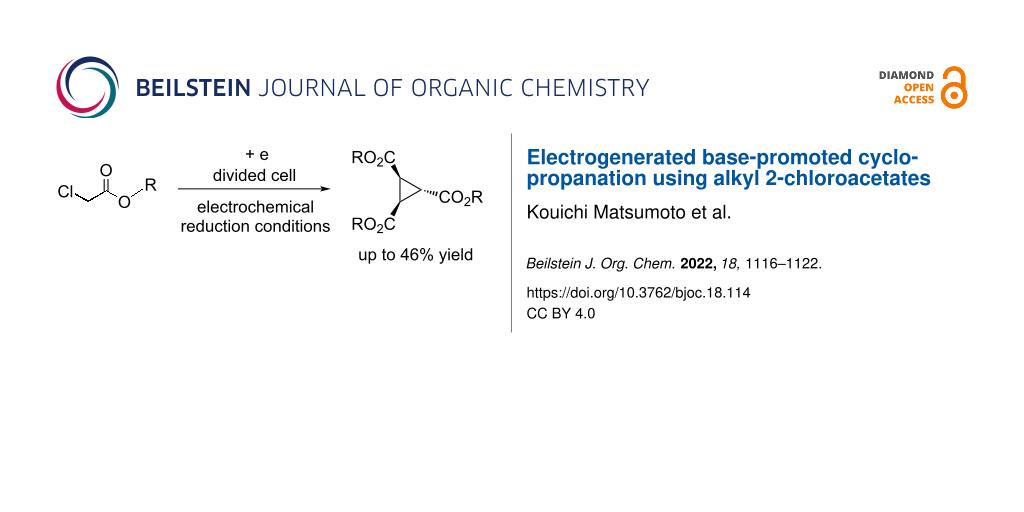
Abstract
The electrochemical reduction conditions of the reaction of alkyl 2-chloroacetates in Bu4NBr/DMF using a divided cell equipped with Pt electrodes to produce the corresponding cyclopropane derivatives in moderate yields were discovered. The reaction conditions were optimized, the scope and limitations, as well as scale-up reactions were investigated. The presented method for the electrochemical production of cyclopropane derivatives is an environmentally friendly and easy to perform synthetic procedure.

Graphical Abstract
Introduction
In organic chemistry, cyclopropanes and their related compounds have been recognized as important molecules. For example, cyclopropane derivatives are found in both natural products and pharmaceutical products. The cyclopropane skeleton is also found in agrochemicals, especially pyrethroid, as an insecticide, is one important compound. Cyclopropanes also play a significant role in organic synthesis as versatile building blocks [1-5]. In general, some synthetic procedures for cyclopropane derivatives have been discovered, e.g., the Simmons–Smith reaction and the use of metal carbenoids being two of the more prominent and reliable methods [6-9].
Aggarwal and colleagues reported in 2000 that the reaction between a Michael acceptor such as diethyl fumarate and a sulfur-ylide, prepared from ethyl 2-diazoacetate and tetrahydro-2H-thiopyran in the presence of Cu(acac)2, yielded triethyl cyclopropane-1,2,3-tricarboxylate in 68% yield (Scheme 1, reaction 1) [10]. The same chemical yield was obtained by using a catalytic amount of tetrahydro-2H-thiopyran (0.2 equiv) in the process [10]. Furthermore, de Meijere and colleagues in 2003 demonstrated that the reaction of diethyl fumarate and ethyl 2-chloroacetate in DMF at 40 °C with K2CO3 and TEBACl (benzyltriethylammonium chloride) produced triethyl cyclopropane-1,2,3-tricarboxylate in 62% yield (Scheme 1, reaction 2) [11]. Both procedures are two-component coupling reactions. In contrast, a method involving a one-component reaction using alkyl 2-haloacetate has been developed by Abushanab in 1967, in which a stoichiometric amount of metal lithium was utilized to reduce ethyl 2-bromoacetate to form the corresponding 1,2,3-trisubstituted cyclopropane derivatives (Scheme 1, reaction 3) [12]. The generation of an anionic intermediate was indicated. During our study, we discovered that 1,2,3-trisubstituted cyclopropane derivatives could be formed in moderate yields through the electrochemical reduction [13-21] of alkyl 2-chloroacetates in a divided cell (Scheme 1, reaction 4). The in Abushanab’s study utilized metal lithium is one of the rarest and most expensive metals. In addition, the treatment of metal lithium is difficult and occasionally dangerous, and the reaction also produces the corresponding Li salt as waste [22,23]. In contrast, in this work, we use basic electricity to make the corresponding cyclopropane derivatives. Herein, we would like to report the details of our investigation.
![[1860-5397-18-114-i1]](/bjoc/content/inline/1860-5397-18-114-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formations of 1,2,3-trialkyl cyclopropanetricarboxylates. Previous reports (reactions 1–3) and this work (reaction 4).
Scheme 1: Formations of 1,2,3-trialkyl cyclopropanetricarboxylates. Previous reports (reactions 1–3) and this...
Results and Discussion
First, we investigated the reaction conditions for the electrochemical reduction to optimize the reaction outcome. The typical procedure is as follows: the electrochemical reduction was carried out in an H-type divided cell. Both electrodes were made from Pt plates. In the cathodic chamber, 1 (0.5 mmol) was dissolved in 0.3 M Bu4NBr in DMF (4.0 mL) and 0.3 M Bu4NBr in DMF (4.0 mL) was introduced to the anodic chamber. Constant current electrolysis at 12 mA until 1.0 F/mol was consumed in the cathode yielded the corresponding compound 2 in a 46% yield (Table 1, entry 1). Various parameters were varied to increase the chemical yield, as shown in Table 1. For example, the use of carbon felt as the cathode produced 2 in ≪25% yield (Table 1, entry 2). The small influence of the anodic electrode material was confirmed in the reaction using carbon felt as the anode. In this reaction compound 2 was obtained in 40% yield (Table 1, entry 3). The use of DMSO instead of DMF resulted in <22% yield of 2 (Table 1, entry 4). However, when MeOH was used 2 could not be obtained at all (Table 1, entry 5). Reactions with Bu4NCl, Bu4NI, and Bu4NBF4 instead of Bu4NBr produced the corresponding compound 2 in 35%, <21%, and 44% yields, respectively (Table 1, entries 6–8). The amount of the supporting electrolyte Bu4NBr, such as 0.8 equiv and 4.0 equiv instead of 2.4 equiv, appeared to have no influence, and 2 was produced at 40% and 43% yields, respectively (Table 1, entries 9 and 10). In terms of temperature and current (Table 1, entries 11–14), 6 mA at room temperature yielded the best result of 46% yield (Table 1, entry 14). The reaction did not take place in the absence of electricity (Table 1, entry 15). Based on the above optimizations, we chose the conditions given in entry 1 of Table 1 as the optimized parameters [24].
Table 1: Reaction optimization.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-114-i3.svg?max-width=637&scale=1.0)
|
||
| Entry | Variation from standard conditionsa | % Yieldb |
| 1 | none | 46 |
| 2 | Pt (+) | C (–) instead of Pt (+) | Pt (–) | ≪25c |
| 3 | C (+) | Pt (–) instead of Pt (+) | Pt (–) | 40 |
| 4 | DMSO as solventd | <22 |
| 5 | MeOH as solventd | n.d.e |
| 6 | Bu4NCl instead of Bu4NBrd | 35 |
| 7 | Bu4NI instead of Bu4NBrd | <21 |
| 8 | Bu4NBF4 instead of Bu4NBrd | 44 |
| 9 | 0.8 equiv Bu4NBr instead of 2.4 equiv Bu4NBrd | 40 |
| 10 | 4.0 equiv Bu4NBr instead of 2.4 equiv Bu4NBrd | 43 |
| 11 | 0 °C instead of rt | 45 |
| 12 | 60 °C instead of rt | <44 |
| 13 | 20 mA instead of 12 mA, 1.0 F/mol | <33 |
| 14 | 6 mA instead of 12 mA, 1.0 F/mol | 46 |
| 15 | no electric current | n.d.c,e |
aStandard conditions: 1 (0.5 mmol), 0.3 M Bu4NBr in DMF (4.0 mL × 2), divided cell, 12 mA, rt, 1.0 F/mol of electricity against 0.5 mmol of substrate 1. bIsolated yields using preparative GPC separation of the crude materials. cObserved from gas chromatography (GC) analysis. dIn both anodic and cathodic chambers. en.d. = no detection.
Next, we investigated the effect of electricity around 1 F/mol on the yield, as shown in Table 2, using the optimized conditions [25]. The yield of 2 was 46% in the case of 1.0 F/mol (Table 2, entry 1), as shown in entry 1 of Table 1. The chemical output of 2 was 44% in the case of 0.90 F/mol (Table 2, entry 2). However, using 1.1 F/mol resulted as well in a lower yield of 2 (<35%, Table 2, entry 3). Thus, 0.90 F/mol or 1.0 F/mol of electricity for the current reaction was found to be sufficient to obtain the product in high yield, and we choose 1.0 F/mol of electricity for the next investigations (Table 3). Finally, the electrolysis using the undivided cell shown in entry 4 of Table 2 yielded 2 in ≪5% yield, indicating that the divided cell is essential for the current reaction. In the undivided cell, the anionic species from the cathode might be consumed on the surface of the anode.
Table 2: Effect of electricity around 1 F/mol and type of electrochemical cell.
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-114-i4.svg?max-width=637&scale=1.0)
|
||
| Entry | F/mol | % Yielda |
| 1 | 1.0 | 46b |
| 2 | 0.90 | 44 |
| 3 | 1.1 | <35 |
| 4c | 1.0 | ≪5 |
aIsolated yields using preparative GPC separation of the crude materials. Compound 2 of entry 3 contained a small amount of impurity. bThis yield is from entry 1 of Table 1. cAn undivided cell was used instead of a divided cell.
To examine the scope and limitations, we carried out electrochemical reductions of various alkyl 2-haloacetates under the optimized conditions. Table 3 summarizes the results. The reaction of methyl 2-chloroacetate (3) afforded the corresponding compound 4 in 28% yield (Table 3, entry 1). The reaction of ethyl 2-chloroacetate (5) produced the corresponding compound 6 in a similar <23% yield (Table 3, entry 2). Ethyl 2-bromoacetate (7) and ethyl 2-iodoacetate (8), in which the leaving groups were changed from Cl to Br and I, showed similar reactivities to produce compound 6 in <24% and <26% yields, respectively (Table 3, entries 3 and 4). n-Propyl 2-chloroacetate (9), with the longer alkyl chain, and tert-butyl 2-chloroacetate (11), with the bulky alkyl group, produced 10 and 12 in <22% and <31% yields, respectively (Table 3, entries 5 and 6). The reaction of 13 with the vinyl group did not occur (Table 3, entry 7), but the reaction of compound 15 with the allyl group formed 16 in 34% yield (Table 3, entry 8). Finally, benzyl 2-chloroacetate (17) produced the corresponding compound 18 in 31% yield (Table 3, entry 9).
Table 3: Scope and limitations.
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-114-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Ester | Product | % Yielda | ||
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-114-i6.svg?max-width=637&scale=1.0)
|
3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-18-114-i7.svg?max-width=637&scale=1.0)
|
4 | 28 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-18-114-i8.svg?max-width=637&scale=1.0)
|
5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-18-114-i9.svg?max-width=637&scale=1.0)
|
6 | <23 (21) |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-18-114-i10.svg?max-width=637&scale=1.0)
|
7 | 6 | <24 (19) | |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-18-114-i11.svg?max-width=637&scale=1.0)
|
8 | 6 | <26 (20) | |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-18-114-i12.svg?max-width=637&scale=1.0)
|
9 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-18-114-i13.svg?max-width=637&scale=1.0)
|
10 | <22 (20) |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-18-114-i14.svg?max-width=637&scale=1.0)
|
11 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-18-114-i15.svg?max-width=637&scale=1.0)
|
12 | <31 (28) |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-18-114-i16.svg?max-width=637&scale=1.0)
|
13 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-18-114-i17.svg?max-width=637&scale=1.0)
|
14 | n.d.b |
| 8 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-18-114-i18.svg?max-width=637&scale=1.0)
|
15 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-18-114-i19.svg?max-width=637&scale=1.0)
|
16 | 34 |
| 9 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-18-114-i20.svg?max-width=637&scale=1.0)
|
17 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-18-114-i21.svg?max-width=637&scale=1.0)
|
18 | 31c |
aIsolated yields using preparative GPC separation of the crude materials. Compound 6 in entries 3 and 4 contained an impurity of non-negligible amount, despite of repeated purification by GPC. Compound 6 in entry 2, 10 in entry 5 and 12 in entry 6 contained a small amount of impurities (see 13C NMR spectra of compounds 6, 10 and 12 in Supporting Information File 1). Values in parentheses in entries 2–6 are estimated yields, calculated from the ratio of isolated compounds and impurities given in the 1H NMR spectra, because the impurities seem to be the corresponding trialkyl propane-1,2,3-carboxylates (vide infra). bn.d. = no detection. cIsolated yield after silica-gel column chromatography.
The current electrolysis reaction can be easily scaled-up with obtaining similar yields of the products. The reaction of 1 (1.2 g, 8.0 mmol) in Bu4NBr/DMF at room temperature with 12 mA and 1.0 F/mol yielded the corresponding compound 2 in <45% yield (Table 4, entry 1). In addition, the reaction of 3 (1.3 g, 12.0 mmol) yielded 4 in 32% yield (Table 4, entry 2).
![[Graphic 20]](/bjoc/content/inline/1860-5397-18-114-i22.svg?max-width=637&scale=1.0)
aIsolated yields using preparative GPC separation of the crude materials. Compound 2 in entry 1 contained a small amount of impurity. The value in parenthesis in entry 1 is an estimated yield, calculated from the ratio of isolated compound and impurity given in the 1H NMR spectrum, because the impurity seems to be tri-n-butyl propane-1,2,3-carboxylate (vide infra).
Scheme 2 depicts a plausible reaction mechanism. We assume that the current reaction follows a similar mechanism as described in Abushanab’s report [12]. In addition, the current reaction indicated the generation of an EGB (electrogenerated base) [26-29]. The electrochemical reduction conditions of the solution containing 1 may generate an EGB, which reacts with 1 to produce anionic A. At the stage of the generation of the EGB, the reduction of 1 may generate an enolate ion such as E or F, which might serve as EGB, although other sources of EGBs cannot be denied [30,31]. Intermediate A may combine with 1 to produce B, which may react with the EGB or another molecule A to produce C, releasing HCl. In Abushanab’s report [12], C can be coupled with A in a similar manner to yield D [12]. Finally, intramolecular cyclization of D may yield 2. In order to obtain a deeper insight in the reaction, we made an analysis of the crude material, which was prepared by passing 0.5 F/mol using the standard conditions and the usual work up procedure. Compounds B and C were confirmed by HRMS analysis shown in Scheme 2, which supported the current mechanism.
![[1860-5397-18-114-i2]](/bjoc/content/inline/1860-5397-18-114-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible reaction mechanism. EGB = electrogenerated base.
Scheme 2: Plausible reaction mechanism. EGB = electrogenerated base.
In addition, one of the impurities seems to be the trialkyl propane-1,2,3-carboxylates [32], because the HRMS analyses of the isolated compounds, such as 6 (Table 3, entry 2), 10 (Table 3, entry 5), and 12 (Table 3, entry 6), which were not of high purity, showed the existence of the corresponding trialkyl propane-1,2,3-carboxylates, together with the signal of the desired products 6, 10, and 12. The formation of trialkyl propane-1,2,3-carboxylate might be through the Michael addition of the electrogenerated base (E in Scheme 2) to C or the electrochemical reduction of 2.
Conclusion
A new electrochemical transformation of alkyl 2-chloroacetates to cyclopropane derivatives has been developed. The reaction has been optimized, the scope and limitations have been investigated. Scale-up reactions were performed and satisfactory yields obtained. The generation of an EGB of the enolate ion from alkyl 2-chloroacetates is indicated. The current method is one of the most environmentally benign and accessible methods for the preparation of 1,2,3-trisubstituted cyclopropane derivatives, notwithstanding the low reaction yields. In our laboratory, further synthetic investigations are in progress.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data of new compounds and copies of 1H NMR and 13C NMR spectra. | ||
| Format: PDF | Size: 1015.9 KB | Download |
Acknowledgements
We are grateful to the Kindai University Joint Research Center for the use of facilities. We also appreciate the reviewers for fruitful suggestion.
Funding
This work was supported in part by JSPS KAKENHI Grants JP 20K05588 (Grant-in-Aid for Scientific Research (C)). K. M. acknowledges the Tokyo Ohka Foundation for The Promotion of Science and Technology for the financial support. This work was also partially supported by 2021 Kindai University Research Enchancement Grants (KD2106 and KD2104).
References
-
Cohen, Y.; Cohen, A.; Marek, I. Chem. Rev. 2021, 121, 140–161. doi:10.1021/acs.chemrev.0c00167
Return to citation in text: [1] -
Pirenne, V.; Muriel, B.; Waser, J. Chem. Rev. 2021, 121, 227–263. doi:10.1021/acs.chemrev.0c00109
Return to citation in text: [1] -
Ebner, C.; Carreira, E. M. Chem. Rev. 2017, 117, 11651–11679. doi:10.1021/acs.chemrev.6b00798
Return to citation in text: [1] -
Augustin, A. U.; Werz, D. B. Acc. Chem. Res. 2021, 54, 1528–1541. doi:10.1021/acs.accounts.1c00023
Return to citation in text: [1] -
Pons, A.; Delion, L.; Poisson, T.; Charette, A. B.; Jubault, P. Acc. Chem. Res. 2021, 54, 2969–2990. doi:10.1021/acs.accounts.1c00261
Return to citation in text: [1] -
Simmons, H. E.; Smith, R. D. J. Am. Chem. Soc. 1958, 80, 5323–5324. doi:10.1021/ja01552a080
Return to citation in text: [1] -
Ye, T.; McKervey, M. A. Chem. Rev. 1994, 94, 1091–1160. doi:10.1021/cr00028a010
Return to citation in text: [1] -
Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Synthesis 2014, 46, 0979–1029. doi:10.1055/s-0033-1340838
Return to citation in text: [1] -
Bos, M.; Poisson, T.; Pannecoucke, X.; Charette, A. B.; Jubault, P. Chem. – Eur. J. 2017, 23, 4950–4961. doi:10.1002/chem.201604564
Return to citation in text: [1] -
Aggarwal, V. K.; Smith, H. W.; Hynd, G.; Jones, R. V. H.; Fieldhouse, R.; Spey, S. E. J. Chem. Soc., Perkin Trans. 1 2000, 3267–3276. doi:10.1039/b004367m
Return to citation in text: [1] [2] -
Kozhushkov, S. I.; Leonov, A.; de Meijere, A. Synthesis 2003, 956–958. doi:10.1055/s-2003-38687
Return to citation in text: [1] -
Abushanab, E. Tetrahedron Lett. 1967, 8, 2833–2836. doi:10.1016/s0040-4039(00)90868-2
Return to citation in text: [1] [2] [3] [4] -
Shida, N.; Nakamura, Y.; Atobe, M. Chem. Rec. 2021, 21, 2164–2177. doi:10.1002/tcr.202100016
Return to citation in text: [1] -
Shimakoshi, H.; Hisaeda, Y. Chem. Rec. 2021, 21, 2080–2094. doi:10.1002/tcr.202100077
Return to citation in text: [1] -
Shibuya, A.; Nokami, T. Chem. Rec. 2021, 21, 2389–2396. doi:10.1002/tcr.202100085
Return to citation in text: [1] -
Krueger, R.; Moeller, K. D. J. Org. Chem. 2021, 86, 15847–15865. doi:10.1021/acs.joc.1c01609
Return to citation in text: [1] -
Nutting, J. E.; Gerken, J. B.; Stamoulis, A. G.; Bruns, D. L.; Stahl, S. S. J. Org. Chem. 2021, 86, 15875–15885. doi:10.1021/acs.joc.1c01520
Return to citation in text: [1] -
Mitsudo, K.; Kurimoto, Y.; Yoshioka, K.; Suga, S. Chem. Rev. 2018, 118, 5985–5999. doi:10.1021/acs.chemrev.7b00532
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Fuchigami, T.; Inagi, S. Acc. Chem. Res. 2020, 53, 322–334. doi:10.1021/acs.accounts.9b00520
Return to citation in text: [1] -
Senboku, H.; Katayama, A. Curr. Opin. Green Sustainable Chem. 2017, 3, 50–54. doi:10.1016/j.cogsc.2016.10.003
Return to citation in text: [1] -
Elinson, M. N.; Feducovich, S. K.; Vereshchagin, A. N.; Gorbunov, S. V.; Belyakov, P. A.; Nikishin, G. I. Tetrahedron Lett. 2006, 47, 9129–9133. doi:10.1016/j.tetlet.2006.10.075
See for a reported electrochemical synthesis of cyclopropanes.
Return to citation in text: [1] -
Tokuda, M.; Hayashi, A.; Suginome, H. Bull. Chem. Soc. Jpn. 1991, 64, 2590–2592. doi:10.1246/bcsj.64.2590
See for a reported electrochemical synthesis of cyclopropanes.
Return to citation in text: [1] -
As the possibility for the generation of an EGB, the electrolysis in the presence of pre-EGB, such as H2O (1 mmol) or 2-pyrrolidone (1 mmol), under the optimized conditions gave 12% yield or trace of 2, respectively, which was estimated by 1H NMR analysis of the crude materials.
Return to citation in text: [1] -
We roughly performed the GC-monitoring of the electrochemical reaction until ca. 2 F/mol were consumed, and found that around 1 F/mol seems to be suitable for the best yield of 2 in the reaction.
Return to citation in text: [1] -
Torii, S.; Hayashi, N.; Kuroboshi, M. Synlett 1998, 599–600. doi:10.1055/s-1998-1752
Return to citation in text: [1] -
Strehl, J.; Hilt, G. Org. Lett. 2019, 21, 5259–5263. doi:10.1021/acs.orglett.9b01866
Return to citation in text: [1] -
Mirza, B.; Samiei, S. S. Asian J. Chem. 2012, 24, 1101–1103.
Return to citation in text: [1] -
William, J. M.; Kuriyama, M.; Onomura, O. Electrochemistry 2013, 81, 374–376. doi:10.5796/electrochemistry.81.374
Return to citation in text: [1] -
Baizer, M. M.; Chruma, J. L. J. Org. Chem. 1972, 37, 1951–1960. doi:10.1021/jo00977a020
Return to citation in text: [1] -
Inesi, A.; Zeppa, A.; Zeuli, E. J. Electroanal. Chem. Interfacial Electrochem. 1982, 137, 103–115. doi:10.1016/0022-0728(82)85070-5
Return to citation in text: [1] -
Ye, Y.; Ma, G.; Yao, K.; Gong, H. Synlett 2021, 32, 1625–1628. doi:10.1055/a-1328-0352
See for NMR spectra of triethyl propane-1, 2, 3-carboxylate. By comparison with NMR spectra of trans-triethyl cyclopropane-1,2,3-tricarboxylate (6) in the Supporting Information, triethyl propane-1,2,3-carboxylate was identified as the impurity.
Return to citation in text: [1]
| 1. | Cohen, Y.; Cohen, A.; Marek, I. Chem. Rev. 2021, 121, 140–161. doi:10.1021/acs.chemrev.0c00167 |
| 2. | Pirenne, V.; Muriel, B.; Waser, J. Chem. Rev. 2021, 121, 227–263. doi:10.1021/acs.chemrev.0c00109 |
| 3. | Ebner, C.; Carreira, E. M. Chem. Rev. 2017, 117, 11651–11679. doi:10.1021/acs.chemrev.6b00798 |
| 4. | Augustin, A. U.; Werz, D. B. Acc. Chem. Res. 2021, 54, 1528–1541. doi:10.1021/acs.accounts.1c00023 |
| 5. | Pons, A.; Delion, L.; Poisson, T.; Charette, A. B.; Jubault, P. Acc. Chem. Res. 2021, 54, 2969–2990. doi:10.1021/acs.accounts.1c00261 |
| 11. | Kozhushkov, S. I.; Leonov, A.; de Meijere, A. Synthesis 2003, 956–958. doi:10.1055/s-2003-38687 |
| 12. | Abushanab, E. Tetrahedron Lett. 1967, 8, 2833–2836. doi:10.1016/s0040-4039(00)90868-2 |
| 10. | Aggarwal, V. K.; Smith, H. W.; Hynd, G.; Jones, R. V. H.; Fieldhouse, R.; Spey, S. E. J. Chem. Soc., Perkin Trans. 1 2000, 3267–3276. doi:10.1039/b004367m |
| 32. |
Ye, Y.; Ma, G.; Yao, K.; Gong, H. Synlett 2021, 32, 1625–1628. doi:10.1055/a-1328-0352
See for NMR spectra of triethyl propane-1, 2, 3-carboxylate. By comparison with NMR spectra of trans-triethyl cyclopropane-1,2,3-tricarboxylate (6) in the Supporting Information, triethyl propane-1,2,3-carboxylate was identified as the impurity. |
| 10. | Aggarwal, V. K.; Smith, H. W.; Hynd, G.; Jones, R. V. H.; Fieldhouse, R.; Spey, S. E. J. Chem. Soc., Perkin Trans. 1 2000, 3267–3276. doi:10.1039/b004367m |
| 30. | Baizer, M. M.; Chruma, J. L. J. Org. Chem. 1972, 37, 1951–1960. doi:10.1021/jo00977a020 |
| 31. | Inesi, A.; Zeppa, A.; Zeuli, E. J. Electroanal. Chem. Interfacial Electrochem. 1982, 137, 103–115. doi:10.1016/0022-0728(82)85070-5 |
| 6. | Simmons, H. E.; Smith, R. D. J. Am. Chem. Soc. 1958, 80, 5323–5324. doi:10.1021/ja01552a080 |
| 7. | Ye, T.; McKervey, M. A. Chem. Rev. 1994, 94, 1091–1160. doi:10.1021/cr00028a010 |
| 8. | Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Synthesis 2014, 46, 0979–1029. doi:10.1055/s-0033-1340838 |
| 9. | Bos, M.; Poisson, T.; Pannecoucke, X.; Charette, A. B.; Jubault, P. Chem. – Eur. J. 2017, 23, 4950–4961. doi:10.1002/chem.201604564 |
| 12. | Abushanab, E. Tetrahedron Lett. 1967, 8, 2833–2836. doi:10.1016/s0040-4039(00)90868-2 |
| 24. | As the possibility for the generation of an EGB, the electrolysis in the presence of pre-EGB, such as H2O (1 mmol) or 2-pyrrolidone (1 mmol), under the optimized conditions gave 12% yield or trace of 2, respectively, which was estimated by 1H NMR analysis of the crude materials. |
| 12. | Abushanab, E. Tetrahedron Lett. 1967, 8, 2833–2836. doi:10.1016/s0040-4039(00)90868-2 |
| 22. |
Elinson, M. N.; Feducovich, S. K.; Vereshchagin, A. N.; Gorbunov, S. V.; Belyakov, P. A.; Nikishin, G. I. Tetrahedron Lett. 2006, 47, 9129–9133. doi:10.1016/j.tetlet.2006.10.075
See for a reported electrochemical synthesis of cyclopropanes. |
| 23. |
Tokuda, M.; Hayashi, A.; Suginome, H. Bull. Chem. Soc. Jpn. 1991, 64, 2590–2592. doi:10.1246/bcsj.64.2590
See for a reported electrochemical synthesis of cyclopropanes. |
| 26. | Torii, S.; Hayashi, N.; Kuroboshi, M. Synlett 1998, 599–600. doi:10.1055/s-1998-1752 |
| 27. | Strehl, J.; Hilt, G. Org. Lett. 2019, 21, 5259–5263. doi:10.1021/acs.orglett.9b01866 |
| 28. | Mirza, B.; Samiei, S. S. Asian J. Chem. 2012, 24, 1101–1103. |
| 29. | William, J. M.; Kuriyama, M.; Onomura, O. Electrochemistry 2013, 81, 374–376. doi:10.5796/electrochemistry.81.374 |
| 13. | Shida, N.; Nakamura, Y.; Atobe, M. Chem. Rec. 2021, 21, 2164–2177. doi:10.1002/tcr.202100016 |
| 14. | Shimakoshi, H.; Hisaeda, Y. Chem. Rec. 2021, 21, 2080–2094. doi:10.1002/tcr.202100077 |
| 15. | Shibuya, A.; Nokami, T. Chem. Rec. 2021, 21, 2389–2396. doi:10.1002/tcr.202100085 |
| 16. | Krueger, R.; Moeller, K. D. J. Org. Chem. 2021, 86, 15847–15865. doi:10.1021/acs.joc.1c01609 |
| 17. | Nutting, J. E.; Gerken, J. B.; Stamoulis, A. G.; Bruns, D. L.; Stahl, S. S. J. Org. Chem. 2021, 86, 15875–15885. doi:10.1021/acs.joc.1c01520 |
| 18. | Mitsudo, K.; Kurimoto, Y.; Yoshioka, K.; Suga, S. Chem. Rev. 2018, 118, 5985–5999. doi:10.1021/acs.chemrev.7b00532 |
| 19. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 20. | Fuchigami, T.; Inagi, S. Acc. Chem. Res. 2020, 53, 322–334. doi:10.1021/acs.accounts.9b00520 |
| 21. | Senboku, H.; Katayama, A. Curr. Opin. Green Sustainable Chem. 2017, 3, 50–54. doi:10.1016/j.cogsc.2016.10.003 |
| 12. | Abushanab, E. Tetrahedron Lett. 1967, 8, 2833–2836. doi:10.1016/s0040-4039(00)90868-2 |
| 25. | We roughly performed the GC-monitoring of the electrochemical reaction until ca. 2 F/mol were consumed, and found that around 1 F/mol seems to be suitable for the best yield of 2 in the reaction. |
© 2022 Matsumoto et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.