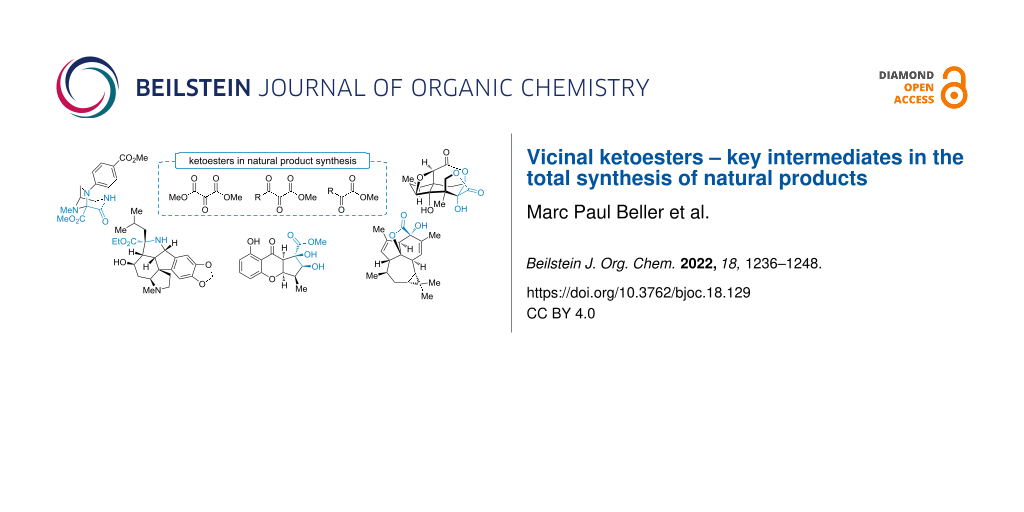
Abstract
This review summarizes examples for the application of vicinal ketoesters such as α-ketoesters, mesoxalic esters, and α,β-diketoesters as key intermediates in the total synthesis of natural products utilizing their electrophilic keto group as reactive site. Suitable key reactions are, e.g., aldol additions, carbonyl ene reactions, Mannich reactions, and additions of organometallic reagents. The vicinal arrangement of carbonyl groups allows the stabilization of reactive conformations by chelation or dipole control.

Graphical Abstract
Introduction
Vicinal ketoesters contain a carbonyl group adjacent to an ester group. One keto group results in α-ketoesters 1 and two vicinal keto groups lead to α,β-diketo esters 2 (Scheme 1). On the other hand, two carboxylic acid functionalities adjacent to a keto group result in mesoxalic diesters 3, or mesoxalic ester amides 4. The increased electrophilicity of the keto group and the high density of these complex functional groups make such structures attractive as key intermediates for the total synthesis of natural products [1]. Thus, the high electrophilicity of the central carbonyl group in α,β-diketoesters 2 allows the formation of stable hydrates 5. In case of an enolizable position enolization (2→6) is facilitated.
![[1860-5397-18-129-i1]](/bjoc/content/inline/1860-5397-18-129-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structures of vicinal ketoesters and examples for their typical reactivity.
Scheme 1: Structures of vicinal ketoesters and examples for their typical reactivity.
The chemistry of vicinal polycarbonyl compounds such as vic-diketoesters has been investigated in depth by Wasserman, Parr [2] and Gleiter, Rubin [3]. Important contributions for the use of α,β-diketoesters in stereoselective transformations came from Doyle’s group [4,5]. One remarkable example is the diastereoselective intramolecular aldol addition of ketones such as 7 (Scheme 2) [5]. Brønsted-acid catalysis leads via a transition state 8 to the aldol 9, while the use of chelating Lewis acids results via 10 in the epimeric aldol 11.
![[1860-5397-18-129-i2]](/bjoc/content/inline/1860-5397-18-129-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Doyle’s diastereoselective intramolecular aldol addition of α,β-diketoester.
Scheme 2: Doyle’s diastereoselective intramolecular aldol addition of α,β-diketoester.
This review is a collection of total syntheses of natural products where vicinal keto esters were used as key intermediates. For reasons of clarity and better comparability all syntheses are strongly summarized highlighting the key step only.
The presentation of the examples is structured in three parts:
- α-Ketoesters as key intermediates: (+)-euphorikanin A, (−)-preussochromone A, (−)-preussochromone D, (−)-jiadifenoxolane A, palau’amine, jatrophen, (−)-hopeanol, (+)-campthotecin, isoretronecanol, corynoxine, (+)-gracilamine, (−)-irofulven.
- Mesoxalic diester and ester amides as key intermediates: (+)-awajanomycin, (−)-aplaminal, cladoniamide G.
- α,β-Diketoesters as key intermediates: preussochromones E and F.
Review
1. α-Ketoesters as key intermediates:
(+)-Euphorikanin A
In the final step of the synthesis of (+)-euphorikanin A (16), an ingenane-derived diterpenoid with a 5/6/7/3-fused tetracyclic carbon skeleton, Carreira et al. used an intramolecular nucleophilic addition of an alkenyl metal species to the α-ketoester 15 (Scheme 3) [6]. The ketoester 15 was synthesized by a chiral pool approach starting from (+)-3-carene derived cycloheptenone 13 [7,8] and aldehyde 12 (accessible from (R)-Roche ester [9]) via the γ-lactone 14. The ketoester moiety was established by an enolate hydroxylation with Davis’ oxaziridine and subsequent oxidation using Dess–Martin periodinane. Initial attempts for the key step (15 → 16) like a Nozaki–Hiyama–Kishi reaction failed, but lithium–halogen exchange using t-BuLi at low temperatures gave the desired vinyllithium intermediate I which successfully added to the desired α-carbonyl group.
![[1860-5397-18-129-i3]](/bjoc/content/inline/1860-5397-18-129-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of euphorikanin A (16) by intramolecular, nucleophilic addition [6].
Scheme 3: Synthesis of euphorikanin A (16) by intramolecular, nucleophilic addition [6].
(−)-Preussochromone A
In 2020, the Koert group disclosed the synthesis of (−)-preussochromone A (24), a fungal metabolite with a highly substituted tetrahydrothiopyrane core annulated to a chromenone [10]. The tetrahydrothiopyrane ring was closed by a Lewis-acid-promoted cycloisomerization of the α-ketoester 22, which can be described as a Friedel–Crafts-type reaction or an aldol reaction of an S,O-ketene acetal (Scheme 4). The required ketoester 22 was synthesized from sulfonylchromenone 20, accessible from dihydroxyacetophenone 19 and thiol 18 derived from known alcohol 17 [11,12]. DMP oxidation of α-hydroxyester 21 and subsequent cycloisomerization led to the desired cyclization product 23 via transition state II in a dr of 5:1. Final deprotection gave preussochromone A (24).
![[1860-5397-18-129-i4]](/bjoc/content/inline/1860-5397-18-129-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Ketoester cycloisomerization for the synthesis of preussochromone A (24) [10].
Scheme 4: Ketoester cycloisomerization for the synthesis of preussochromone A (24) [10].
(−)-Preussochromone D
A similar approach was chosen in the synthesis of the structurally related natural product preussochromone D (30) reported by Koert et al. [13]. The synthesis commenced with the efficient production of alcohol 26 from 5-hydroxy-4H-chromen-4-one (25, Scheme 5) [14]. The ketoester moiety was build up via oxidation and nucleophilic addition of methyl diazoacetate, yielding alcohol 27. Subsequent oxidation gave α-ketoester 28 which was used in an intramolecular, Lewis acid-mediated aldol reaction, presumably via tridentate complex transition state III, to give diol 29 as a single diastereomer. Inversion of the secondary alcohol and deprotection gave preussochromone D (30).
![[1860-5397-18-129-i5]](/bjoc/content/inline/1860-5397-18-129-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Diastereoselective, intramolecular aldol reaction of an α-ketoester 28 in the synthesis of (−)-preussochromone D (30) [13,14].
Scheme 5: Diastereoselective, intramolecular aldol reaction of an α-ketoester 28 in the synthesis of (−)-preu...
(−)-Jiadifenoxolane A
The Illicium sesquiterpenes containing a seco-prezizaane carbon framework are highly oxidized, structurally complex natural products. Maimone et al. published a remarkable synthesis of the Illicium sesquiterpene (−)-jiadifenoxolane A (36), starting from the abundant sesquiterpene (+)-cedrol (31, Scheme 6) [15]. Through a series of finely tuned CH oxidations, cedrol (31) was converted to the lactone 32. In a single step, using Riley oxidation conditions, the methyl ketone moiety was transferred to the α-ketoester 33. Reduction, lactonization, and elimination gave the ketoesters-derived enol 34. Oxidation of the latter compound to the α-keto-β-hydroxy ester IV using DMDO and subsequent heating in PhCF3 triggered an α-ketol rearrangement which led to ketol V. Diastereoselective reduction gave α,β-dihydroxyester 35 which was converted to (−)-jiadifenoxolane A (36) in five further steps.
![[1860-5397-18-129-i6]](/bjoc/content/inline/1860-5397-18-129-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of an α-ketoester through Riley oxidation and its use in an α-ketol rearrangement in the synthesis of (−)-jiadifenoxolane A (36) [15].
Scheme 6: Synthesis of an α-ketoester through Riley oxidation and its use in an α-ketol rearrangement in the ...
Palau’amine
Palau’amine (45), a dimeric pyrrole-imidazole-bisguanidine alkaloid, was first isolated from the marine sponge Stylotella aurantium in 1993 [16,17]. It received considerable attention from the synthetic community because of its broad range of biological activities and complex structure. In an early endeavour of L. Overman et al. in 1997 [18] towards the originally proposed structure of palau’amine (44), a [3 + 2]-dipolar cycloaddition of α-ketoester 41 and the thiosemicarbazide 42-derived azomethine imine VI to the triazacyclopenta[cd]pentalene 43 was utilized as a key step (Scheme 7) [18-21].
![[1860-5397-18-129-i7]](/bjoc/content/inline/1860-5397-18-129-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Azomethine imine cycloaddition towards the synthesis of the proposed structure of palau’amine (44) [19].
Scheme 7: Azomethine imine cycloaddition towards the synthesis of the proposed structure of palau’amine (44) [19]....
The α-ketoester 41 was accessible from amide 38, which in turn was obtained from allylic alcohol 37. Oxidation and Horner–Wadsworth–Emmons reaction with phosphonate 39 delivered the silyl enol ether 40, which was deprotected and cyclized via a Grubbs metathesis to α-ketoester 41. Subsequent cycloaddition delivered the advanced intermediate 43 in an efficient and elegant way.
Jatropha-5,12-diene
Towards the total synthesis of natural and unnatural jatrophane diterpenes, Hiersemann et al. used a highly efficient, intramolecular carbonyl-ene reaction of α-ketoester 49 (Scheme 8) [22]. The ketoester was synthesized by a Horner–Wadsworth–Emmons reaction of phosphonate 48 with aldehyde 47. Enantiopure aldehyde 47 was easily accessible from oxazolidinone 46 via Evans-aldol chemistry [23]. Heating of the α-ketoester 49 led to the highly substituted cyclopentanol 50 in a good dr of ≈5:1 (minor diastereomer not shown) via transition state VII where pseudo-1,3-strain is minimized. Nineteen further steps were necessary to give the naturally occurring jatrophen 51.
![[1860-5397-18-129-i8]](/bjoc/content/inline/1860-5397-18-129-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Intramolecular diastereoselective carbonyl-ene reaction of an α-ketoester in the synthesis of jatrophane diterpenoids [22].
Scheme 8: Intramolecular diastereoselective carbonyl-ene reaction of an α-ketoester in the synthesis of jatro...
(−)-Hopeanol
In the synthesis of the polyphenolic natural product (−)-hopeanol (59), Nicolaou et al. used an α-ketoester moiety as a precursor for an intramolecular Friedel–Crafts cyclization (Scheme 9) [24]. Therefore, phenylacetaldehyde 52 was converted to the alcohol 53, which was esterified with the α-ketoacid 54 to give ketoester 55. Grignard addition to the keto carbonyl and subsequent TBS deprotection delivered the tertiary alcohol 56, which was dehydroxylated to the diastereomeric cations VIII and IX. Friedel–Crafts reaction gave diastereomeric lactones 57 and 58. The major diastereomer 58 could be converted to the complex polyphenol (−)-hopeanol (59) in seven further steps.
![[1860-5397-18-129-i9]](/bjoc/content/inline/1860-5397-18-129-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Grignard addition to an α-ketoester and subsequent Friedel–Crafts cyclization in the synthesis of (−)-hopeanol (59) [24].
Scheme 9: Grignard addition to an α-ketoester and subsequent Friedel–Crafts cyclization in the synthesis of (...
(+)-Camptothecin
In the formal synthesis of the pentacyclic, antiproliferative quinoline alkaloid camptothecin (65), Peters et al. used an α-ketoester moiety in an auxiliary controlled approach towards the only stereogenic center present in the natural product (Scheme 10) [25]. First, the ketoacid 60 was esterified with 8-phenylmenthol (61) to yield the α-ketoester 62, followed by nucleophilic addition of isopropenylmagnesium bromide to give α-hydroxyester 63 in excellent yield and diastereoselectivity. Eight additional steps gave the bicyclic compound 64 which was already known from previous camptothecin syntheses.
![[1860-5397-18-129-i10]](/bjoc/content/inline/1860-5397-18-129-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Diastereoselective addition to an auxiliary modified α-ketoester in the formal synthesis of (+)-campthotecin (65) [25].
Scheme 10: Diastereoselective addition to an auxiliary modified α-ketoester in the formal synthesis of (+)-cam...
Isoretronecanol
The α-ketoester moiety can also be used in photochemical reactions, as shown by Gramain et al. in the synthesis of the pyrrolizidine alkaloid (rac)-isoretronecanol (69, Scheme 11) [26]. A Claisen condensation of the lithium enolate of N-acetylpyrrolidine (66) with diethyl oxalate gave the ketoester 67. Irradiation of compound 67 with a medium pressure mercury lamp in Pyrex® glassware triggered a 1,6-HAT leading to biradical X which combined to the racemic pyrrolizidine 68 as a 1:1 mixture of diastereomers. Three more steps gave the target compound 69 in 31% overall yield.
![[1860-5397-18-129-i11]](/bjoc/content/inline/1860-5397-18-129-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Intramolecular photoreduction of an α-ketoester in the synthesis of (rac)-isoretronecanol (69) [26].
Scheme 11: Intramolecular photoreduction of an α-ketoester in the synthesis of (rac)-isoretronecanol (69) [26].
Corynoxine
Hiemstra et al. used the α-ketoester moiety for different purposes in the syntheses of a range of oxindole alkaloids. The start of the synthesis of (rac)-corynoxine (76) was the conversion of tryptamine (70) to oxindole 71, which was used in a chemoselective Mannich reaction with aldehyde 72, introducing the α-ketoester moiety (Scheme 12) [27].
![[1860-5397-18-129-i12]](/bjoc/content/inline/1860-5397-18-129-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: α-Ketoester as nucleophile in a Tsuji–Trost reaction in the synthesis of (rac)-corynoxine (76) [27].
Scheme 12: α-Ketoester as nucleophile in a Tsuji–Trost reaction in the synthesis of (rac)-corynoxine (76) [27].
The major trans-isomer 73 was further converted to the natural products corynoxine and rychnophylline. The minor cis-isomer 74 was used in an intramolecular Tsuji–Trost reaction, where the ketoester served as a nucleophile, which build up the piperidine ring and selectively set the desired cis-substitution. Subsequent transesterification gave the α-ketoester 75, which was used in a Wittig reaction. The undesired Z-configured double bond was isomerized to the E-alkene and final hydrogenation delivered corynoxine (76).
(+)-Gracilamine
The Mannich reaction was also used by Nagasawa et al. as a key step in the synthesis of (+)-gracilamine (83), a pentacyclic alkaloid isolated from the plant Galanthus gracilis, (Scheme 13) [28]. The synthesis started from readily available sesamol (79) and imine 78 which gave the advanced intermediate 80 in ten steps. An intramolecular Mannich reaction of compound 80 with α-ketoester 81 furnished compound 82 with the last ring of the target (+)-gracilamine (83), which was accessible after two further steps.
![[1860-5397-18-129-i13]](/bjoc/content/inline/1860-5397-18-129-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Mannich reaction of an α-ketoester in the synthesis of (+)-gracilamine (83) [28].
Scheme 13: Mannich reaction of an α-ketoester in the synthesis of (+)-gracilamine (83) [28].
(−)-Irofulven
Irofulven (87) is a highly cytotoxic, semisynthetic drug obtained from the illudin sesquiterpene family. In a de novo synthesis towards (−)-irofulven (87), Movassaghi et al. used a CuII-catalyzed asymmetric aldol reaction of O-silyl ketene S,O-acetal 84 with methyl pyruvate (85) to enantioselectively install the crucial tertiary TMS-protected alcohol in ester 86 (Scheme 14) [29]. Eleven further steps gave (−)-irofulven (87).
![[1860-5397-18-129-i14]](/bjoc/content/inline/1860-5397-18-129-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Enantioselective aldol reaction using an α-ketoester in the synthesis of (−)-irofulven (87) [29].
Scheme 14: Enantioselective aldol reaction using an α-ketoester in the synthesis of (−)-irofulven (87) [29].
2. Mesoxalic diesters and ester amides as key intermediates
(+)-Awajanomycin
Diethyl mesoxalate (90a) is a valuable building block due to the high density of carbon atoms in high oxidation states. As a vic-tricarbonyl compound, its central keto group is an especially potent electrophile. The Koert group used this reactivity in their synthesis of (+)-awajanomycin (92), a marine natural product with a γ-lactone-δ-lactam core structure (Scheme 15) [30,31]. Key step was an asymmetric allylboration of diethyl mesoxalate (90a) with boronate 89, which was easily accessible through a Matteson homologation of dichloromethyl boronate 88. The reaction of (Z)-alkenyl boronate 89 with mesoxalate 90a delivered product 91 through the six-membered transition state XI. Eight further steps accomplished the total synthesis of (+)-awajanomycin (92).
![[1860-5397-18-129-i15]](/bjoc/content/inline/1860-5397-18-129-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Allylboration of a mesoxalic acid ester in the synthesis of (+)-awajanomycin (92) [30,31].
Scheme 15: Allylboration of a mesoxalic acid ester in the synthesis of (+)-awajanomycin (92) [30,31].
(−)-Aplaminal
Dimethyl mesoxalate (90b) was used by Smith and Liu in the synthesis of the cytotoxic metabolite (−)-aplaminal (96), which was isolated from the sea hare Aplysia kurodai [32]. The natural product is characterized by a triazabicyclo[3.2.1]octane, where each bridge possesses a nitrogen atom. The synthesis commenced with N-Boc-serine (93) which was converted to secondary aniline 94 in three steps (Scheme 16). Subsequent deprotection and condensation with dimethyl mesoxalate (90b) gave imidazolidine 95. With compound 95 at hands, five further steps gave (−)-aplaminal (96) in a good overall yield of 19%.
![[1860-5397-18-129-i16]](/bjoc/content/inline/1860-5397-18-129-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Condensation of a diamine with mesoxolate in the synthesis of (−)-aplaminal (96) [32].
Scheme 16: Condensation of a diamine with mesoxolate in the synthesis of (−)-aplaminal (96) [32].
Cladoniamide G
The unsymmetrical mesoxalic acid amide 102 was used by Koert et al. in the racemic synthesis of the bisindole alkaloid (rac)-cladoniamide G (103, Scheme 17) [33]. The synthesis started with benzaldehyde 97 and indole 99 which were converted to the indole building blocks 98 and 100, respectively. These were connected to bisindole 101, which reacted with mesoxalic ester amide 102 in a Friedel–Crafts reaction followed by a spontaneous lactamization to give (rac)-cladoniamide G (103). The mesoxalic ester amide 102 was synthesized from malonyl chloride 104 through amidation and Regitz diazotransfer, yielding diazo compound 105. Subsequent oxidation and dehydration of the resulting hydrate through short-patch distillation gave the desired vic-tricarbonyl compound 102.
![[1860-5397-18-129-i17]](/bjoc/content/inline/1860-5397-18-129-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of mesoxalic ester amide 102 and its use in the synthesis of (rac)-cladoniamide G (103) [33].
Scheme 17: Synthesis of mesoxalic ester amide 102 and its use in the synthesis of (rac)-cladoniamide G (103) [33].
3. α,β-Diketoesters as key intermediates
Preussochromone E and F
In a short and enantioselective total synthesis of preussochromone E (110) and F (109), Koert et al. used the complex vic-tricarbonyl compound 108 to set two stereogenic centers and correct one via an intramolecular aldol addition (108 → 109; Scheme 18) [34]. The vic-tricarbonyl compound 108 was synthesized via DMDO oxidation from α-diazo-β-ketoester 107, which was easily accessible from 5-methoxy-4H-chromen-4-one (106). The thermodynamically controlled basic intramolecular aldol addition of compound 108 using the bulky amine base 2,6-di-tert-butyl-4-methylpyridine (DTBMP) led to epimerization of the methyl group and cyclization, giving preussochromone F (109) as single isolable diastereomer probably via transition state XII. The subsequent reduction of compound 109 gave preussochromone E (110).
![[1860-5397-18-129-i18]](/bjoc/content/inline/1860-5397-18-129-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: The thermodynamically controlled, intramolecular aldol addition of a vic-tricarbonyl compound in the synthesis of preussochromones E (110) and F (109) [34].
Scheme 18: The thermodynamically controlled, intramolecular aldol addition of a vic-tricarbonyl compound in th...
Conclusion
The variety of examples prove that vicinal ketoesters are valuable synthetic intermediates for the synthesis of complex target structures such as natural products. α-Ketoesters, mesoxalic esters, and α,β-diketoesters can be used bearing an electrophilic keto group as reactive site. The vicinal arrangement of carbonyl groups allows the stabilization of reactive conformations by chelation or dipole control. Suitable key reactions are e.g., aldol additions, carbonyl ene reactions, Mannich reactions, and additions of organometallic reagents. The presented examples may encourage the use of vicinal ketoesters in future applications, in particular in the field of natural product synthesis.
References
-
Selter, L.; Zygalski, L.; Kerste, E.; Koert, U. Synthesis 2016, 49, 17–28. doi:10.1055/s-0035-1562623
Return to citation in text: [1] -
Wasserman, H. H.; Parr, J. Acc. Chem. Res. 2004, 37, 687–701. doi:10.1021/ar0300221
Return to citation in text: [1] -
Rubin, M. B.; Gleiter, R. Chem. Rev. 2000, 100, 1121–1164. doi:10.1021/cr960079j
Return to citation in text: [1] -
Truong, P. M.; Zavalij, P. Y.; Doyle, M. P. Angew. Chem., Int. Ed. 2014, 53, 6468–6472. doi:10.1002/anie.201402233
Return to citation in text: [1] -
Truong, P.; Shanahan, C. S.; Doyle, M. P. Org. Lett. 2012, 14, 3608–3611. doi:10.1021/ol301317a
Return to citation in text: [1] [2] -
Classen, M. J.; Böcker, M. N. A.; Roth, R.; Amberg, W. M.; Carreira, E. M. J. Am. Chem. Soc. 2021, 143, 8261–8265. doi:10.1021/jacs.1c04210
Return to citation in text: [1] [2] -
Satoh, T.; Kaneko, Y.; Okuda, T.; Uwaya, S.; Yamakawa, K. Chem. Pharm. Bull. 1984, 32, 3452–3460. doi:10.1248/cpb.32.3452
Return to citation in text: [1] -
Jackson, R. K., III; Wood, J. L. Org. Lett. 2021, 23, 1243–1246. doi:10.1021/acs.orglett.0c04219
Return to citation in text: [1] -
Chen, T.; Altmann, K.-H. Chem. – Eur. J. 2015, 21, 8403–8407. doi:10.1002/chem.201501252
Return to citation in text: [1] -
Beller, M. P.; Harms, K.; Koert, U. Org. Lett. 2020, 22, 6127–6131. doi:10.1021/acs.orglett.0c02197
Return to citation in text: [1] [2] -
Pavan Kumar, C.; Ravinder, M.; Kumar, S.; Rao, V. Synthesis 2011, 1320. doi:10.1055/s-0030-1259985
Return to citation in text: [1] -
Ramulu, U.; Ramesh, D.; Rajaram, S.; Reddy, S. P.; Venkatesham, K.; Venkateswarlu, Y. Tetrahedron: Asymmetry 2012, 23, 117–123. doi:10.1016/j.tetasy.2012.01.014
Return to citation in text: [1] -
Kerste, E.; Harms, K.; Koert, U. Org. Lett. 2019, 21, 4374–4377. doi:10.1021/acs.orglett.9b01594
Return to citation in text: [1] [2] -
Kerste, E.; Beller, M. P.; Koert, U. Eur. J. Org. Chem. 2020, 3699–3711. doi:10.1002/ejoc.202000465
Return to citation in text: [1] [2] -
Condakes, M. L.; Hung, K.; Harwood, S. J.; Maimone, T. J. J. Am. Chem. Soc. 2017, 139, 17783–17786. doi:10.1021/jacs.7b11493
Return to citation in text: [1] [2] -
Kinnel, R. B.; Gehrken, H. P.; Scheuer, P. J. J. Am. Chem. Soc. 1993, 115, 3376–3377. doi:10.1021/ja00061a065
Return to citation in text: [1] -
Kinnel, R. B.; Gehrken, H.-P.; Swali, R.; Skoropowski, G.; Scheuer, P. J. J. Org. Chem. 1998, 63, 3281–3286. doi:10.1021/jo971987z
Return to citation in text: [1] -
Overman, L. E.; Rogers, B. N.; Tellew, J. E.; Trenkle, W. C. J. Am. Chem. Soc. 1997, 119, 7159–7160. doi:10.1021/ja9712985
Return to citation in text: [1] [2] -
Bélanger, G.; Hong, F.-T.; Overman, L. E.; Rogers, B. N.; Tellew, J. E.; Trenkle, W. C. J. Org. Chem. 2002, 67, 7880–7883. doi:10.1021/jo026282y
Return to citation in text: [1] [2] -
Katz, J. D.; Overman, L. E. Tetrahedron 2004, 60, 9559–9568. doi:10.1016/j.tet.2004.06.140
Return to citation in text: [1] -
Lanman, B. A.; Overman, L. E.; Paulini, R.; White, N. S. J. Am. Chem. Soc. 2007, 129, 12896–12900. doi:10.1021/ja074939x
Return to citation in text: [1] -
Schnabel, C.; Sterz, K.; Müller, H.; Rehbein, J.; Wiese, M.; Hiersemann, M. J. Org. Chem. 2011, 76, 512–522. doi:10.1021/jo1019738
Return to citation in text: [1] [2] -
Helmboldt, H.; Köhler, D.; Hiersemann, M. Org. Lett. 2006, 8, 1573–1576. doi:10.1021/ol060115t
Return to citation in text: [1] -
Nicolaou, K. C.; Kang, Q.; Wu, T. R.; Lim, C. S.; Chen, D. Y.-K. J. Am. Chem. Soc. 2010, 132, 7540–7548. doi:10.1021/ja102623j
Return to citation in text: [1] [2] -
Peters, R.; Althaus, M.; Nagy, A.-L. Org. Biomol. Chem. 2006, 4, 498–509. doi:10.1039/b514147h
Return to citation in text: [1] [2] -
Gramain, J. C.; Remuson, R.; Vallee, D. J. Org. Chem. 1985, 50, 710–712. doi:10.1021/jo00205a037
Return to citation in text: [1] [2] -
Wanner, M. J.; Ingemann, S.; van Maarseveen, J. H.; Hiemstra, H. Eur. J. Org. Chem. 2013, 1100–1106. doi:10.1002/ejoc.201201505
Return to citation in text: [1] [2] -
Odagi, M.; Yamamoto, Y.; Nagasawa, K. Angew. Chem., Int. Ed. 2018, 57, 2229–2232. doi:10.1002/anie.201708575
Return to citation in text: [1] [2] -
Movassaghi, M.; Piizzi, G.; Siegel, D. S.; Piersanti, G. Angew. Chem., Int. Ed. 2006, 45, 5859–5863. doi:10.1002/anie.200602011
Return to citation in text: [1] [2] -
Wohlfahrt, M.; Harms, K.; Koert, U. Angew. Chem., Int. Ed. 2011, 50, 8404–8406. doi:10.1002/anie.201103679
Return to citation in text: [1] [2] -
Wohlfahrt, M.; Harms, K.; Koert, U. Eur. J. Org. Chem. 2012, 2260–2265. doi:10.1002/ejoc.201200059
Return to citation in text: [1] [2] -
Smith, A. B., III; Liu, Z. Org. Lett. 2008, 10, 4363–4365. doi:10.1021/ol801794f
Return to citation in text: [1] [2] -
Schütte, J.; Kilgenstein, F.; Fischer, M.; Koert, U. Eur. J. Org. Chem. 2014, 5302–5311. doi:10.1002/ejoc.201402531
Return to citation in text: [1] [2] -
Beller, M. P.; Ivlev, S.; Koert, U. Org. Lett. 2022, 24, 912–915. doi:10.1021/acs.orglett.1c04261
Return to citation in text: [1] [2]
| 25. | Peters, R.; Althaus, M.; Nagy, A.-L. Org. Biomol. Chem. 2006, 4, 498–509. doi:10.1039/b514147h |
| 26. | Gramain, J. C.; Remuson, R.; Vallee, D. J. Org. Chem. 1985, 50, 710–712. doi:10.1021/jo00205a037 |
| 26. | Gramain, J. C.; Remuson, R.; Vallee, D. J. Org. Chem. 1985, 50, 710–712. doi:10.1021/jo00205a037 |
| 1. | Selter, L.; Zygalski, L.; Kerste, E.; Koert, U. Synthesis 2016, 49, 17–28. doi:10.1055/s-0035-1562623 |
| 5. | Truong, P.; Shanahan, C. S.; Doyle, M. P. Org. Lett. 2012, 14, 3608–3611. doi:10.1021/ol301317a |
| 13. | Kerste, E.; Harms, K.; Koert, U. Org. Lett. 2019, 21, 4374–4377. doi:10.1021/acs.orglett.9b01594 |
| 14. | Kerste, E.; Beller, M. P.; Koert, U. Eur. J. Org. Chem. 2020, 3699–3711. doi:10.1002/ejoc.202000465 |
| 30. | Wohlfahrt, M.; Harms, K.; Koert, U. Angew. Chem., Int. Ed. 2011, 50, 8404–8406. doi:10.1002/anie.201103679 |
| 31. | Wohlfahrt, M.; Harms, K.; Koert, U. Eur. J. Org. Chem. 2012, 2260–2265. doi:10.1002/ejoc.201200059 |
| 4. | Truong, P. M.; Zavalij, P. Y.; Doyle, M. P. Angew. Chem., Int. Ed. 2014, 53, 6468–6472. doi:10.1002/anie.201402233 |
| 5. | Truong, P.; Shanahan, C. S.; Doyle, M. P. Org. Lett. 2012, 14, 3608–3611. doi:10.1021/ol301317a |
| 15. | Condakes, M. L.; Hung, K.; Harwood, S. J.; Maimone, T. J. J. Am. Chem. Soc. 2017, 139, 17783–17786. doi:10.1021/jacs.7b11493 |
| 30. | Wohlfahrt, M.; Harms, K.; Koert, U. Angew. Chem., Int. Ed. 2011, 50, 8404–8406. doi:10.1002/anie.201103679 |
| 31. | Wohlfahrt, M.; Harms, K.; Koert, U. Eur. J. Org. Chem. 2012, 2260–2265. doi:10.1002/ejoc.201200059 |
| 3. | Rubin, M. B.; Gleiter, R. Chem. Rev. 2000, 100, 1121–1164. doi:10.1021/cr960079j |
| 13. | Kerste, E.; Harms, K.; Koert, U. Org. Lett. 2019, 21, 4374–4377. doi:10.1021/acs.orglett.9b01594 |
| 29. | Movassaghi, M.; Piizzi, G.; Siegel, D. S.; Piersanti, G. Angew. Chem., Int. Ed. 2006, 45, 5859–5863. doi:10.1002/anie.200602011 |
| 2. | Wasserman, H. H.; Parr, J. Acc. Chem. Res. 2004, 37, 687–701. doi:10.1021/ar0300221 |
| 14. | Kerste, E.; Beller, M. P.; Koert, U. Eur. J. Org. Chem. 2020, 3699–3711. doi:10.1002/ejoc.202000465 |
| 29. | Movassaghi, M.; Piizzi, G.; Siegel, D. S.; Piersanti, G. Angew. Chem., Int. Ed. 2006, 45, 5859–5863. doi:10.1002/anie.200602011 |
| 6. | Classen, M. J.; Böcker, M. N. A.; Roth, R.; Amberg, W. M.; Carreira, E. M. J. Am. Chem. Soc. 2021, 143, 8261–8265. doi:10.1021/jacs.1c04210 |
| 11. | Pavan Kumar, C.; Ravinder, M.; Kumar, S.; Rao, V. Synthesis 2011, 1320. doi:10.1055/s-0030-1259985 |
| 12. | Ramulu, U.; Ramesh, D.; Rajaram, S.; Reddy, S. P.; Venkatesham, K.; Venkateswarlu, Y. Tetrahedron: Asymmetry 2012, 23, 117–123. doi:10.1016/j.tetasy.2012.01.014 |
| 28. | Odagi, M.; Yamamoto, Y.; Nagasawa, K. Angew. Chem., Int. Ed. 2018, 57, 2229–2232. doi:10.1002/anie.201708575 |
| 9. | Chen, T.; Altmann, K.-H. Chem. – Eur. J. 2015, 21, 8403–8407. doi:10.1002/chem.201501252 |
| 10. | Beller, M. P.; Harms, K.; Koert, U. Org. Lett. 2020, 22, 6127–6131. doi:10.1021/acs.orglett.0c02197 |
| 28. | Odagi, M.; Yamamoto, Y.; Nagasawa, K. Angew. Chem., Int. Ed. 2018, 57, 2229–2232. doi:10.1002/anie.201708575 |
| 7. | Satoh, T.; Kaneko, Y.; Okuda, T.; Uwaya, S.; Yamakawa, K. Chem. Pharm. Bull. 1984, 32, 3452–3460. doi:10.1248/cpb.32.3452 |
| 8. | Jackson, R. K., III; Wood, J. L. Org. Lett. 2021, 23, 1243–1246. doi:10.1021/acs.orglett.0c04219 |
| 27. | Wanner, M. J.; Ingemann, S.; van Maarseveen, J. H.; Hiemstra, H. Eur. J. Org. Chem. 2013, 1100–1106. doi:10.1002/ejoc.201201505 |
| 6. | Classen, M. J.; Böcker, M. N. A.; Roth, R.; Amberg, W. M.; Carreira, E. M. J. Am. Chem. Soc. 2021, 143, 8261–8265. doi:10.1021/jacs.1c04210 |
| 10. | Beller, M. P.; Harms, K.; Koert, U. Org. Lett. 2020, 22, 6127–6131. doi:10.1021/acs.orglett.0c02197 |
| 27. | Wanner, M. J.; Ingemann, S.; van Maarseveen, J. H.; Hiemstra, H. Eur. J. Org. Chem. 2013, 1100–1106. doi:10.1002/ejoc.201201505 |
| 18. | Overman, L. E.; Rogers, B. N.; Tellew, J. E.; Trenkle, W. C. J. Am. Chem. Soc. 1997, 119, 7159–7160. doi:10.1021/ja9712985 |
| 15. | Condakes, M. L.; Hung, K.; Harwood, S. J.; Maimone, T. J. J. Am. Chem. Soc. 2017, 139, 17783–17786. doi:10.1021/jacs.7b11493 |
| 32. | Smith, A. B., III; Liu, Z. Org. Lett. 2008, 10, 4363–4365. doi:10.1021/ol801794f |
| 16. | Kinnel, R. B.; Gehrken, H. P.; Scheuer, P. J. J. Am. Chem. Soc. 1993, 115, 3376–3377. doi:10.1021/ja00061a065 |
| 17. | Kinnel, R. B.; Gehrken, H.-P.; Swali, R.; Skoropowski, G.; Scheuer, P. J. J. Org. Chem. 1998, 63, 3281–3286. doi:10.1021/jo971987z |
| 32. | Smith, A. B., III; Liu, Z. Org. Lett. 2008, 10, 4363–4365. doi:10.1021/ol801794f |
| 33. | Schütte, J.; Kilgenstein, F.; Fischer, M.; Koert, U. Eur. J. Org. Chem. 2014, 5302–5311. doi:10.1002/ejoc.201402531 |
| 24. | Nicolaou, K. C.; Kang, Q.; Wu, T. R.; Lim, C. S.; Chen, D. Y.-K. J. Am. Chem. Soc. 2010, 132, 7540–7548. doi:10.1021/ja102623j |
| 25. | Peters, R.; Althaus, M.; Nagy, A.-L. Org. Biomol. Chem. 2006, 4, 498–509. doi:10.1039/b514147h |
| 22. | Schnabel, C.; Sterz, K.; Müller, H.; Rehbein, J.; Wiese, M.; Hiersemann, M. J. Org. Chem. 2011, 76, 512–522. doi:10.1021/jo1019738 |
| 24. | Nicolaou, K. C.; Kang, Q.; Wu, T. R.; Lim, C. S.; Chen, D. Y.-K. J. Am. Chem. Soc. 2010, 132, 7540–7548. doi:10.1021/ja102623j |
| 22. | Schnabel, C.; Sterz, K.; Müller, H.; Rehbein, J.; Wiese, M.; Hiersemann, M. J. Org. Chem. 2011, 76, 512–522. doi:10.1021/jo1019738 |
| 34. | Beller, M. P.; Ivlev, S.; Koert, U. Org. Lett. 2022, 24, 912–915. doi:10.1021/acs.orglett.1c04261 |
| 23. | Helmboldt, H.; Köhler, D.; Hiersemann, M. Org. Lett. 2006, 8, 1573–1576. doi:10.1021/ol060115t |
| 18. | Overman, L. E.; Rogers, B. N.; Tellew, J. E.; Trenkle, W. C. J. Am. Chem. Soc. 1997, 119, 7159–7160. doi:10.1021/ja9712985 |
| 19. | Bélanger, G.; Hong, F.-T.; Overman, L. E.; Rogers, B. N.; Tellew, J. E.; Trenkle, W. C. J. Org. Chem. 2002, 67, 7880–7883. doi:10.1021/jo026282y |
| 20. | Katz, J. D.; Overman, L. E. Tetrahedron 2004, 60, 9559–9568. doi:10.1016/j.tet.2004.06.140 |
| 21. | Lanman, B. A.; Overman, L. E.; Paulini, R.; White, N. S. J. Am. Chem. Soc. 2007, 129, 12896–12900. doi:10.1021/ja074939x |
| 33. | Schütte, J.; Kilgenstein, F.; Fischer, M.; Koert, U. Eur. J. Org. Chem. 2014, 5302–5311. doi:10.1002/ejoc.201402531 |
| 19. | Bélanger, G.; Hong, F.-T.; Overman, L. E.; Rogers, B. N.; Tellew, J. E.; Trenkle, W. C. J. Org. Chem. 2002, 67, 7880–7883. doi:10.1021/jo026282y |
| 34. | Beller, M. P.; Ivlev, S.; Koert, U. Org. Lett. 2022, 24, 912–915. doi:10.1021/acs.orglett.1c04261 |
© 2022 Beller and Koert; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.