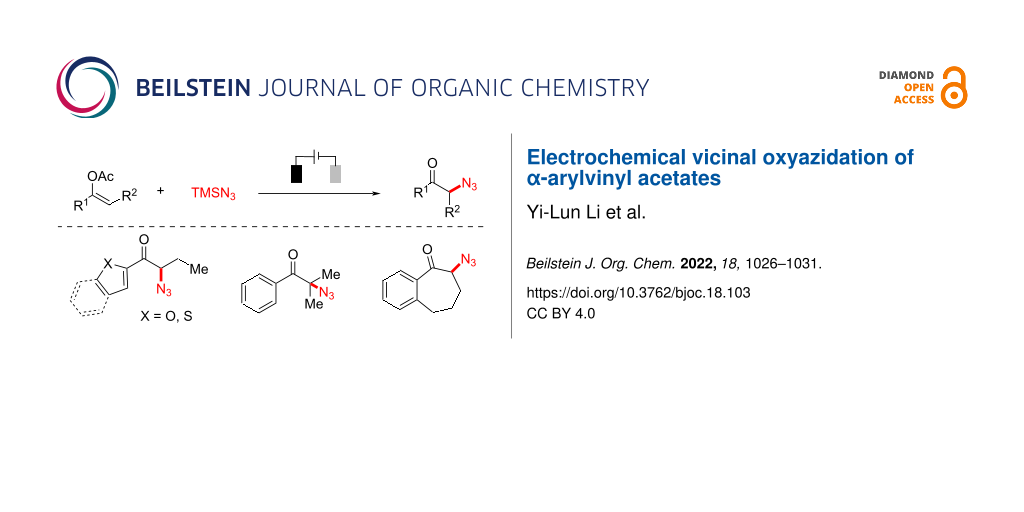
Abstract
α-Azidoketones are valuable and versatile building blocks in the synthesis of various bioactive small molecules. Herein, we describe an environmentally friendly and efficient electrochemical vicinal oxyazidation protocol of α-arylvinyl acetates to afford diverse α-azidoketones in good yields without the use of a stoichiometric amount of chemical oxidant. A range of functionality is shown to be compatible with this transformation, and further applications are demonstrated.

Graphical Abstract
Introduction
Organoazides play important roles in pharmaceutical, bioorthogonal chemistry, and many other interdisciplinary research areas [1-3]. Among them, azidoketones are also very versatile building blocks in organic synthesis, pharmaceutical, and materials science [4-6]. Therefore, the development of a green, efficient, and sustainable protocol for the synthesis of azidoketones is of great significance [7-9].
Retrosynthetically, the nucleophilic substitution of α-bromoketones by sodium azide [10] and oxidation of the azido alcohols [11] are among the most straightforward methods to generate azidoketones. Remarkably, photoredox [12] and electrochemical [13] oxyazidation of vinylarenes are also becoming competent synthetic approaches. Besides vinylarenes, vinyl acetates are potentially versatile precursors for the anticipated vicinal oxyazidation.
For instance, Singh and co-workers have reported a manganese dioxide-catalyzed radical azidation of enol acetates to afford the corresponding azidoketones using dioxygen as the oxidant (Scheme 1A) [14]. The adoption of electrosynthesis in green and sustainable redox transformations has been experiencing a dynamic renaissance [15-22] not only because it employs the passage of charge instead of chemical oxidants or reductants but also offers opportunities for the precise control of reactivity by "dialing-in" the specific potential on demand [23-25].
![[1860-5397-18-103-i1]](/bjoc/content/inline/1860-5397-18-103-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: From vinyl acetates to α-azidoketones.
Scheme 1: From vinyl acetates to α-azidoketones.
Specifically, the electrochemical oxyfunctionalization of vinyl acetates has been developed to afford the corresponding α-fluorinated [26], -arylated [27], and -sulfenylated [28] ketones (Scheme 1B). In addition, electrochemical azidation [29-33] has also become a robust and reliable synthetic tool to incorporate azido functionality [34,35] into diverse organic frameworks. Herein, we report that the electrochemical oxyfunctionalization strategy could be well applied to the synthesis of α-azidoketone using readily available α-arylvinyl acetates, and azidotrimethylsilane (Scheme 1C).
Results and Discussion
The constant cell potential electrolysis (Ecell = 2.3 V, carbon cloth anode, and Pt cathode) of 1-phenylvinyl acetate (1) with azidotrimethylsilane was performed and the desired α-azidoketone (2) was obtained in 68% yield (Table 1, entry 1, for details of the reaction optimization see Supporting Information File 1). The cyclic voltammetry studies showed while there was no obvious oxidation peak for TMSN3, 1-phenylvinyl acetate (1) exhibits two oxidation peaks. The first peak (Ep/2 = 1.51 V vs Fc+/0) was assigned to be the oxidation of the vinyl acetate moiety. The control experiment demonstrated that there was no conversion without an electric current (Table 1, entry 2). In addition, in the absence of the electrolyte only a low yield of the desired product was obtained (Table 1, entry 3). The use of n-Bu4NPF6 was crucial because lower yields were generally obtained with other electrolytes, such as n-Bu4NOAc or n-Bu4NBF4 (Table 1, entries 4 and 5). Note that the yield decreased without the addition of water suggesting water may facilitate the formation of the azidoketone (Table 1, entry 6). Interestingly, the H218O labeling experiment confirmed that there was no 18O incorporation in the obtained α-azidoketone (2, for details see Supporting Information File 1). Therefore, the oxygen source of the newly formed carbonyl moiety may originate directly from the vinyl acetate. This conclusion is also consistent with the fact that even in the absence of water, the desired α-azidoketone was still obtained.
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-103-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Variation from the standard reaction conditions | Yield (%) |
| 1 | none | 65 (68)b |
| 2 | no current | n.d. |
| 3 | without electrolyte | 9 |
| 4 | n-Bu4NOAc instead of n-Bu4NPF6 | 12 |
| 5 | n-Bu4NBF4 instead of n-Bu4NPF6 | 35 |
| 6 | without H2O | 34 |
aReaction conditions: 1 (0.5 mmol), TMSN3 (1.0 mmol), n-Bu4NPF6 (0.5 mmol), H2O (2.5 mmol), MeCN (5 mL), carbon cloth anode, platinum cathode, undivided cell, Ecell = 2.3 V, room temperature, 6 h, yields were determined using 1H NMR with dibromomethane as the internal standard. bIsolated yield.
Under the optimal conditions, the substrate scope of this electrochemical oxyazidation reaction was investigated (Scheme 2). Enol acetates derived from various alkyl-substituted phenylacetones were generally well tolerated (3–9, 40–76% yields). The relatively low yield of the isopropyl-substituted one (7) was attributed to the competing oxidation of the isopropylbenzene moiety [31,36], which was consistent with its relative low oxidation potential (Eonset = 1.68 V vs Fc+/0). Other electron-donating substituents, such as the OMe (10–12), OPh (13), and OAc (14) were also well tolerated.
![[1860-5397-18-103-i2]](/bjoc/content/inline/1860-5397-18-103-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope. Reaction conditions: α-arylvinyl acetate (0.5 mmol), TMSN3 (1.0 mmol), n-Bu4NPF6 (0.5 mmol), H2O (2.5 mmol), MeCN (5 mL), carbon cloth anode, platinum cathode, undivided cell, Ecell = 2.3 V, room temperature, 6 h. a2 h.
Scheme 2: Substrate scope. Reaction conditions: α-arylvinyl acetate (0.5 mmol), TMSN3 (1.0 mmol), n-Bu4NPF6 (...
However, halogenated substrates, including fluoro (15), chloro (16), and bromo (17), proceeded with the anticipated reactivity less efficiently (30–48% yields). The presence of other electron-withdrawing groups, such as CO2Me (18) and OCF3 (19), exhibited similar negative effects on the reaction yields. Naphthalene (20), thiophene (21), benzothiophene (22), and benzofuran (23) were all amenable in this transformation. In addition, various linear- (24, 25) and cyclic enol acetates (26, 27) also readily underwent the anticipated oxyazidation. Unfortunately, the current protocol was not applicable to the oxyazidation of enol acetate deriving from aliphatic ketones, such as cyclohexanone (see Supporting Information File 1 for details). As illustrated in Scheme 3, the synthetic utility of α-azidoketone was further evaluated [37,38]. Click reaction between 2-azido-1-phenylethan-1-one (2) and ethisterone (28) [39-41] readily afforded the target triazole product (29) in 84% yield. Upon treatment with piperidinium acetate and ethyl 3-(2-formylphenyl)acrylate (30), α-azidoketone (2) was transformed into isoquinoline product (31) in 58% yield [42].
![[1860-5397-18-103-i3]](/bjoc/content/inline/1860-5397-18-103-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Derivatization of α-azidoketone 2.
Scheme 3: Derivatization of α-azidoketone 2.
Based on our reaction results and the known literature [13,14], a possible mechanism is proposed (Scheme 4). The enol acetate A first undergoes anodic oxidation to form a radical cation intermediate B, which is then intercepted by azidotrimethylsilane to afford the benzyl radical C. Subsequently, this radical is further anodically oxidized to its oxocarbenium ion intermediate D, which finally reacts with water to form the desired product E. According to our 18O labeling experiment, the oxygen source of the newly installed carbonyl group probably originates from the vinyl acetate, not from H2O.
![[1860-5397-18-103-i4]](/bjoc/content/inline/1860-5397-18-103-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have developed an environmentally friendly and efficient electrochemical oxyazidation of α-arylvinyl acetates to access diverse vicinal α-azidoketones. The protocol employs the experimentally simple undivided electrochemical cell and tolerates a broad substrate scope. The obtained α-azidoketones have been shown to be versatile building blocks for the preparation of biologically relevant heterocycles.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 3.4 MB | Download |
References
-
Sletten, E. M.; Bertozzi, C. R. Acc. Chem. Res. 2011, 44, 666–676. doi:10.1021/ar200148z
Return to citation in text: [1] -
Li, J.; Chen, P. R. Nat. Chem. Biol. 2016, 12, 129–137. doi:10.1038/nchembio.2024
Return to citation in text: [1] -
Prescher, J. A.; Bertozzi, C. R. Nat. Chem. Biol. 2005, 1, 13–21. doi:10.1038/nchembio0605-13
Return to citation in text: [1] -
Kádár, Z.; Frank, É.; Schneider, G.; Molnár, J.; Zupkó, I.; Kóti, J.; Schönecker, B.; Wölfling, J. ARKIVOC 2012, No. 3, 279–296. doi:10.3998/ark.5550190.0013.320
Return to citation in text: [1] -
Brusis, T.; Grofe, H. J.; Förster, D.; Weuta, H. Infection (Munich, Ger.) 1977, 5, 26–31. doi:10.1007/bf01639106
Return to citation in text: [1] -
Zhao, R.; Chen, B.-C.; Bednarz, M. S.; Wang, B.; Skoumbourdis, A. P.; Sundeen, J. E.; Dhar, T. G. M.; Iwanowicz, E. J.; Balasubramanian, B.; Barrish, J. C. ARKIVOC 2007, No. 12, 36–42. doi:10.3998/ark.5550190.0008.c03
Return to citation in text: [1] -
Erythropel, H. C.; Zimmerman, J. B.; de Winter, T. M.; Petitjean, L.; Melnikov, F.; Lam, C. H.; Lounsbury, A. W.; Mellor, K. E.; Janković, N. Z.; Tu, Q.; Pincus, L. N.; Falinski, M. M.; Shi, W.; Coish, P.; Plata, D. L.; Anastas, P. T. Green Chem. 2018, 20, 1929–1961. doi:10.1039/c8gc00482j
Return to citation in text: [1] -
Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d
Return to citation in text: [1] -
Kim, Y.; Li, C.-J. Green Synth. Catal. 2020, 1, 1–11. doi:10.1016/j.gresc.2020.06.002
Return to citation in text: [1] -
Patonay, T.; Hoffman, R. V. J. Org. Chem. 1994, 59, 2902–2905. doi:10.1021/jo00089a043
Return to citation in text: [1] -
Yang, B.; Lu, Z. ACS Catal. 2017, 7, 8362–8365. doi:10.1021/acscatal.7b02892
Return to citation in text: [1] -
Hossain, A.; Vidyasagar, A.; Eichinger, C.; Lankes, C.; Phan, J.; Rehbein, J.; Reiser, O. Angew. Chem., Int. Ed. 2018, 57, 8288–8292. doi:10.1002/anie.201801678
Return to citation in text: [1] -
Ye, Z.; Zhu, R.; Wang, F.; Jiang, H.; Zhang, F. Org. Lett. 2021, 23, 8240–8245. doi:10.1021/acs.orglett.1c02991
Return to citation in text: [1] [2] -
Panday, P.; Garg, P.; Singh, A. Asian J. Org. Chem. 2018, 7, 111–115. doi:10.1002/ajoc.201700508
Return to citation in text: [1] [2] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Kingston, C.; Palkowitz, M. D.; Takahira, Y.; Vantourout, J. C.; Peters, B. K.; Kawamata, Y.; Baran, P. S. Acc. Chem. Res. 2020, 53, 72–83. doi:10.1021/acs.accounts.9b00539
Return to citation in text: [1] -
Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492–2521. doi:10.1039/c3cs60464k
Return to citation in text: [1] -
Yoshida, J.-i.; Shimizu, A.; Hayashi, R. Chem. Rev. 2018, 118, 4702–4730. doi:10.1021/acs.chemrev.7b00475
Return to citation in text: [1] -
Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339–3350. doi:10.1021/acs.accounts.9b00472
Return to citation in text: [1] -
Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300–310. doi:10.1021/acs.accounts.9b00603
Return to citation in text: [1] -
Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Chem. Soc. Rev. 2021, 50, 7941–8002. doi:10.1039/d1cs00223f
Return to citation in text: [1] -
Shi, S.-H.; Liang, Y.; Jiao, N. Chem. Rev. 2021, 121, 485–505. doi:10.1021/acs.chemrev.0c00335
Return to citation in text: [1] -
Aoyama, M.; Fukuhara, T.; Hara, S. J. Org. Chem. 2008, 73, 4186–4189. doi:10.1021/jo8004759
Return to citation in text: [1] -
Lam, K.; Markó, I. E. Org. Lett. 2009, 11, 2752–2755. doi:10.1021/ol900828x
Return to citation in text: [1] -
Yu, Y.; Jiang, Y.; Wu, S.; Shi, Z.; Wu, J.; Yuan, Y.; Ye, K. Chin. Chem. Lett. 2022, 33, 2009–2014. doi:10.1016/j.cclet.2021.10.016
Return to citation in text: [1] -
Vil’, V. A.; Merkulova, V. M.; Ilovaisky, A. I.; Paveliev, S. A.; Nikishin, G. I.; Terent’ev, A. O. Org. Lett. 2021, 23, 5107–5112. doi:10.1021/acs.orglett.1c01643
Return to citation in text: [1] -
de Souza, A. A. N.; Bartolomeu, A. d. A.; Brocksom, T. J.; Noël, T.; de Oliveira, K. T. J. Org. Chem. 2022, 87, 5856–5865. doi:10.1021/acs.joc.2c00147
Return to citation in text: [1] -
Zhou, P.; Liu, Y.; Xu, Y.; Wang, D. Org. Chem. Front. 2022, 9, 2215–2219. doi:10.1039/d1qo01765a
Return to citation in text: [1] -
Wu, J.; Dou, Y.; Guillot, R.; Kouklovsky, C.; Vincent, G. J. Am. Chem. Soc. 2019, 141, 2832–2837. doi:10.1021/jacs.8b13371
Return to citation in text: [1] -
Meyer, T. H.; Samanta, R. C.; Del Vecchio, A.; Ackermann, L. Chem. Sci. 2021, 12, 2890–2897. doi:10.1039/d0sc05924b
Return to citation in text: [1] -
Niu, L.; Jiang, C.; Liang, Y.; Liu, D.; Bu, F.; Shi, R.; Chen, H.; Chowdhury, A. D.; Lei, A. J. Am. Chem. Soc. 2020, 142, 17693–17702. doi:10.1021/jacs.0c08437
Return to citation in text: [1] [2] -
Siu, J. C.; Parry, J. B.; Lin, S. J. Am. Chem. Soc. 2019, 141, 2825–2831. doi:10.1021/jacs.8b13192
Return to citation in text: [1] -
Fu, N.; Sauer, G. S.; Saha, A.; Loo, A.; Lin, S. Science 2017, 357, 575–579. doi:10.1126/science.aan6206
Return to citation in text: [1] -
Wu, K.; Liang, Y.; Jiao, N. Molecules 2016, 21, 352. doi:10.3390/molecules21030352
Return to citation in text: [1] -
Ge, L.; Chiou, M.-F.; Li, Y.; Bao, H. Green Synth. Catal. 2020, 1, 86–120. doi:10.1016/j.gresc.2020.07.001
Return to citation in text: [1] -
Sharma, A.; Hartwig, J. F. Nature 2015, 517, 600–604. doi:10.1038/nature14127
Return to citation in text: [1] -
Bangalore, P. K.; Vagolu, S. K.; Bollikanda, R. K.; Veeragoni, D. K.; Choudante, P. C.; Misra, S.; Sriram, D.; Sridhar, B.; Kantevari, S. J. Nat. Prod. 2020, 83, 26–35. doi:10.1021/acs.jnatprod.9b00475
Return to citation in text: [1] -
Lv, G.; Qiu, L.; Li, K.; Liu, Q.; Li, X.; Peng, Y.; Wang, S.; Lin, J. New J. Chem. 2019, 43, 3419–3427. doi:10.1039/c8nj04159h
Return to citation in text: [1] -
Buil, M. L.; Esteruelas, M. A.; Garcés, K.; Oñate, E. Organometallics 2009, 28, 5691–5696. doi:10.1021/om900647a
Return to citation in text: [1] -
Levine, P. M.; Lee, E.; Greenfield, A.; Bonneau, R.; Logan, S. K.; Garabedian, M. J.; Kirshenbaum, K. ACS Chem. Biol. 2012, 7, 1693–1701. doi:10.1021/cb300332w
Return to citation in text: [1] -
Levine, P. M.; Imberg, K.; Garabedian, M. J.; Kirshenbaum, K. J. Am. Chem. Soc. 2012, 134, 6912–6915. doi:10.1021/ja300170n
Return to citation in text: [1] -
Prasad, B.; Phanindrudu, M.; Tiwari, D. K.; Kamal, A. J. Org. Chem. 2019, 84, 12334–12343. doi:10.1021/acs.joc.9b01534
Return to citation in text: [1]
| 42. | Prasad, B.; Phanindrudu, M.; Tiwari, D. K.; Kamal, A. J. Org. Chem. 2019, 84, 12334–12343. doi:10.1021/acs.joc.9b01534 |
| 37. | Bangalore, P. K.; Vagolu, S. K.; Bollikanda, R. K.; Veeragoni, D. K.; Choudante, P. C.; Misra, S.; Sriram, D.; Sridhar, B.; Kantevari, S. J. Nat. Prod. 2020, 83, 26–35. doi:10.1021/acs.jnatprod.9b00475 |
| 38. | Lv, G.; Qiu, L.; Li, K.; Liu, Q.; Li, X.; Peng, Y.; Wang, S.; Lin, J. New J. Chem. 2019, 43, 3419–3427. doi:10.1039/c8nj04159h |
| 39. | Buil, M. L.; Esteruelas, M. A.; Garcés, K.; Oñate, E. Organometallics 2009, 28, 5691–5696. doi:10.1021/om900647a |
| 40. | Levine, P. M.; Lee, E.; Greenfield, A.; Bonneau, R.; Logan, S. K.; Garabedian, M. J.; Kirshenbaum, K. ACS Chem. Biol. 2012, 7, 1693–1701. doi:10.1021/cb300332w |
| 41. | Levine, P. M.; Imberg, K.; Garabedian, M. J.; Kirshenbaum, K. J. Am. Chem. Soc. 2012, 134, 6912–6915. doi:10.1021/ja300170n |
| 1. | Sletten, E. M.; Bertozzi, C. R. Acc. Chem. Res. 2011, 44, 666–676. doi:10.1021/ar200148z |
| 2. | Li, J.; Chen, P. R. Nat. Chem. Biol. 2016, 12, 129–137. doi:10.1038/nchembio.2024 |
| 3. | Prescher, J. A.; Bertozzi, C. R. Nat. Chem. Biol. 2005, 1, 13–21. doi:10.1038/nchembio0605-13 |
| 34. | Wu, K.; Liang, Y.; Jiao, N. Molecules 2016, 21, 352. doi:10.3390/molecules21030352 |
| 35. | Ge, L.; Chiou, M.-F.; Li, Y.; Bao, H. Green Synth. Catal. 2020, 1, 86–120. doi:10.1016/j.gresc.2020.07.001 |
| 10. | Patonay, T.; Hoffman, R. V. J. Org. Chem. 1994, 59, 2902–2905. doi:10.1021/jo00089a043 |
| 31. | Niu, L.; Jiang, C.; Liang, Y.; Liu, D.; Bu, F.; Shi, R.; Chen, H.; Chowdhury, A. D.; Lei, A. J. Am. Chem. Soc. 2020, 142, 17693–17702. doi:10.1021/jacs.0c08437 |
| 36. | Sharma, A.; Hartwig, J. F. Nature 2015, 517, 600–604. doi:10.1038/nature14127 |
| 7. | Erythropel, H. C.; Zimmerman, J. B.; de Winter, T. M.; Petitjean, L.; Melnikov, F.; Lam, C. H.; Lounsbury, A. W.; Mellor, K. E.; Janković, N. Z.; Tu, Q.; Pincus, L. N.; Falinski, M. M.; Shi, W.; Coish, P.; Plata, D. L.; Anastas, P. T. Green Chem. 2018, 20, 1929–1961. doi:10.1039/c8gc00482j |
| 8. | Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d |
| 9. | Kim, Y.; Li, C.-J. Green Synth. Catal. 2020, 1, 1–11. doi:10.1016/j.gresc.2020.06.002 |
| 28. | Zhou, P.; Liu, Y.; Xu, Y.; Wang, D. Org. Chem. Front. 2022, 9, 2215–2219. doi:10.1039/d1qo01765a |
| 4. | Kádár, Z.; Frank, É.; Schneider, G.; Molnár, J.; Zupkó, I.; Kóti, J.; Schönecker, B.; Wölfling, J. ARKIVOC 2012, No. 3, 279–296. doi:10.3998/ark.5550190.0013.320 |
| 5. | Brusis, T.; Grofe, H. J.; Förster, D.; Weuta, H. Infection (Munich, Ger.) 1977, 5, 26–31. doi:10.1007/bf01639106 |
| 6. | Zhao, R.; Chen, B.-C.; Bednarz, M. S.; Wang, B.; Skoumbourdis, A. P.; Sundeen, J. E.; Dhar, T. G. M.; Iwanowicz, E. J.; Balasubramanian, B.; Barrish, J. C. ARKIVOC 2007, No. 12, 36–42. doi:10.3998/ark.5550190.0008.c03 |
| 29. | Wu, J.; Dou, Y.; Guillot, R.; Kouklovsky, C.; Vincent, G. J. Am. Chem. Soc. 2019, 141, 2832–2837. doi:10.1021/jacs.8b13371 |
| 30. | Meyer, T. H.; Samanta, R. C.; Del Vecchio, A.; Ackermann, L. Chem. Sci. 2021, 12, 2890–2897. doi:10.1039/d0sc05924b |
| 31. | Niu, L.; Jiang, C.; Liang, Y.; Liu, D.; Bu, F.; Shi, R.; Chen, H.; Chowdhury, A. D.; Lei, A. J. Am. Chem. Soc. 2020, 142, 17693–17702. doi:10.1021/jacs.0c08437 |
| 32. | Siu, J. C.; Parry, J. B.; Lin, S. J. Am. Chem. Soc. 2019, 141, 2825–2831. doi:10.1021/jacs.8b13192 |
| 33. | Fu, N.; Sauer, G. S.; Saha, A.; Loo, A.; Lin, S. Science 2017, 357, 575–579. doi:10.1126/science.aan6206 |
| 15. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 16. | Kingston, C.; Palkowitz, M. D.; Takahira, Y.; Vantourout, J. C.; Peters, B. K.; Kawamata, Y.; Baran, P. S. Acc. Chem. Res. 2020, 53, 72–83. doi:10.1021/acs.accounts.9b00539 |
| 17. | Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492–2521. doi:10.1039/c3cs60464k |
| 18. | Yoshida, J.-i.; Shimizu, A.; Hayashi, R. Chem. Rev. 2018, 118, 4702–4730. doi:10.1021/acs.chemrev.7b00475 |
| 19. | Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339–3350. doi:10.1021/acs.accounts.9b00472 |
| 20. | Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300–310. doi:10.1021/acs.accounts.9b00603 |
| 21. | Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Chem. Soc. Rev. 2021, 50, 7941–8002. doi:10.1039/d1cs00223f |
| 22. | Shi, S.-H.; Liang, Y.; Jiao, N. Chem. Rev. 2021, 121, 485–505. doi:10.1021/acs.chemrev.0c00335 |
| 26. | Vil’, V. A.; Merkulova, V. M.; Ilovaisky, A. I.; Paveliev, S. A.; Nikishin, G. I.; Terent’ev, A. O. Org. Lett. 2021, 23, 5107–5112. doi:10.1021/acs.orglett.1c01643 |
| 14. | Panday, P.; Garg, P.; Singh, A. Asian J. Org. Chem. 2018, 7, 111–115. doi:10.1002/ajoc.201700508 |
| 27. | de Souza, A. A. N.; Bartolomeu, A. d. A.; Brocksom, T. J.; Noël, T.; de Oliveira, K. T. J. Org. Chem. 2022, 87, 5856–5865. doi:10.1021/acs.joc.2c00147 |
| 13. | Ye, Z.; Zhu, R.; Wang, F.; Jiang, H.; Zhang, F. Org. Lett. 2021, 23, 8240–8245. doi:10.1021/acs.orglett.1c02991 |
| 13. | Ye, Z.; Zhu, R.; Wang, F.; Jiang, H.; Zhang, F. Org. Lett. 2021, 23, 8240–8245. doi:10.1021/acs.orglett.1c02991 |
| 14. | Panday, P.; Garg, P.; Singh, A. Asian J. Org. Chem. 2018, 7, 111–115. doi:10.1002/ajoc.201700508 |
| 12. | Hossain, A.; Vidyasagar, A.; Eichinger, C.; Lankes, C.; Phan, J.; Rehbein, J.; Reiser, O. Angew. Chem., Int. Ed. 2018, 57, 8288–8292. doi:10.1002/anie.201801678 |
| 23. | Aoyama, M.; Fukuhara, T.; Hara, S. J. Org. Chem. 2008, 73, 4186–4189. doi:10.1021/jo8004759 |
| 24. | Lam, K.; Markó, I. E. Org. Lett. 2009, 11, 2752–2755. doi:10.1021/ol900828x |
| 25. | Yu, Y.; Jiang, Y.; Wu, S.; Shi, Z.; Wu, J.; Yuan, Y.; Ye, K. Chin. Chem. Lett. 2022, 33, 2009–2014. doi:10.1016/j.cclet.2021.10.016 |
© 2022 Li et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.