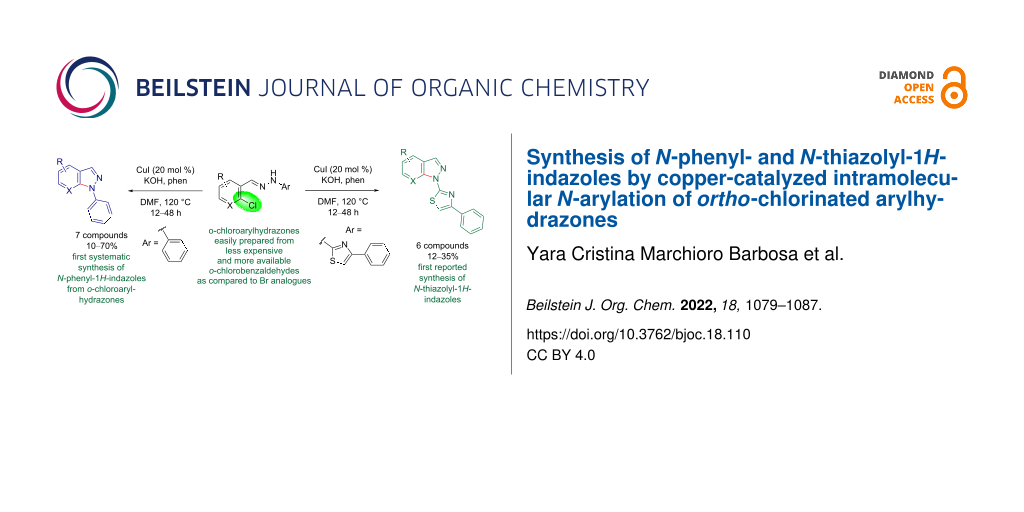
Abstract
The broad application of 1H-indazoles has prompted the development of several approaches for the synthesis of such compounds, including metal-free, palladium-, or copper-promoted intramolecular N-arylation of in situ-generated or isolated o-haloarylhydrazones. Such methods mainly start from o-bromo derivatives due to the better yield observed when compared to those obtained from o-chloroarylhydrazones. However, the o-chloroarylaldehydes and o-chloroarylketones used to prepare the arylhydrazones are more commercially available and less expensive than brominated analogs. Seeking to cover a lack in the literature, this work reports a convenient protocol for the synthesis of N-phenyl- and N-thiazolyl-1H-indazoles by copper-catalyzed intramolecular N-arylation of o-chlorinated arylhydrazones. Therefore, a series of seven N-phenyl derivatives and a series of six novel N-thiazolyl derivatives was obtained in 10–70% and 12–35% yield, respectively, after stirring the o-chlorinated arylhydrazones, CuI, KOH, and 1,10-phenantroline for 12–48 hours in DMF at 120 °C. The products were isolated by column chromatography on silica gel. All products were fully characterized by HRMS as well as 1H and 13C NMR spectroscopy. Thus, this approach is valuable for promoting the synthesis of N-phenyl-1H-indazoles in a higher yield than that reported in the literature using copper catalysis and the same substrates. This study also prompted the first reported synthesis of pharmacologically interesting N-thiazolyl derivatives.

Graphical Abstract
Introduction
1H-Indazoles are important scaffolds due to the prevalence in compounds with biological activity [1], such as anticancer [2], anti-HIV [3], anti-inflammatory [4], antiprotozoal [5], antifungal [6], antibacterial [7], antiplatelet [8], and antihypertensive [9] properties. The relevance to medicinal chemistry is also demonstrated by the presence of the 1H-indazole core in the structure of drugs. The anticataract agent bendazac [10], the anti-inflammatory agent benzydamine [11], and the antiemetic agent granisetron [12] are commercially available examples.
In view of the abovementioned interest, an increasing number of approaches for the synthesis of 1H-indazoles has been recently reported, including 1,3-dipolar cycloaddition reaction of α-diazomethylphosphonates with o-(trimethylsilyl)phenyl triflate in the presence of CsF [13], Cu2+-mediated N−N bond formation from ketimines in the presence of oxygen [14], Pd2+-mediated oxidative benzannulation from pyrazoles and internal alkynes [15], Pd-catalyzed Aza–Nenitzescu reaction of hydrazones and p-benzoquinones [16], and Co3+/Cu2+-catalyzed C−N/N−N coupling of imidates with anthranils as both aminating reagents and organic oxidants [17]. Additional established routes to 1H-indazoles comprise transition metal‐catalyzed [18-20] and metal-free [21,22] intramolecular amination by oxidative C−H bond functionalizations. These methods showed significant improvement with respect to the substrate scope and reaction conditions. However, mostly they are restricted to substrates containing hydrogen [13,14], alkyl [14,15], or (substituted) phenyl moieties [14,16,17] as N‐substituents, and others suffer from poor regioselectivity for substrates without a directing group [18,19,21,22]. On the other hand, methods based on metal-free [23], palladium- [24,25], and copper-promoted [26-31] intramolecular N-arylation of in situ-generated or isolated o-haloarylhydrazones are advantageous concerning regioselectivity. However, they also present limitations, such as costly Pd catalysts, a scope of the N-substituent limited to alkyl [30], (substituted) phenyl [23-29], and tosyl moieties [31], and low to moderate yield when o-chloroarylaldehydes or o-chloroarylketones are the starting material [27,28]. The preference for the use of o-bromo analogues is due to to the better yield obtained compared to using o-chloroarylhydrazones. However, the availability of commercial o-chloroarylaldehydes and o-chloroaryl ketones is greater than that of brominated analogs, and the cost of the former is significantly advantageous, even when the lower yield is considered.
Since the cost and availability of the starting material are crucial issues during the synthetic planning, we envisioned that it would be possible to employ o-chlorobenzaldehydes as starting material for the synthesis of N-phenyl-1H-indazoles if the reaction conditions are carefully chosen. In addition, another target of the work was the application of the optimized reaction conditions to the cyclization of thiazolylhydrazones, aiming for the synthesis of novel N-thiazolyl-1H-indazoles.
Results and Discussion
First, optimization of the reaction conditions was conducted using substrate 1a as a model according to Table 1. The molar ratio of 1a, the catalyst, the base, and the ligand used in Table 1, entry 1 was adapted from the literature [25]. The reaction mixture was stirred for 5 h at 120 °C, which gave the product 2a in 32% yield. Increasing the time to 12 h led to a better yield of 40% (Table 1, entry 2). The best yield (60%) was reached after 24 h (Table 1, entry 3). The reduction of the temperature to 100 °C drastically decreased the yield of 2a (Table 1, entry 4). The solvent effect on the reaction yield was evaluated employing other aprotic solvents, but no improvement was observed compared to when DMF was used (Table 1, entries 5–7). When the reaction was carried out in NMP (N-methyl-2-pyrrolidone), the 1H NMR spectrum of the crude mixture obtained after extraction did not show the presence of the product 2a, but it showed the presence of 91% of the starting hydrazone (Table 1, entry 8). To evaluate the effect of the base, K3PO4 or Cs2CO3 was used, but the yield was lower than 60% (Table 1, entries 9 and 10). The influence of the catalyst/ligand molar ratio (Table 1, entries 11 and 12) showed that in both cases, the yield was not better than that in Table 1, entry 3. Further, the increase in the reaction concentration from 0.2 M to 0.4 M did not allow for better results (Table 1, entry 13). The copper source is a less explored aspect, considering previous literature on the synthesis of 1H-indazoles. As such, this work devoted great attention to the nature of the catalyst and evaluated several copper sources other than CuI, such as CuBr, CuCl, CuO, and Cu0 in powder form (Table 1, entries 14–17). Among them, CuI provided the best performance. Finally, N,N'-dimethylethanolamine (DMEA) and trans-1,2-diaminocyclohexane (DACH) were evaluated as ligands (Table 1, entries 18 and 19). However, the yield was lower compared to that obtained with 1,10-phenanthroline (phen). Control experiments were performed without catalyst (Table 1, entry 20), base (Table 1, entry 21), or ligand (Table 1, entry 22), but the product could not be detected by TLC analysis.
Table 1: Optimization of the reaction conditions for the synthesis of 1-phenyl-1H-indazole (2a).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-110-i3.svg?max-width=637&scale=1.0)
|
|||||||
| entry | catalyst | base | ligand | solvent | T (°C) | t (h) | yield (%)b |
| 1 | CuI | KOH | phen | DMF | 120 | 5 | 32 |
| 2 | CuI | KOH | phen | DMF | 120 | 12 | 40 |
| 3 | CuI | KOH | phen | DMF | 120 | 24 | 60 |
| 4 | CuI | KOH | phen | DMF | 100 | 24 | —c |
| 5 | CuI | KOH | phen | DMSO | 120 | 24 | 27 |
| 6 | CuI | KOH | phen | dioxane | 120 | 24 | 42 |
| 7 | CuI | KOH | phen | toluene | 120 | 24 | 46 |
| 8 | CuI | KOH | phen | NMP | 120 | 24 | —d |
| 9 | CuI | K3PO4 | phen | DMF | 120 | 24 | 40 |
| 10 | CuI | Cs2CO3 | phen | DMF | 120 | 24 | 26 |
| 11 | CuI | KOH | phene | DMF | 120 | 24 | 39 |
| 12 | CuI | KOH | phenf | DMF | 120 | 24 | 47 |
| 13 | CuI | KOH | phen | DMFg | 120 | 24 | 57 |
| 14 | CuCl | KOH | phen | DMF | 120 | 24 | 47 |
| 15 | CuBr | KOH | phen | DMF | 120 | 24 | 50 |
| 16 | CuO | KOH | phen | DMF | 120 | 24 | 34 |
| 17 | Cu0 | KOH | phen | DMF | 120 | 24 | 36 |
| 18 | CuI | KOH | DMEA | DMF | 120 | 24 | 36 |
| 19 | CuI | KOH | DACH | DMF | 120 | 24 | 29 |
| 20 | — | KOH | phen | DMF | 120 | 24 | —h |
| 21 | CuI | — | phen | DMF | 120 | 24 | —h |
| 22 | CuI | KOH | — | DMF | 120 | 24 | —h |
aUnless otherwise noted, the reactions were carried out with 1a (0.5 mmol), catalyst (20 mol %), base (200 mol %), ligand (22 mol %), and solvent (2.5 mL) and heated at 120 °C for the indicated time. bIsolated yield. c≈23% of the product 2a and ≈53% of the starting hydrazone 1a were detected by 1H NMR spectroscopy (Figures S1 and S2, Supporting Information File 1). dNo product and ≈91% of the starting hydrazone 1a were detected by 1H NMR (Figures S3 and S4, Supporting Information File 1). e40 mol % of phen was used. f20 mol % of phen and 10 mol % of CuI were used. g1.25 mL of DMF (0.4 M) was used. hProduct 2a was not detected by TLC analysis. phen = 1,10-phenanthroline, NMP = N-methyl-2-pyrrolidone, DMEA = N,N’-dimethylethanolamine, DACH = trans-1,2-diaminocyclohexane.
In order to compare the efficiency of the optimized conditions with the one-pot approach, a reaction was carried out starting from o-chlorobenzaldehyde and phenylhydrazine under the conditions shown in Table 1, entry 3 (Scheme 1). In the 1H NMR spectrum of the crude reaction mixture, only 3% of the desired product 2a was detected, together with an undefined amount of the intermediate hydrazone 1a (Figures S5 and S6, Supporting Information File 1).
![[1860-5397-18-110-i1]](/bjoc/content/inline/1860-5397-18-110-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-pot approach for the synthesis of 2a. aYield calculated vs trichloroethylene by 1H NMR spectroscopy.
Scheme 1: One-pot approach for the synthesis of 2a. aYield calculated vs trichloroethylene by 1H NMR spectros...
Next, the most effective conditions as determined from the optimization were applied to prepare a series of seven N-phenyl-1H-indazole derivatives, 2a,b,d–h, from o-chlorinated arylhydrazones 1a,b,d–h. The structure of the starting materials and products as well as the reaction conditions and yields are shown in Table 2. In general, the reactions were highly impacted by the structure of the starting materials. The best yield was obtained from the unsubstituted arylhydrazones 1a (60%) and 1h (70%). On the other hand, the method was inefficient in converting arylhydrazones containing electron-donating groups at the 4-position. Accordingly, the reaction of 4-methyl-substituted arylhydrazone 1b led to 2b in only 10% yield and 4-methoxy-substituted arylhydrazone 1c was not converted to the desired 1H-indazole 2c. Arylhydrazones 1d–f substituted with halogen atoms were converted in 23–53% yield, but for the 4-fluoro- and 5-fluoro-substituted substrates 1d and 1f, a longer reaction time was needed in order to increase the rather low yield observed after 24 h. In turn, to obtain the product 2g derived from 5-nitro-substituted arylhydrazone 1g, the reaction time had to be decreased.
Table 2: Selected experimental data for the synthesis of N-phenyl-1H-indazoles 2a,b,d–h.
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-110-i4.svg?max-width=637&scale=1.0)
|
|||
| hydrazone 1 | product 2 | t (h) | yield (%)a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-110-i5.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-110-i6.svg?max-width=637&scale=1.0)
2a |
24 | 60 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-18-110-i7.svg?max-width=637&scale=1.0)
1b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-18-110-i8.svg?max-width=637&scale=1.0)
2b |
24 | 10 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-18-110-i9.svg?max-width=637&scale=1.0)
1c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-18-110-i10.svg?max-width=637&scale=1.0)
2c |
24 | —b |
![[Graphic 9]](/bjoc/content/inline/1860-5397-18-110-i11.svg?max-width=637&scale=1.0)
1d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-18-110-i12.svg?max-width=637&scale=1.0)
2d |
39 | 23 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-18-110-i13.svg?max-width=637&scale=1.0)
1e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-18-110-i14.svg?max-width=637&scale=1.0)
2e |
24 | 39 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-18-110-i15.svg?max-width=637&scale=1.0)
1f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-18-110-i16.svg?max-width=637&scale=1.0)
2f |
48 | 53 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-18-110-i17.svg?max-width=637&scale=1.0)
1g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-18-110-i18.svg?max-width=637&scale=1.0)
2g |
12 | 16 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-18-110-i19.svg?max-width=637&scale=1.0)
1h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-18-110-i20.svg?max-width=637&scale=1.0)
2h |
24 | 70 |
aIsolated yield. bProduct not obtained.
Aiming to expand the scope of 1H-indazoles synthetized by copper-catalyzed intramolecular N-arylation of arylhydrazones, which was limited to derivatives with alkyl, (substituted) phenyl, and tosyl N-substituents, as detailed in the Introduction section, the same conditions were applied to convert N-thiazolyl-substituted arylhydrazones 3a–h to the corresponding N-thiazolyl-1H-indazoles 4a–h. Unfortunately, the conditions were not suitable to convert arylhydrazones containing an electron-donating group at the 3-position, such as Me in 3b and OMe in 3c. In general, the yield was lower than for the products in the N-phenyl-1H-indazole series. The structure of the starting materials impacted the yield of the products in a similar manner as observed for the previous series. Products 4a and 4h were obtained with the best yield of 35% and 34%, respectively. The yield of products 4d–g ranged from 12–24%. Table 3 shows the structures of the starting materials and products as well as the reaction conditions and yields.
Table 3: Selected experimental data for the synthesis of N-thiazolyl-1H-indazoles 4a,d–h.
![[Graphic 19]](/bjoc/content/inline/1860-5397-18-110-i21.svg?max-width=637&scale=1.0)
|
|||
| hydrazone 3 | product 4 | t (h) | yield (%)a |
![[Graphic 20]](/bjoc/content/inline/1860-5397-18-110-i22.svg?max-width=637&scale=1.0)
3a |
![[Graphic 21]](/bjoc/content/inline/1860-5397-18-110-i23.svg?max-width=637&scale=1.0)
4a |
24 | 35 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-18-110-i24.svg?max-width=637&scale=1.0)
3b |
![[Graphic 23]](/bjoc/content/inline/1860-5397-18-110-i25.svg?max-width=637&scale=1.0)
4b |
24 | —b |
![[Graphic 24]](/bjoc/content/inline/1860-5397-18-110-i26.svg?max-width=637&scale=1.0)
3c |
![[Graphic 25]](/bjoc/content/inline/1860-5397-18-110-i27.svg?max-width=637&scale=1.0)
4c |
48 | —b |
![[Graphic 26]](/bjoc/content/inline/1860-5397-18-110-i28.svg?max-width=637&scale=1.0)
3d |
![[Graphic 27]](/bjoc/content/inline/1860-5397-18-110-i29.svg?max-width=637&scale=1.0)
4d |
24 | 12 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-18-110-i30.svg?max-width=637&scale=1.0)
3e |
![[Graphic 29]](/bjoc/content/inline/1860-5397-18-110-i31.svg?max-width=637&scale=1.0)
4e |
24 | 23 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-18-110-i32.svg?max-width=637&scale=1.0)
3f |
![[Graphic 31]](/bjoc/content/inline/1860-5397-18-110-i33.svg?max-width=637&scale=1.0)
4f |
24 | 24 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-18-110-i34.svg?max-width=637&scale=1.0)
3g |
![[Graphic 33]](/bjoc/content/inline/1860-5397-18-110-i35.svg?max-width=637&scale=1.0)
4g |
12 | 19 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-18-110-i36.svg?max-width=637&scale=1.0)
3h |
![[Graphic 35]](/bjoc/content/inline/1860-5397-18-110-i37.svg?max-width=637&scale=1.0)
4h |
24 | 34 |
aIsolated yield. bProduct not obtained.
When arylhydrazone 1i or 3i, respectively, was the starting material, competition between attack of the nitrogen atom on position 2 (C−Cl) and 6 (C−F) was observed (Scheme 2). The analysis of the 1H NMR (Figure S31, Supporting Information File 1), 13C NMR (Figure S32, Supporting Information File 1), and HRMS spectra (Figures S63 and S64, Supporting Information File 1) of the product obtained from N-phenylhydrazone 1i showed a mixture of 2i and 2i’, with 2i’ being the main product, in the ratio 10:1, as calculated from the 1H NMR spectrum. The starting material 3i also afforded a mixture of 4i and 4i’, as concluded from the analysis of the 1H NMR (Figure S45, Supporting Information File 1), 13C NMR (Figure S46, Supporting Information File 1), and HRMS spectra (Figures S79 and S80, Supporting Information File 1). However, 4i was detected as the major product in the ratio 10:6, as seen in the 1H NMR spectrum. This inversion of the regioselectivity can be attributed to electronic and steric effects of the N-substituent but needs additional investigation for full comprehension. When arylhydrazone 1i or 3i, respectively, was subjected to the same reaction conditions in the absence of CuI, only the product 2i’ or 4i’, respectively, was detected by 1H NMR spectroscopy in the crude reaction mixture (Figures S7–S10 and Figures S11–S14, respectively, Supporting Information File 1). This result shows that o-fluorinated arylhydrazones may be suitable as starting materials to prepare the title compounds by a SNAr approach.
![[1860-5397-18-110-i2]](/bjoc/content/inline/1860-5397-18-110-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Regioselectivity of the reaction of arylhydrazones 1i and 3i, respectively.
Scheme 2: Regioselectivity of the reaction of arylhydrazones 1i and 3i, respectively.
Conclusion
A novel set of conditions to convert o-chlorinated arylhydrazones into 1H-indazoles by a copper-catalyzed intramolecular N-arylation approach has been determined. A series of seven N-phenyl-1H-indazoles was obtained in 10–70% yield. Although this yield is lower than that reached using o-brominated arylhydrazones by methods described in the literature, they surpass the yield obtained from the same substrates using copper catalysis. This approach also prompted the synthesis of six novel and interesting N-thiazolyl-1H-indazoles in 12–35% yield. Despite the lower yield, this is the first reported method for the preparation of such compounds. In summary, N-phenyl- and N-thiazolyl-1H-indazoles were conveniently obtained from the less expensive and more available o-chlorinated arylhydrazones.
Experimental
General
All chemicals were purchased from Sigma-Aldrich or Oakwood Chemicals and were used as supplied without further purification. The solvents were purchased from Synth. DMF was distilled over 4 Å molecular sieves and degassed by sparging with nitrogen for 30 min before use. Reactions for the synthesis of 1H-indazoles were performed in a resealable screw-cap Schlenk flask (≈10 mL volume) in the presence of a Teflon-coated magnetic stirring bar. TLC analyses were performed with Al plates covered with silica gel (SiliCycle, F254) and visualized by UV detection. Silica gel (Vetec, 0.063–0.200 mm, 70–230 mesh) was used for column chromatography. Melting points were determined with a PMF-II MS Tecnopon melting point apparatus using open capillaries. 1H (300 or 500 MHz) and 13C (75 or 126 MHz) NMR spectra were recorded at 25 °C with a Bruker Avance III HD or Ascend 500 spectrometer, respectively. The samples were dissolved in CDCl3/TMS or DMSO-d6. Chemical shifts (δ) were recorded in ppm. 1H NMR spectra recorded in CDCl3/TMS were calibrated using the TMS peak, and those recorded in DMSO-d6 were calibrated using the DMSO-d5 peak. 13C NMR spectra recorded in CDCl3/TMS and DMSO-d6 were calibrated using the CDCl3 and DMSO-d6 peak, respectively. The determination of the amount of product and/or starting material in the crude mixture by 1H NMR spectroscopy was conducted by adding 1 equiv of trichloroethylene as calibration compound after diluting the crude mixture in CDCl3. The amounts were estimated by comparing the integral value of the product C3–H signal at 8.21 ppm and the hydrazone triplet at 6.89 ppm, respectively, vs the integral value of the calibration compound singlet at 6.46 ppm (see Supporting Information File 1). HRMS spectra were acquired on a hybrid high-resolution and high-accuracy microTof (Q-TOF, Bruker Scientific) spectrometer with electrospray ionization (ESI) source (MicrOTOF-QII, Bruker Scientific) in positive mode. The compounds were individually dissolved in a solution of 50% chromatographic grade MeCN and 50% deionized H2O + 0.1% formic acid.
General experimental procedure for the synthesis of hydrazones 1a–i
In a 100 mL round-bottom flask, the o-chlorinated aromatic aldehyde (5 mmol) and phenylhydrazine (0.540 g, 5 mmol) were dissolved in methanol (25 mL). Glacial acetic acid (0.060 g, 20 mol %) and sodium acetate (0.082 g, 20 mol %) were added, and the solution was stirred for 4 h at room temperature. The resulting mixture was cooled in an ice bath and cold water (30 mL) was added to give a precipitate. In a Büchner funnel, the solid was filtered off and washed with an additional amount of cold water. After drying the solid in a desiccator, it was recrystallized from methanol/water 1:1 to give the products 1a–i with good purity. The identity of the compounds 1a–i was confirmed by HRMS analysis (see Supporting Information File 1).
General experimental procedure for the synthesis of indazoles 2a,b,d–i,i’
To a dried Schlenk tube, an arylhydrazone 1a–i (0.5 mmol), KOH (0.056 g, 200 mol %), phen (0.020 g, 22 mol %), CuI (0.019 g, 20 mol %), and DMF (2.5 mL) were successively added under N2 atmosphere. The reaction mixture was stirred and heated at 120 °C for 12–48 h (see Table 1). After cooling to room temperature, AcOEt (10 mL) was added to the mixture. This was passed through a short column with silica gel 60, and then the eluate was washed with water (1 × 10 mL) and a saturated aqueous NaCl solution (2 × 10 mL). The resulting organic solution was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Each product 2a,b,d–i,i’ was isolated using column chromatography with silica gel 60 using the following eluent: hexane/AcOEt 98:2 for 2a,b,d,e, petroleum ether/AcOEt 98:2 for 2f, hexane/AcOEt 90:10 for 2g,i,i’, and hexane/AcOEt 80:20 for 2h.
General experimental procedure for the synthesis of hydrazones 3a–i
The o-chlorinated aromatic aldehyde (1.5 mmol), thiosemicarbazide (0.136 g, 1.5 mmol), 2-bromoacetophenone (0.298 g, 1.5 mmol), and absolute ethanol (2 mL) were added to a 10 mL round bottom flask. The mixture was stirred at room temperature for 5–10 min. The solid was filtered off, washed with ethyl ether, and dried in a desiccator to give the product 3a–i with adequate purity to be used without further purification. The identity of the compounds 3a–i was confirmed by HRMS analysis (see Supporting Information File 1).
General experimental procedure for the synthesis of indazoles 4a,d–i,i’
Each arylhydrazone 3a–i was allowed to react following the same procedure reported above for 1a–i. The reaction time ranged from 12 to 48 h (see Table 2). Each product 4a,d–i,i’ was isolated by column chromatography with silica gel 60 using the following eluent: hexane/AcOEt 90:10 for 4a, petroleum ether/AcOEt 95:5 for 4d, petroleum ether/AcOEt 98:2 for 4e, hexane/AcOEt 95:5 for 4f, hexane/AcOEt 80:20 for 4g, hexane/AcOEt 98:2 for 4h, and hexane/AcOEt 85:15 for 4i and 4i’.
Supporting Information
| Supporting Information File 1: Reaction analysis by 1H and 13C NMR spectroscopy, characterization data, NMR spectra for the 1H-indazoles and HRMS spectra for the arylhydrazones and 1H-indazoles. | ||
| Format: PDF | Size: 4.1 MB | Download |
References
-
Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Molecules 2018, 23, 2783. doi:10.3390/molecules23112783
Return to citation in text: [1] -
Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. ChemMedChem 2018, 13, 1490–1507. doi:10.1002/cmdc.201800253
Return to citation in text: [1] -
Kim, S.-H.; Markovitz, B.; Trovato, R.; Murphy, B. R.; Austin, H.; Willardsen, A. J.; Baichwal, V.; Morham, S.; Bajji, A. Bioorg. Med. Chem. Lett. 2013, 23, 2888–2892. doi:10.1016/j.bmcl.2013.03.075
Return to citation in text: [1] -
Cheekavolu, C.; Muniappan, M. J. Clin. Diagn. Res. 2016, 10, FF01–FF06. doi:10.7860/jcdr/2016/19338.8465
Return to citation in text: [1] -
Gerpe, A.; Aguirre, G.; Boiani, L.; Cerecetto, H.; González, M.; Olea-Azar, C.; Rigol, C.; Maya, J. D.; Morello, A.; Piro, O. E.; Arán, V. J.; Azqueta, A.; de Ceráin, A. L.; Monge, A.; Rojas, M. A.; Yaluff, G. Bioorg. Med. Chem. 2006, 14, 3467–3480. doi:10.1016/j.bmc.2006.01.007
Return to citation in text: [1] -
Rodríguez-Villar, K.; Hernández-Campos, A.; Yépez-Mulia, L.; Sainz-Espuñes, T. d. R.; Soria-Arteche, O.; Palacios-Espinosa, J. F.; Cortés-Benítez, F.; Leyte-Lugo, M.; Varela-Petrissans, B.; Quintana-Salazar, E. A.; Pérez-Villanueva, J. Pharmaceuticals 2021, 14, 176. doi:10.3390/ph14030176
Return to citation in text: [1] -
Reddy, G. S.; Viswanath, I. V. K.; Rao, A. T. Indian J. Heterocycl. Chem. 2018, 28, 467–475. doi:10.13140/rg.2.2.32357.24802
Return to citation in text: [1] -
Chen, H.-S.; Kuo, S.-C.; Teng, C.-M.; Lee, F.-Y.; Wang, J.-P.; Lee, Y.-C.; Kuo, C.-W.; Huang, C.-C.; Wu, C.-C.; Huang, L.-J. Bioorg. Med. Chem. 2008, 16, 1262–1278. doi:10.1016/j.bmc.2007.10.070
Return to citation in text: [1] -
Goodman, K. B.; Cui, H.; Dowdell, S. E.; Gaitanopoulos, D. E.; Ivy, R. L.; Sehon, C. A.; Stavenger, R. A.; Wang, G. Z.; Viet, A. Q.; Xu, W.; Ye, G.; Semus, S. F.; Evans, C.; Fries, H. E.; Jolivette, L. J.; Kirkpatrick, R. B.; Dul, E.; Khandekar, S. S.; Yi, T.; Jung, D. K.; Wright, L. L.; Smith, G. K.; Behm, D. J.; Bentley, R.; Doe, C. P.; Hu, E.; Lee, D. J. Med. Chem. 2007, 50, 6–9. doi:10.1021/jm0609014
Return to citation in text: [1] -
Heruye, S. H.; Maffofou Nkenyi, L. N.; Singh, N. U.; Yalzadeh, D.; Ngele, K. K.; Njie-Mbye, Y.-F.; Ohia, S. E.; Opere, C. A. Pharmaceuticals 2020, 13, 15. doi:10.3390/ph13010015
Return to citation in text: [1] -
Quane, P. A.; Graham, G. G.; Ziegler, J. B. Inflammopharmacology 1998, 6, 95–107. doi:10.1007/s10787-998-0026-0
Return to citation in text: [1] -
Yarker, Y. E.; McTavish, D. Drugs 1994, 48, 761–793. doi:10.2165/00003495-199448050-00008
Return to citation in text: [1] -
Chen, G.; Hu, M.; Peng, Y. J. Org. Chem. 2018, 83, 1591–1597. doi:10.1021/acs.joc.7b02857
Return to citation in text: [1] [2] -
Chen, C.-y.; Tang, G.; He, F.; Wang, Z.; Jing, H.; Faessler, R. Org. Lett. 2016, 18, 1690–1693. doi:10.1021/acs.orglett.6b00611
Return to citation in text: [1] [2] [3] [4] -
Kim, O. S.; Jang, J. H.; Kim, H. T.; Han, S. J.; Tsui, G. C.; Joo, J. M. Org. Lett. 2017, 19, 1450–1453. doi:10.1021/acs.orglett.7b00410
Return to citation in text: [1] [2] -
Janardhanan, J. C.; Mishra, R. K.; Das, G.; Sini, S.; Jayamurthy, P.; Suresh, C. H.; Praveen, V. K.; Manoj, N.; Babu, B. P. Asian J. Org. Chem. 2018, 7, 2094–2104. doi:10.1002/ajoc.201800413
Return to citation in text: [1] [2] -
Li, L.; Wang, H.; Yu, S.; Yang, X.; Li, X. Org. Lett. 2016, 18, 3662–3665. doi:10.1021/acs.orglett.6b01716
Return to citation in text: [1] [2] -
Inamoto, K.; Saito, T.; Katsuno, M.; Sakamoto, T.; Hiroya, K. Org. Lett. 2007, 9, 2931–2934. doi:10.1021/ol0711117
Return to citation in text: [1] [2] -
Li, X.; He, L.; Chen, H.; Wu, W.; Jiang, H. J. Org. Chem. 2013, 78, 3636–3646. doi:10.1021/jo400162d
Return to citation in text: [1] [2] -
Park, A.; Jeong, K.-S.; Lee, H.; Kim, H. ACS Omega 2021, 6, 6498–6508. doi:10.1021/acsomega.1c00025
Return to citation in text: [1] -
Wei, W.; Wang, Z.; Yang, X.; Yu, W.; Chang, J. Adv. Synth. Catal. 2017, 359, 3378–3387. doi:10.1002/adsc.201700824
Return to citation in text: [1] [2] -
Zhang, Z.; Huang, Y.; Huang, G.; Zhang, G.; Liu, Q. J. Heterocycl. Chem. 2017, 54, 2426–2433. doi:10.1002/jhet.2839
Return to citation in text: [1] [2] -
Annor-Gyamfi, J. K.; Gnanasekaran, K. K.; Bunce, R. A. Molecules 2018, 23, 674. doi:10.3390/molecules23030674
Return to citation in text: [1] [2] -
Cho, C. S.; Lim, D. K.; Heo, N. H.; Kim, T.-J.; Shim, S. C. Chem. Commun. 2004, 104–105. doi:10.1039/b312154m
Return to citation in text: [1] [2] -
Lebedev, A. Y.; Khartulyari, A. S.; Voskoboynikov, A. Z. J. Org. Chem. 2005, 70, 596–602. doi:10.1021/jo048671t
Return to citation in text: [1] [2] [3] -
Pabba, C.; Wang, H.-J.; Mulligan, S. R.; Chen, Z.-J.; Stark, T. M.; Gregg, B. T. Tetrahedron Lett. 2005, 46, 7553–7557. doi:10.1016/j.tetlet.2005.08.143
Return to citation in text: [1] [2] -
Liu, R.; Zhu, Y.; Qin, L.; Ji, S. Synth. Commun. 2008, 38, 249–254. doi:10.1080/00397910701750250
Return to citation in text: [1] [2] [3] -
Gao, M.; Liu, X.; Wang, X.; Cai, Q.; Ding, K. Chin. J. Chem. 2011, 29, 1199–1204. doi:10.1002/cjoc.201190223
Return to citation in text: [1] [2] [3] -
Lee, H. K.; Cho, C. S. Synth. Commun. 2013, 43, 915–921. doi:10.1080/00397911.2011.614714
Return to citation in text: [1] [2] -
Veerareddy, A.; Gogireddy, S.; Dubey, P. K. J. Heterocycl. Chem. 2014, 51, 1311–1321. doi:10.1002/jhet.1717
Return to citation in text: [1] [2] -
Kylmälä, T.; Udd, S.; Tois, J.; Franzén, R. Tetrahedron Lett. 2010, 51, 3613–3615. doi:10.1016/j.tetlet.2010.05.024
Return to citation in text: [1] [2]
| 23. | Annor-Gyamfi, J. K.; Gnanasekaran, K. K.; Bunce, R. A. Molecules 2018, 23, 674. doi:10.3390/molecules23030674 |
| 24. | Cho, C. S.; Lim, D. K.; Heo, N. H.; Kim, T.-J.; Shim, S. C. Chem. Commun. 2004, 104–105. doi:10.1039/b312154m |
| 25. | Lebedev, A. Y.; Khartulyari, A. S.; Voskoboynikov, A. Z. J. Org. Chem. 2005, 70, 596–602. doi:10.1021/jo048671t |
| 26. | Pabba, C.; Wang, H.-J.; Mulligan, S. R.; Chen, Z.-J.; Stark, T. M.; Gregg, B. T. Tetrahedron Lett. 2005, 46, 7553–7557. doi:10.1016/j.tetlet.2005.08.143 |
| 27. | Liu, R.; Zhu, Y.; Qin, L.; Ji, S. Synth. Commun. 2008, 38, 249–254. doi:10.1080/00397910701750250 |
| 28. | Gao, M.; Liu, X.; Wang, X.; Cai, Q.; Ding, K. Chin. J. Chem. 2011, 29, 1199–1204. doi:10.1002/cjoc.201190223 |
| 29. | Lee, H. K.; Cho, C. S. Synth. Commun. 2013, 43, 915–921. doi:10.1080/00397911.2011.614714 |
| 31. | Kylmälä, T.; Udd, S.; Tois, J.; Franzén, R. Tetrahedron Lett. 2010, 51, 3613–3615. doi:10.1016/j.tetlet.2010.05.024 |
| 27. | Liu, R.; Zhu, Y.; Qin, L.; Ji, S. Synth. Commun. 2008, 38, 249–254. doi:10.1080/00397910701750250 |
| 28. | Gao, M.; Liu, X.; Wang, X.; Cai, Q.; Ding, K. Chin. J. Chem. 2011, 29, 1199–1204. doi:10.1002/cjoc.201190223 |
| 1. | Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Molecules 2018, 23, 2783. doi:10.3390/molecules23112783 |
| 5. | Gerpe, A.; Aguirre, G.; Boiani, L.; Cerecetto, H.; González, M.; Olea-Azar, C.; Rigol, C.; Maya, J. D.; Morello, A.; Piro, O. E.; Arán, V. J.; Azqueta, A.; de Ceráin, A. L.; Monge, A.; Rojas, M. A.; Yaluff, G. Bioorg. Med. Chem. 2006, 14, 3467–3480. doi:10.1016/j.bmc.2006.01.007 |
| 15. | Kim, O. S.; Jang, J. H.; Kim, H. T.; Han, S. J.; Tsui, G. C.; Joo, J. M. Org. Lett. 2017, 19, 1450–1453. doi:10.1021/acs.orglett.7b00410 |
| 4. | Cheekavolu, C.; Muniappan, M. J. Clin. Diagn. Res. 2016, 10, FF01–FF06. doi:10.7860/jcdr/2016/19338.8465 |
| 16. | Janardhanan, J. C.; Mishra, R. K.; Das, G.; Sini, S.; Jayamurthy, P.; Suresh, C. H.; Praveen, V. K.; Manoj, N.; Babu, B. P. Asian J. Org. Chem. 2018, 7, 2094–2104. doi:10.1002/ajoc.201800413 |
| 3. | Kim, S.-H.; Markovitz, B.; Trovato, R.; Murphy, B. R.; Austin, H.; Willardsen, A. J.; Baichwal, V.; Morham, S.; Bajji, A. Bioorg. Med. Chem. Lett. 2013, 23, 2888–2892. doi:10.1016/j.bmcl.2013.03.075 |
| 13. | Chen, G.; Hu, M.; Peng, Y. J. Org. Chem. 2018, 83, 1591–1597. doi:10.1021/acs.joc.7b02857 |
| 2. | Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. ChemMedChem 2018, 13, 1490–1507. doi:10.1002/cmdc.201800253 |
| 14. | Chen, C.-y.; Tang, G.; He, F.; Wang, Z.; Jing, H.; Faessler, R. Org. Lett. 2016, 18, 1690–1693. doi:10.1021/acs.orglett.6b00611 |
| 9. | Goodman, K. B.; Cui, H.; Dowdell, S. E.; Gaitanopoulos, D. E.; Ivy, R. L.; Sehon, C. A.; Stavenger, R. A.; Wang, G. Z.; Viet, A. Q.; Xu, W.; Ye, G.; Semus, S. F.; Evans, C.; Fries, H. E.; Jolivette, L. J.; Kirkpatrick, R. B.; Dul, E.; Khandekar, S. S.; Yi, T.; Jung, D. K.; Wright, L. L.; Smith, G. K.; Behm, D. J.; Bentley, R.; Doe, C. P.; Hu, E.; Lee, D. J. Med. Chem. 2007, 50, 6–9. doi:10.1021/jm0609014 |
| 11. | Quane, P. A.; Graham, G. G.; Ziegler, J. B. Inflammopharmacology 1998, 6, 95–107. doi:10.1007/s10787-998-0026-0 |
| 8. | Chen, H.-S.; Kuo, S.-C.; Teng, C.-M.; Lee, F.-Y.; Wang, J.-P.; Lee, Y.-C.; Kuo, C.-W.; Huang, C.-C.; Wu, C.-C.; Huang, L.-J. Bioorg. Med. Chem. 2008, 16, 1262–1278. doi:10.1016/j.bmc.2007.10.070 |
| 12. | Yarker, Y. E.; McTavish, D. Drugs 1994, 48, 761–793. doi:10.2165/00003495-199448050-00008 |
| 7. | Reddy, G. S.; Viswanath, I. V. K.; Rao, A. T. Indian J. Heterocycl. Chem. 2018, 28, 467–475. doi:10.13140/rg.2.2.32357.24802 |
| 25. | Lebedev, A. Y.; Khartulyari, A. S.; Voskoboynikov, A. Z. J. Org. Chem. 2005, 70, 596–602. doi:10.1021/jo048671t |
| 6. | Rodríguez-Villar, K.; Hernández-Campos, A.; Yépez-Mulia, L.; Sainz-Espuñes, T. d. R.; Soria-Arteche, O.; Palacios-Espinosa, J. F.; Cortés-Benítez, F.; Leyte-Lugo, M.; Varela-Petrissans, B.; Quintana-Salazar, E. A.; Pérez-Villanueva, J. Pharmaceuticals 2021, 14, 176. doi:10.3390/ph14030176 |
| 10. | Heruye, S. H.; Maffofou Nkenyi, L. N.; Singh, N. U.; Yalzadeh, D.; Ngele, K. K.; Njie-Mbye, Y.-F.; Ohia, S. E.; Opere, C. A. Pharmaceuticals 2020, 13, 15. doi:10.3390/ph13010015 |
| 21. | Wei, W.; Wang, Z.; Yang, X.; Yu, W.; Chang, J. Adv. Synth. Catal. 2017, 359, 3378–3387. doi:10.1002/adsc.201700824 |
| 22. | Zhang, Z.; Huang, Y.; Huang, G.; Zhang, G.; Liu, Q. J. Heterocycl. Chem. 2017, 54, 2426–2433. doi:10.1002/jhet.2839 |
| 17. | Li, L.; Wang, H.; Yu, S.; Yang, X.; Li, X. Org. Lett. 2016, 18, 3662–3665. doi:10.1021/acs.orglett.6b01716 |
| 18. | Inamoto, K.; Saito, T.; Katsuno, M.; Sakamoto, T.; Hiroya, K. Org. Lett. 2007, 9, 2931–2934. doi:10.1021/ol0711117 |
| 19. | Li, X.; He, L.; Chen, H.; Wu, W.; Jiang, H. J. Org. Chem. 2013, 78, 3636–3646. doi:10.1021/jo400162d |
| 20. | Park, A.; Jeong, K.-S.; Lee, H.; Kim, H. ACS Omega 2021, 6, 6498–6508. doi:10.1021/acsomega.1c00025 |
| 26. | Pabba, C.; Wang, H.-J.; Mulligan, S. R.; Chen, Z.-J.; Stark, T. M.; Gregg, B. T. Tetrahedron Lett. 2005, 46, 7553–7557. doi:10.1016/j.tetlet.2005.08.143 |
| 27. | Liu, R.; Zhu, Y.; Qin, L.; Ji, S. Synth. Commun. 2008, 38, 249–254. doi:10.1080/00397910701750250 |
| 28. | Gao, M.; Liu, X.; Wang, X.; Cai, Q.; Ding, K. Chin. J. Chem. 2011, 29, 1199–1204. doi:10.1002/cjoc.201190223 |
| 29. | Lee, H. K.; Cho, C. S. Synth. Commun. 2013, 43, 915–921. doi:10.1080/00397911.2011.614714 |
| 30. | Veerareddy, A.; Gogireddy, S.; Dubey, P. K. J. Heterocycl. Chem. 2014, 51, 1311–1321. doi:10.1002/jhet.1717 |
| 31. | Kylmälä, T.; Udd, S.; Tois, J.; Franzén, R. Tetrahedron Lett. 2010, 51, 3613–3615. doi:10.1016/j.tetlet.2010.05.024 |
| 30. | Veerareddy, A.; Gogireddy, S.; Dubey, P. K. J. Heterocycl. Chem. 2014, 51, 1311–1321. doi:10.1002/jhet.1717 |
| 23. | Annor-Gyamfi, J. K.; Gnanasekaran, K. K.; Bunce, R. A. Molecules 2018, 23, 674. doi:10.3390/molecules23030674 |
| 24. | Cho, C. S.; Lim, D. K.; Heo, N. H.; Kim, T.-J.; Shim, S. C. Chem. Commun. 2004, 104–105. doi:10.1039/b312154m |
| 25. | Lebedev, A. Y.; Khartulyari, A. S.; Voskoboynikov, A. Z. J. Org. Chem. 2005, 70, 596–602. doi:10.1021/jo048671t |
| 14. | Chen, C.-y.; Tang, G.; He, F.; Wang, Z.; Jing, H.; Faessler, R. Org. Lett. 2016, 18, 1690–1693. doi:10.1021/acs.orglett.6b00611 |
| 16. | Janardhanan, J. C.; Mishra, R. K.; Das, G.; Sini, S.; Jayamurthy, P.; Suresh, C. H.; Praveen, V. K.; Manoj, N.; Babu, B. P. Asian J. Org. Chem. 2018, 7, 2094–2104. doi:10.1002/ajoc.201800413 |
| 17. | Li, L.; Wang, H.; Yu, S.; Yang, X.; Li, X. Org. Lett. 2016, 18, 3662–3665. doi:10.1021/acs.orglett.6b01716 |
| 18. | Inamoto, K.; Saito, T.; Katsuno, M.; Sakamoto, T.; Hiroya, K. Org. Lett. 2007, 9, 2931–2934. doi:10.1021/ol0711117 |
| 19. | Li, X.; He, L.; Chen, H.; Wu, W.; Jiang, H. J. Org. Chem. 2013, 78, 3636–3646. doi:10.1021/jo400162d |
| 21. | Wei, W.; Wang, Z.; Yang, X.; Yu, W.; Chang, J. Adv. Synth. Catal. 2017, 359, 3378–3387. doi:10.1002/adsc.201700824 |
| 22. | Zhang, Z.; Huang, Y.; Huang, G.; Zhang, G.; Liu, Q. J. Heterocycl. Chem. 2017, 54, 2426–2433. doi:10.1002/jhet.2839 |
| 13. | Chen, G.; Hu, M.; Peng, Y. J. Org. Chem. 2018, 83, 1591–1597. doi:10.1021/acs.joc.7b02857 |
| 14. | Chen, C.-y.; Tang, G.; He, F.; Wang, Z.; Jing, H.; Faessler, R. Org. Lett. 2016, 18, 1690–1693. doi:10.1021/acs.orglett.6b00611 |
| 14. | Chen, C.-y.; Tang, G.; He, F.; Wang, Z.; Jing, H.; Faessler, R. Org. Lett. 2016, 18, 1690–1693. doi:10.1021/acs.orglett.6b00611 |
| 15. | Kim, O. S.; Jang, J. H.; Kim, H. T.; Han, S. J.; Tsui, G. C.; Joo, J. M. Org. Lett. 2017, 19, 1450–1453. doi:10.1021/acs.orglett.7b00410 |
© 2022 Barbosa et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.