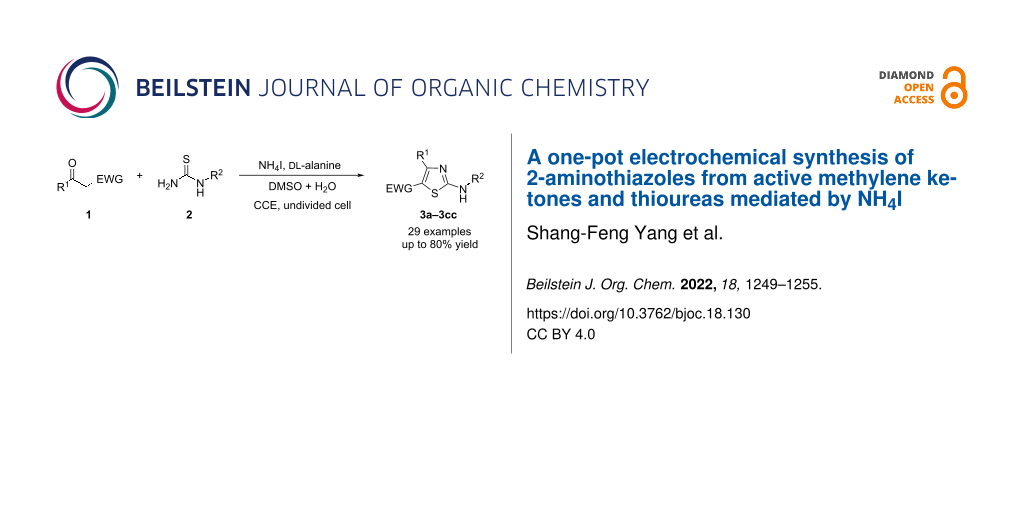
Abstract
The electrochemical preparation of 2-aminothiazoles has been achieved by the reaction of active methylene ketones with thioureas assisted by ᴅʟ-alanine using NH4I as a redox mediator. The electrochemical protocol proceeds in an undivided cell equipped with graphite plate electrodes under constant current conditions. Various active methylene ketones, including β-keto ester, β-keto amide, β-keto nitrile, β-keto sulfone and 1,3-diketones, can be converted to the corresponding 2-aminothiazoles. Mechanistically, the in situ generated α-iodoketone was proposed to be the key active species.

Graphical Abstract
Introduction
Thiazoles are prevalent structural motifs in a wide range of natural products [1] and synthetic molecules possessing various pharmaceutical activities such as antimicrobial [2,3], antiviral [4], antitumor [5,6], anti-inflammatory [7,8] and so on. Moreover, as a type of important intermediates, thiazole is of prime importance in organic synthesis [9,10] which is used extensively in the preparation of flavors [11], polymers [12], dyes [13], etc. These important features of thiazoles have driven intense interests in their facile synthesis [14-17]. Among various synthetic routes to the thiazole unit, the Hantzsch condensation of α-halo ketones (dielectrophiles) with various thioureas (dinucleophiles) should be the most well-known method (Scheme 1a) [18]. Since active methylene ketones are able to be in situ α-halogenated, the modified Hantzsch condensation of active methylene ketones with thioureas has attracted increasing attention in thiazoles’ synthesis, thereby saving costs and time needed to prepare the required α-halogenated dielectrophiles. Along this line, in situ α-halogenation strategies have been developed, using various halogenating reagents including Br2 [19,20], I2 [21,22], NBS [23-25], tribromoisocyanuric acid [26], 1,3-dichloro-5,5-dimethylhydantoin [27], HBr or HI, DMSO [28] etc. (Scheme 1b). Alternatively, the oxidation of α-C–H of active methylene ketones generate α-carbon-centered radicals, thus providing another way to obtain thiazoles. Recently, Sun et al. reported a tert-butyl hydroperoxide/azodiisobutyronitrile-mediated synthesis of 2-aminothiazoles from active methylene ketones and thiourea via an oxidative cyclization initiated by a radical process and a following condensation reaction (Scheme 1c) [29]. Although these methods are practical, most of these strategies require stoichiometric or excess amounts of halogenating reagents or oxidants, which are toxic, hazardous and would lead to a large quantity of waste. Considering the importance of thiazoles in synthetic and medicinal chemistry, the development of greener and more atom economic processes for the thiazole synthesis has become increasingly necessary with growing awareness of environmental constraints.
![[1860-5397-18-130-i1]](/bjoc/content/inline/1860-5397-18-130-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Methods for the synthesis of thiazoles using active methylene ketones as starting materials.
Scheme 1: Methods for the synthesis of thiazoles using active methylene ketones as starting materials.
Organic electrosynthesis has been recognized as a green, modern, and safe technique, since electrons can be used as an alternative for oxidants or reductants [30-38]. During our continuous interests in halide-mediated indirect electrolysis [39-42], we have achieved the in situ generation of α-iodocarbonyl ketones from constant current electrolysis (CCE) of ketones in the presence of iodide ions. It is worth noting that we have also reported an electrochemical method for the synthesis of 2-aminothiazoles via the one-pot direct α-C–H functionalization of ketones with thioureas [42]. However, the reported method only tolerates aromatic and aliphatic ketones; the active methylene ketones were not suitable. Given that amino acids have been reported to work as green organocatalysts for the synthesis of 2-aminothiazole heterocycles [43,44]. We herein report a ᴅʟ-alanine-assisted one-pot electrochemical synthesis of 2-aminothiazoles from active methylene ketones and thioureas mediated by NH4I (Scheme 1d). This electrochemical method features external-oxidant-free conditions and avoids the prefunctionalization of the substrates.
Results and Discussion
To demonstrate the feasibility of our idea, ethyl acetoacetate (1a) and thiourea (2a) were chosen as model substrates for the optimization of reaction conditions. Based on our previous studies on the halide-mediated α-C–H functionalization of carbonyl compounds [32-35], graphite was chosen as the working electrode rather than exploring other options. As shown in Table 1, when the electrolysis of 1a with 2a was performed in aqueous DMSO at a constant current of 5 mA/cm2 using NH4I as the mediator, LiClO4 as electrolyte and asparagine as additive in an undivided cell, the desired product 3a was isolated in 51% after passing 6 F/mol of charge (Table 1, entry 1). The yield of 3a decreased obviously when the ratio of 1a to 2a changed from 2:1 to 1:1 or 1:2 (Table 1, entries 2 and 3). Subsequent solvent screening disclosed that aqueous DMSO was the optimal solvent and lower yields were obtained in other solvents, such as aqueous DMF, aqueous MeCN or aqueous EtOH (Table 1, entries 4–6). Owing to the tedious workup process in using a large amount of DMSO as solvent, we decided to increase the proportion of H2O and disclosed that the yield of 3a improved from 52% to 61% when the ratio of DMSO to H2O changed from 2:1 to 1:14 (Table 1, entry 7). When the reaction was performed in H2O, 3a was obtained in 51% yield (Table 1, entry 8). It was observed that the additive plays an important role, among which ᴅʟ-alanine was proved to be the best, although asparagine, ʟ-phenylalanine, ʟ-aspartic acid and ʟ-allysine also gave comparable yields of 3a (Table 1, entries 9–12). It is interesting to note that 3a was achieved in higher yields (75%) when the temperature decreased from 70 °C to 30 °C (Table 1, entry 14). The effect of current density was also examined. It was found that a slightly lower yield of 3a was produced when 4 mA/cm2 (Table 1, entry 15) or 6 mA/cm2 (Table 1, entry 16) was used instead of the optimal 5 mA/cm2. In addition, 6 F/mol of charge was preferable because less yield of 3a was obtained when 4 F/mol (Table 1, entry 17) and 8 F/mol (Table 1, entry 18) charge were consumed.
Table 1: Optimization of the reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-130-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | 1a:2a | Mediator | Solvent (15 mL) | Additive | T [°C] | F [mol] | Yield [%]b |
| 1 | 2:1 | NH4I | DMSO/H2O (2:1) | asparagine | 70 | 6 | 51 |
| 2 | 1:1 | NH4I | DMSO/H2O (2:1) | asparagine | 70 | 6 | 17 |
| 3 | 1:2 | NH4I | DMSO/H2O (2:1) | asparagine | 70 | 6 | 15 |
| 4 | 2:1 | NH4I | DMF/H2O (2:1) | asparagine | 70 | 6 | 41 |
| 5 | 2:1 | NH4I | MeCN/H2O (2:1) | asparagine | 70 | 6 | 33 |
| 6 | 2:1 | NH4I | EtOH/H2O (2:1) | asparagine | 70 | 6 | 46 |
| 7 | 2:1 | NH4I | DMSO/H2O (1:14) | asparagine | 70 | 6 | 61 |
| 8 | 2:1 | NH4I | H2O | asparagine | 70 | 6 | 51 |
| 9 | 2:1 | NH4I | DMSO/H2O (1:14) | ʟ-phenylalanine | 70 | 6 | 60 |
| 10 | 2:1 | NH4I | DMSO/H2O (1:14) | ʟ-aspartic acid | 70 | 6 | 62 |
| 11 | 2:1 | NH4I | DMSO/H2O (1:14) | ʟ-allysine | 70 | 6 | 60 |
| 12 | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 70 | 6 | 65 |
| 13 | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 50 | 6 | 65 |
| 14 | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 75 |
| 15c | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 65 |
| 16d | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 73 |
| 17 | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 4 | 66 |
| 18 | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 8 | 75 |
| 19 | 2:1 | NH4Cl | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 35 |
| 20 | 2:1 | NH4Br | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 55 |
| 21 | 2:1 | Et4NI | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 64 |
| 22 | 2:1 | Bu4NI | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 59 |
| 23 | 2:1 | NaI | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 72 |
| 24e | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 73 |
| 25f | 2:1 | NH4I | DMSO/H2O (1:14) | ᴅʟ-alanine | 30 | 6 | 70 |
aReaction conditions: ethyl acetoacetate (1a, 2 mmol), thiourea (2a, 1 mmol), NH4I (1 mmol), acid (1 mmol), LiClO4 (0.5 mmol), DMSO/H2O (v/v), undivided cell, graphite plate anode and cathode, 5 mA/cm2. bIsolated yield. c4 mA/cm2. d6 mA/cm2. eNH4I (0.1 mmol). fWithout LiClO4.
Next, the effect of halide-based mediators on the reaction was examined. When NH4Cl or NH4Br was used as the mediator, the yield of 3a decreased to 35% (Table 1, entry 19) and 55% (Table 1, entry 20), respectively. Slightly lower yields of 3a were obtained when the mediator NH4I was replaced by Et4NI, Bu4NI or NaI (Table 1, entries 21–23). Interestingly, almost same yield of 3a was isolated when the amount of NH4I was reduced to 0.1 mmol (Table 1, entry 24). In addition, without conductive salt (LiClO4) the electrochemical reaction also proceeded smoothly with a comparable yield (Table 1, entry 25).
Based on the results mentioned above, the optimal reaction conditions are as follows: constant current electrolysis was performed in an undivided cell equipped with a graphite plate as working electrode and counter electrode, using 0.1 mmol of NH4I as the mediator and DMSO/H2O (1 mL + 14 mL) as the solvent in the presence of ᴅʟ-alanine (Table 1, entry 24).
Under the optimized reaction conditions (entry 24, Table 1), the scope of the electrochemical reaction was studied using a series of active methylene ketones (Scheme 2). Various linear and branched alkyl acetoacetates including methyl, ethyl, tert-butyl, and amyl reacted smoothly with thiourea 2a under the optimized conditions, giving the corresponding products in 30–80% yields (3a–d). In addition, allyl and benzyl acetoacetate were also suitable substrates to give the desired 2-aminothiazoles 3e and 3f in 78% and 52% yields, respectively. β-ketoesters containing ethyl, n-propyl, isopropyl, n-butyl, tert-butyl and cyclohexyl moieties were also compatible with the optimized conditions, providing the corresponding 2-aminothiazoles in 24% to 65% yields (3g–l).
![[1860-5397-18-130-i2]](/bjoc/content/inline/1860-5397-18-130-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope. Reaction conditions: 1 (2 mmol), 2 (1 mmol), NH4I (0.1 mmol), ᴅʟ-alanine (1 mmol), LiClO4 (0.5 mmol) in DMSO 1 mL + H2O 14 mL, undivided cell, graphite plate anode and cathode, 30 °C, 5 mA/cm2, 6 F/mol; isolated yields are given. a8 F/mol.
Scheme 2: Substrate scope. Reaction conditions: 1 (2 mmol), 2 (1 mmol), NH4I (0.1 mmol), ᴅʟ-alanine (1 mmol),...
As shown in Scheme 2, when the R1 group changed from alkyl to aryl, the target products could also be obtained. For example, ethyl 3-oxo-3-phenylpropanoate and ethyl 3-oxo-3-(p-tolyl)propanoate afforded the corresponding 2-aminothiazoles 3m and 3n in 52% and 41% isolated yields under the standard conditions. When an electron-donating methoxy substituent was introduced to the ortho-, meta-, or para-position of the phenyl moiety, the corresponding 2-aminothiazoles, 3o–q, were given in 17–53% yields. With electron-withdrawing groups (F, Cl, Br, NO2), the desired products 3r–u were obtained in moderate yields. Other heteroaryl ketones, ethyl 3-(furan-2-yl)-3-oxopropanoate (1v) reacted with thiourea to give 3v in 23% yield. In addition to β-keto esters, other active methylene derivatives, including β-ketoamide, β-keto nitrile and β-keto sulfone were also suitable coupling partners with relatively lower yields (3w–y) and most of the starting materials were recovered. The method could also be applied to 1,3-diones. For example, in the cases of 1,3-diphenylpropane-1,3-dione and acetoacetone, the corresponding products 3z and 3aa were given in 4% and 25% yields, respectively.
Different N-substituted thioureas were also screened for the electrochemical reactions. N-methylthiourea and N-(2-phenylethyl)thiourea reacted smoothly with ethyl acetoacetate 1a under the optimized conditions, providing the corresponding 2-aminothiazoles, 3bb and 3cc, in 77% and 44% yields, respectively. It is regretful that thioacetamide is not the best suitable partner due to its low reactivity under the optimized conditions.
To demonstrate the practicability of the reaction, a scale-up reaction of ethyl acetoacetate (1a, 28 mmol, 3.64 g) and thiourea (2a,14 mmol, 1.05 g) was carried out under the optimized conditions to give 3a in 50% yield (Scheme 3).
![[1860-5397-18-130-i3]](/bjoc/content/inline/1860-5397-18-130-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
In order to better understand the iodide-mediated reaction mechanism and to determine the possible active intermediates involved, several control experiments were carried out. As shown in Scheme 4, when molecular iodine was employed as oxidant, the desired product 3a was obtained in a 67% yield under otherwise identical conditions (Scheme 4a), but without passing electricity. Therefore, the in situ-generated I2 was one of the active species. Further investigation revealed that the condensation of ethyl 2-iodo-3-oxobutanoate (4a) with thiourea 2a could give the target product 3a in 70% yield (Scheme 4b). Therefore, the in situ-generated ethyl 2-iodo-3-oxobutanoate (4a) should be a key intermediate for this tandem reaction.
![[1860-5397-18-130-i4]](/bjoc/content/inline/1860-5397-18-130-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
On the basis of the above mechanistic studies and the previous works on iodide-mediated electrochemical transformation [37-40], a possible mechanism for this electrochemical reaction was proposed (Scheme 5). It is well known that amino acid can act as a bi-functional organocatalyst due to the existence of both Lewis base (NH2) and Brønsted acidic (COOH) sites. In the suggested mechanism, the carboxy group may polarize the carbonyl group of the active methylene ketone and the amino group as a Lewis base serves the formation of enolate to produce α-iodo ketone with the molecular I2 produced by anodic oxidation. Subsequently, the nucleophilic substitution between intermediate 4 and thiourea tautomer gives α-sulfur substituted ketone 5. Intermediate 5 undergoes intramolecular nucleophilic addition to the carbonyl group and followed by dehydration to give the heterocyclic product 3. At the cathode, protons are reduced to release H2.
![[1860-5397-18-130-i5]](/bjoc/content/inline/1860-5397-18-130-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The proposed mechanism for the one-pot electrochemical synthesis of 2-aminothiazoles mediated by NH4I.
Scheme 5: The proposed mechanism for the one-pot electrochemical synthesis of 2-aminothiazoles mediated by NH4...
Conclusion
In conclusion, we have developed a one-pot electrochemical strategy for the synthesis of 2-amniothiazoles by the reaction of active methylene ketones with thioureas. The electrochemical synthesis was performed in an undivided cell with NH4I as the mediator and cheap graphite plate as the working electrode. Various active methylene ketones, including β-keto ester, β-keto amide, β-keto nitrile, β-keto sulfone and 1,3-diones proved to be compatible with the protocol. Since external oxidants and prefunctionalization of substrates are avoided, the present electrochemical protocol represents an appealing alternative for the synthesis of 2-aminothiazoles. Gram-scale synthesis also highlighted the synthetic practicability of this electrochemical strategy. Mechanistically, the in situ-generated α-iodoketone was proposed to be a key active species. Further application of this method is underway in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of spectra of the all synthesized compounds (1H NMR, 13C NMR and HRMS). | ||
| Format: PDF | Size: 3.7 MB | Download |
References
-
de Souza, M. V. N. J. Sulfur Chem. 2005, 26, 429–449. doi:10.1080/17415990500322792
Return to citation in text: [1] -
Samadhiya, P.; Sharma, R.; Srivastava, S. K.; Srivastava, S. D. J. Serb. Chem. Soc. 2012, 77, 599–605. doi:10.2298/jsc110616002s
Return to citation in text: [1] -
Sarıgüney, A. B.; Kocabaş, E.; Erci, F.; Torlak, E.; Coşkun, A. J. Heterocycl. Chem. 2018, 55, 2107–2110. doi:10.1002/jhet.3254
Return to citation in text: [1] -
Jeong, K.-w.; Lee, J.-h.; Park, S.-m.; Choi, J.-H.; Jeong, D.-Y.; Choi, D.-H.; Nam, Y.; Park, J.-H.; Lee, K.-N.; Kim, S.-M.; Ku, J.-M. Eur. J. Med. Chem. 2015, 102, 387–397. doi:10.1016/j.ejmech.2015.08.020
Return to citation in text: [1] -
Hutchinson, I.; Jennings, S. A.; Vishnuvajjala, B. R.; Westwell, A. D.; Stevens, M. F. G. J. Med. Chem. 2002, 45, 744–747. doi:10.1021/jm011025r
Return to citation in text: [1] -
Ottmann, O.; Saglio, G.; Apperley, J. F.; Arthur, C.; Bullorsky, E.; Charbonnier, A.; Dipersio, J. F.; Kantarjian, H.; Khoury, H. J.; Kim, D.-W.; Healey, D.; Strauss, L.; Cortes, J. E. Blood Cancer J. 2018, 8, 88. doi:10.1038/s41408-018-0122-3
Return to citation in text: [1] -
Kalkhambkar, R. G.; Kulkarni, G. M.; Shivkumar, H.; Rao, R. N. Eur. J. Med. Chem. 2007, 42, 1272–1276. doi:10.1016/j.ejmech.2007.01.023
Return to citation in text: [1] -
Singh, N.; Bhati, S. K.; Kumar, A. Eur. J. Med. Chem. 2008, 43, 2597–2609. doi:10.1016/j.ejmech.2007.12.024
Return to citation in text: [1] -
Kaupp, G.; Amer, F. A.; Metwally, M. A.; Abdel-Latif, E. J. Heterocycl. Chem. 2003, 40, 963–971. doi:10.1002/jhet.5570400603
Return to citation in text: [1] -
Alajarín, M.; Cabrera, J.; Pastor, A.; Sánchez-Andrada, P.; Bautista, D. J. Org. Chem. 2006, 71, 5328–5339. doi:10.1021/jo060664c
Return to citation in text: [1] -
Liu, Y.; Chen, H.; Yin, D.; Sun, B. Molecules 2010, 15, 5104–5111. doi:10.3390/molecules15085104
Return to citation in text: [1] -
Al-Dujaili, A. H.; Atto, A. T.; Al-Kurde, A. M. Eur. Polym. J. 2001, 37, 927–932. doi:10.1016/s0014-3057(00)00221-4
Return to citation in text: [1] -
Abu-Melha, S. Pigm. Resin Technol. 2019, 48, 375–382. doi:10.1108/prt-09-2018-0102
Return to citation in text: [1] -
Kashyap, S. J.; Garg, V. K.; Sharma, P. K.; Kumar, N.; Dudhe, R.; Gupta, J. K. Med. Chem. Res. 2012, 21, 2123–2132. doi:10.1007/s00044-011-9685-2
Return to citation in text: [1] -
Das, D.; Sikdar, P.; Bairagi, M. Eur. J. Med. Chem. 2016, 109, 89–98. doi:10.1016/j.ejmech.2015.12.022
Return to citation in text: [1] -
Gümüş, M.; Yakan, M.; Koca, İ. Future Med. Chem. 2019, 11, 1979–1998. doi:10.4155/fmc-2018-0196
Return to citation in text: [1] -
Jadhav, P. M.; Kantevari, S.; Tekale, A. B.; Bhosale, S. V.; Pawar, R. P.; Tekale, S. U. Phosphorus, Sulfur Silicon Relat. Elem. 2021, 196, 879–895. doi:10.1080/10426507.2021.1945601
Return to citation in text: [1] -
Hantzsch, A.; Weber, J. H. Ber. Dtsch. Chem. Ges. 1887, 20, 3118–3132. doi:10.1002/cber.188702002200
Return to citation in text: [1] -
Zav'yalov, S. I.; Kravchenko, N. E.; Ezhova, G. I.; Kulikova, L. B.; Zavozin, A. G.; Dorofeeva, O. V. Pharm. Chem. J. 2007, 41, 105–108. doi:10.1007/s11094-007-0023-4
Return to citation in text: [1] -
Dziuk, B.; Kyzioł, J. B.; Zaleski, J.; Ejsmont, K.; Zarychta, B. J. Heterocycl. Chem. 2018, 55, 763–768. doi:10.1002/jhet.3086
Return to citation in text: [1] -
Abedi-Jazini, Z.; Safari, J.; Zarnegar, Z.; Sadeghi, M. Polycyclic Aromat. Compd. 2018, 38, 231–235. doi:10.1080/10406638.2016.1200104
Return to citation in text: [1] -
Safari, J.; Shokrani, Z.; Zarnegar, Z. Polycyclic Aromat. Compd. 2020, 40, 1105–1111. doi:10.1080/10406638.2018.1528287
Return to citation in text: [1] -
Narender, M.; Reddy, M. S.; Kumar, V. P.; Srinivas, B.; Sridhar, R.; Nageswar, Y. V. D.; Rao, K. R. Synthesis 2007, 3469–3472. doi:10.1055/s-2007-990849
Return to citation in text: [1] -
Meng, G.; Wang, M.; Zheng, A.; Dou, J.; Guo, Z. Green Chem. Lett. Rev. 2014, 7, 46–49. doi:10.1080/17518253.2014.895858
Return to citation in text: [1] -
Kuarm, B. S.; Madhav, J. V.; Rajitha, B. Lett. Org. Chem. 2011, 8, 549–553. doi:10.2174/157017811797249443
Return to citation in text: [1] -
de Andrade, V. S. C.; de Mattos, M. C. S. Synthesis 2018, 4867–4874. doi:10.1055/s-0037-1610243
Return to citation in text: [1] -
Sadeghi, M.; Safari, J.; Zarnegar, Z. RSC Adv. 2016, 6, 64749–64755. doi:10.1039/c6ra11175k
Return to citation in text: [1] -
Zarnegar, Z.; Sadeghi, M.; Alizadeh, R.; Safai, J. J. Mol. Liq. 2018, 255, 76–79. doi:10.1016/j.molliq.2018.01.119
Return to citation in text: [1] -
Sun, J.; Ge, H.; Zhen, X.; An, X.; Zhang, G.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Tetrahedron 2018, 74, 2107–2114. doi:10.1016/j.tet.2018.02.064
Return to citation in text: [1] -
Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d
Return to citation in text: [1] -
Francke, R. Beilstein J. Org. Chem. 2014, 10, 2858–2873. doi:10.3762/bjoc.10.303
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] [2] -
Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271
Return to citation in text: [1] [2] -
Liang, S.; Zeng, C.-C. Curr. Opin. Electrochem. 2020, 24, 31–43. doi:10.1016/j.coelec.2020.06.005
Return to citation in text: [1] [2] -
Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Chem. Soc. Rev. 2021, 50, 7941–8002. doi:10.1039/d1cs00223f
Return to citation in text: [1] [2] -
Wang, Z.-Q.; Meng, X.-J.; Li, Q.-Y.; Tang, H.-T.; Wang, H.-S.; Pan, Y.-M. Adv. Synth. Catal. 2018, 360, 4043–4048. doi:10.1002/adsc.201800871
Return to citation in text: [1] -
Li, J.-S.; Xie, X.-Y.; Yang, P.-P.; Jiang, S.; Tao, L.; Li, Z.-W.; Lu, C.-H.; Liu, W.-D. Adv. Synth. Catal. 2020, 362, 771–775. doi:10.1002/adsc.201901290
Return to citation in text: [1] [2] -
Ahdenov, R.; Mohammadi, A. A.; Makarem, S.; Taheri, S.; Mollabagher, H. Heterocycl. Commun. 2022, 28, 67–74. doi:10.1515/hc-2022-0008
Return to citation in text: [1] [2] -
Liang, S.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G.; Luo, X.-G.; Ren, F.-z. J. Org. Chem. 2016, 81, 11565–11573. doi:10.1021/acs.joc.6b01595
Return to citation in text: [1] [2] -
Liang, S.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G.; Luo, X.-G.; Ren, F.-z. Adv. Synth. Catal. 2018, 360, 1444–1452. doi:10.1002/adsc.201701401
Return to citation in text: [1] [2] -
Liang, S.; Xu, K.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G. Adv. Synth. Catal. 2018, 360, 4266–4292. doi:10.1002/adsc.201800519
Return to citation in text: [1] -
Li, P.; Yang, S.-F.; Fang, Z.-L.; Cui, H.-R.; Liang, S.; Tian, H.-Y.; Sun, B.-G.; Zeng, C.-C. J. Environ. Chem. Eng. 2022, 10, 107487. doi:10.1016/j.jece.2022.107487
Return to citation in text: [1] [2] -
Shaikh, I. R. J. Catal. 2014, 402860. doi:10.1155/2014/402860
Return to citation in text: [1] -
Safari, J.; Shokrani, Z.; Zarnegar, Z. Polycyclic Aromat. Compd. 2020, 40, 1105–1111. doi:10.1080/10406638.2018.1528287
Return to citation in text: [1]
| 30. | Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d |
| 31. | Francke, R. Beilstein J. Org. Chem. 2014, 10, 2858–2873. doi:10.3762/bjoc.10.303 |
| 32. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 33. | Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271 |
| 34. | Liang, S.; Zeng, C.-C. Curr. Opin. Electrochem. 2020, 24, 31–43. doi:10.1016/j.coelec.2020.06.005 |
| 35. | Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Chem. Soc. Rev. 2021, 50, 7941–8002. doi:10.1039/d1cs00223f |
| 36. | Wang, Z.-Q.; Meng, X.-J.; Li, Q.-Y.; Tang, H.-T.; Wang, H.-S.; Pan, Y.-M. Adv. Synth. Catal. 2018, 360, 4043–4048. doi:10.1002/adsc.201800871 |
| 37. | Li, J.-S.; Xie, X.-Y.; Yang, P.-P.; Jiang, S.; Tao, L.; Li, Z.-W.; Lu, C.-H.; Liu, W.-D. Adv. Synth. Catal. 2020, 362, 771–775. doi:10.1002/adsc.201901290 |
| 38. | Ahdenov, R.; Mohammadi, A. A.; Makarem, S.; Taheri, S.; Mollabagher, H. Heterocycl. Commun. 2022, 28, 67–74. doi:10.1515/hc-2022-0008 |
| 28. | Zarnegar, Z.; Sadeghi, M.; Alizadeh, R.; Safai, J. J. Mol. Liq. 2018, 255, 76–79. doi:10.1016/j.molliq.2018.01.119 |
| 29. | Sun, J.; Ge, H.; Zhen, X.; An, X.; Zhang, G.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Tetrahedron 2018, 74, 2107–2114. doi:10.1016/j.tet.2018.02.064 |
| 1. | de Souza, M. V. N. J. Sulfur Chem. 2005, 26, 429–449. doi:10.1080/17415990500322792 |
| 7. | Kalkhambkar, R. G.; Kulkarni, G. M.; Shivkumar, H.; Rao, R. N. Eur. J. Med. Chem. 2007, 42, 1272–1276. doi:10.1016/j.ejmech.2007.01.023 |
| 8. | Singh, N.; Bhati, S. K.; Kumar, A. Eur. J. Med. Chem. 2008, 43, 2597–2609. doi:10.1016/j.ejmech.2007.12.024 |
| 26. | de Andrade, V. S. C.; de Mattos, M. C. S. Synthesis 2018, 4867–4874. doi:10.1055/s-0037-1610243 |
| 5. | Hutchinson, I.; Jennings, S. A.; Vishnuvajjala, B. R.; Westwell, A. D.; Stevens, M. F. G. J. Med. Chem. 2002, 45, 744–747. doi:10.1021/jm011025r |
| 6. | Ottmann, O.; Saglio, G.; Apperley, J. F.; Arthur, C.; Bullorsky, E.; Charbonnier, A.; Dipersio, J. F.; Kantarjian, H.; Khoury, H. J.; Kim, D.-W.; Healey, D.; Strauss, L.; Cortes, J. E. Blood Cancer J. 2018, 8, 88. doi:10.1038/s41408-018-0122-3 |
| 27. | Sadeghi, M.; Safari, J.; Zarnegar, Z. RSC Adv. 2016, 6, 64749–64755. doi:10.1039/c6ra11175k |
| 4. | Jeong, K.-w.; Lee, J.-h.; Park, S.-m.; Choi, J.-H.; Jeong, D.-Y.; Choi, D.-H.; Nam, Y.; Park, J.-H.; Lee, K.-N.; Kim, S.-M.; Ku, J.-M. Eur. J. Med. Chem. 2015, 102, 387–397. doi:10.1016/j.ejmech.2015.08.020 |
| 21. | Abedi-Jazini, Z.; Safari, J.; Zarnegar, Z.; Sadeghi, M. Polycyclic Aromat. Compd. 2018, 38, 231–235. doi:10.1080/10406638.2016.1200104 |
| 22. | Safari, J.; Shokrani, Z.; Zarnegar, Z. Polycyclic Aromat. Compd. 2020, 40, 1105–1111. doi:10.1080/10406638.2018.1528287 |
| 37. | Li, J.-S.; Xie, X.-Y.; Yang, P.-P.; Jiang, S.; Tao, L.; Li, Z.-W.; Lu, C.-H.; Liu, W.-D. Adv. Synth. Catal. 2020, 362, 771–775. doi:10.1002/adsc.201901290 |
| 38. | Ahdenov, R.; Mohammadi, A. A.; Makarem, S.; Taheri, S.; Mollabagher, H. Heterocycl. Commun. 2022, 28, 67–74. doi:10.1515/hc-2022-0008 |
| 39. | Liang, S.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G.; Luo, X.-G.; Ren, F.-z. J. Org. Chem. 2016, 81, 11565–11573. doi:10.1021/acs.joc.6b01595 |
| 40. | Liang, S.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G.; Luo, X.-G.; Ren, F.-z. Adv. Synth. Catal. 2018, 360, 1444–1452. doi:10.1002/adsc.201701401 |
| 2. | Samadhiya, P.; Sharma, R.; Srivastava, S. K.; Srivastava, S. D. J. Serb. Chem. Soc. 2012, 77, 599–605. doi:10.2298/jsc110616002s |
| 3. | Sarıgüney, A. B.; Kocabaş, E.; Erci, F.; Torlak, E.; Coşkun, A. J. Heterocycl. Chem. 2018, 55, 2107–2110. doi:10.1002/jhet.3254 |
| 23. | Narender, M.; Reddy, M. S.; Kumar, V. P.; Srinivas, B.; Sridhar, R.; Nageswar, Y. V. D.; Rao, K. R. Synthesis 2007, 3469–3472. doi:10.1055/s-2007-990849 |
| 24. | Meng, G.; Wang, M.; Zheng, A.; Dou, J.; Guo, Z. Green Chem. Lett. Rev. 2014, 7, 46–49. doi:10.1080/17518253.2014.895858 |
| 25. | Kuarm, B. S.; Madhav, J. V.; Rajitha, B. Lett. Org. Chem. 2011, 8, 549–553. doi:10.2174/157017811797249443 |
| 13. | Abu-Melha, S. Pigm. Resin Technol. 2019, 48, 375–382. doi:10.1108/prt-09-2018-0102 |
| 18. | Hantzsch, A.; Weber, J. H. Ber. Dtsch. Chem. Ges. 1887, 20, 3118–3132. doi:10.1002/cber.188702002200 |
| 43. | Shaikh, I. R. J. Catal. 2014, 402860. doi:10.1155/2014/402860 |
| 44. | Safari, J.; Shokrani, Z.; Zarnegar, Z. Polycyclic Aromat. Compd. 2020, 40, 1105–1111. doi:10.1080/10406638.2018.1528287 |
| 12. | Al-Dujaili, A. H.; Atto, A. T.; Al-Kurde, A. M. Eur. Polym. J. 2001, 37, 927–932. doi:10.1016/s0014-3057(00)00221-4 |
| 19. | Zav'yalov, S. I.; Kravchenko, N. E.; Ezhova, G. I.; Kulikova, L. B.; Zavozin, A. G.; Dorofeeva, O. V. Pharm. Chem. J. 2007, 41, 105–108. doi:10.1007/s11094-007-0023-4 |
| 20. | Dziuk, B.; Kyzioł, J. B.; Zaleski, J.; Ejsmont, K.; Zarychta, B. J. Heterocycl. Chem. 2018, 55, 763–768. doi:10.1002/jhet.3086 |
| 32. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 33. | Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271 |
| 34. | Liang, S.; Zeng, C.-C. Curr. Opin. Electrochem. 2020, 24, 31–43. doi:10.1016/j.coelec.2020.06.005 |
| 35. | Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Chem. Soc. Rev. 2021, 50, 7941–8002. doi:10.1039/d1cs00223f |
| 11. | Liu, Y.; Chen, H.; Yin, D.; Sun, B. Molecules 2010, 15, 5104–5111. doi:10.3390/molecules15085104 |
| 39. | Liang, S.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G.; Luo, X.-G.; Ren, F.-z. J. Org. Chem. 2016, 81, 11565–11573. doi:10.1021/acs.joc.6b01595 |
| 40. | Liang, S.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G.; Luo, X.-G.; Ren, F.-z. Adv. Synth. Catal. 2018, 360, 1444–1452. doi:10.1002/adsc.201701401 |
| 41. | Liang, S.; Xu, K.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G. Adv. Synth. Catal. 2018, 360, 4266–4292. doi:10.1002/adsc.201800519 |
| 42. | Li, P.; Yang, S.-F.; Fang, Z.-L.; Cui, H.-R.; Liang, S.; Tian, H.-Y.; Sun, B.-G.; Zeng, C.-C. J. Environ. Chem. Eng. 2022, 10, 107487. doi:10.1016/j.jece.2022.107487 |
| 9. | Kaupp, G.; Amer, F. A.; Metwally, M. A.; Abdel-Latif, E. J. Heterocycl. Chem. 2003, 40, 963–971. doi:10.1002/jhet.5570400603 |
| 10. | Alajarín, M.; Cabrera, J.; Pastor, A.; Sánchez-Andrada, P.; Bautista, D. J. Org. Chem. 2006, 71, 5328–5339. doi:10.1021/jo060664c |
| 14. | Kashyap, S. J.; Garg, V. K.; Sharma, P. K.; Kumar, N.; Dudhe, R.; Gupta, J. K. Med. Chem. Res. 2012, 21, 2123–2132. doi:10.1007/s00044-011-9685-2 |
| 15. | Das, D.; Sikdar, P.; Bairagi, M. Eur. J. Med. Chem. 2016, 109, 89–98. doi:10.1016/j.ejmech.2015.12.022 |
| 16. | Gümüş, M.; Yakan, M.; Koca, İ. Future Med. Chem. 2019, 11, 1979–1998. doi:10.4155/fmc-2018-0196 |
| 17. | Jadhav, P. M.; Kantevari, S.; Tekale, A. B.; Bhosale, S. V.; Pawar, R. P.; Tekale, S. U. Phosphorus, Sulfur Silicon Relat. Elem. 2021, 196, 879–895. doi:10.1080/10426507.2021.1945601 |
| 42. | Li, P.; Yang, S.-F.; Fang, Z.-L.; Cui, H.-R.; Liang, S.; Tian, H.-Y.; Sun, B.-G.; Zeng, C.-C. J. Environ. Chem. Eng. 2022, 10, 107487. doi:10.1016/j.jece.2022.107487 |
© 2022 Yang et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.