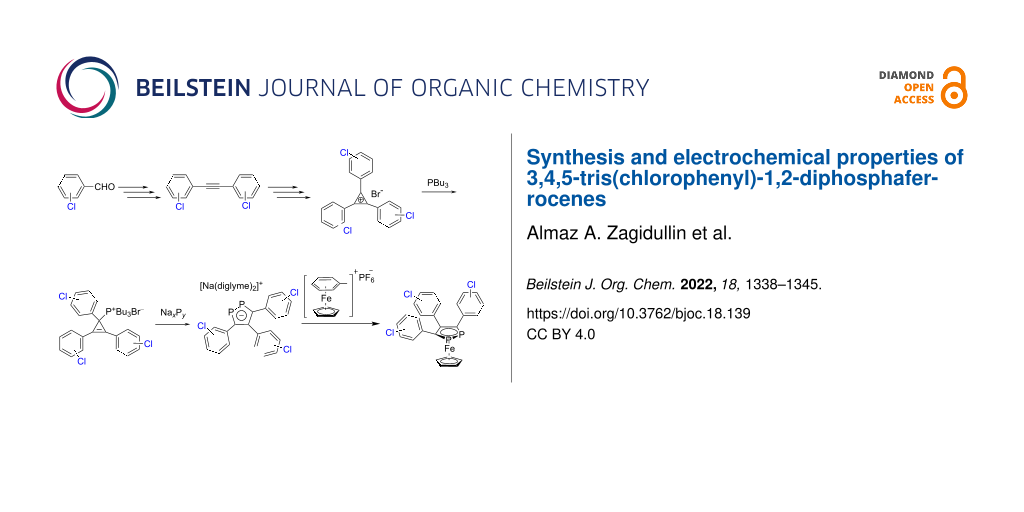
Abstract
A novel representative of sodium 3,4,5-triaryl-1,2-diphosphacyclopentadienide containing a chloro substituent in the meta-position of the aryl groups was obtained with a high yield based on the reaction of tributyl(1,2,3-triarylcyclopropenyl)phosphonium bromide and sodium polyphosphides. Further reaction of sodium 3,4,5-tris(3-chlorophenyl)-1,2-diphosphacyclopentadienide with [FeCp(η6-C6H5CH3)][PF6] complex gives a new 3,4,5-tris(3-chlorophenyl)-1,2-diphosphaferrocene. The electrochemical properties of 3,4,5-tris(3-chlorophenyl)-1,2-diphosphaferrocene were studied and compared to 3,4,5-tris(4-chlorophenyl)-1,2-diphosphaferrocene. It was found that the position of the chlorine atom on the aryl fragment has an influence on the reduction potential of 1,2-diphosphaferrocenes, while the oxidation potentials do not change.

Graphical Abstract
Introduction
Among the various heterometallocenes reported to date, phosphaferrocenes are by far the most investigated because of their structural and electronic features [1,2] and remain the objects of growing interest in the fields of coordination chemistry [3-5] and asymmetric catalysis [6,7]. Due to the sp2-hybridization of the phosphorus atom, phosphaferrocenes are commonly regarded as phosphorus ligands with weaker σ-donor character than classical tertiary phosphines and stronger π-acceptor properties closer to phosphites P(OR)3 [8,9]. Since the P atom in phosphaferrocenes retains an electron lone pair, phosphaferrocenes have been used as P-donor ligands [10-12] as well as nucleophilic catalysts [13,14]. Recently, the pentaphosphaferrocene Cp*Fe(η5-P5) has been used as a mediator in the synthesis of asymmetric phosphines starting from white phosphorus [15]. Moreover, the presence of the lone pair of the P atom opens the route to polynuclear complexes [16-18] and coordination polymers [19-21] with the mixed σ-/π-coordination mode, which is not typical for classical ferrocene species.
Various effective synthetic approaches were developed for 1-mono- [22-24], 1,2,3-tri- [25-27], 1,2,4-tri- [28-30], and pentaphosphaferrocenes [31-33], whereby the chemistry of these compounds is most investigated and well represented at present time. In contrast, very limited data are available concerning 1,2-diphosphaferrocenes due to the absence of simple and effective synthetic routes [34-36]. Recently, we have reported a convenient synthesis of 3,4,5-triaryl-1,2-diphosphaferrocenes with various substituents at the para-positions of aryl groups [37]. Based on this method, herein we report on the complete multistep synthesis of new sodium 3,4,5-tris(3-chlorophenyl)-1,2-diphosphacyclopentadienide and corresponding 1,2-diphosphaferrocene with meta-chlorophenyl substituents and the influence of the position of the Cl atom on aryl moiety on the electrochemical properties.
Results and Discussion
Synthesis of tris(chlorophenyl)cyclopropenyl bromides and derivatives
Cyclopropenium (cyclopropenylium) ions have always attracted attention of the synthetic chemists because of the unique combination of stability and reactivity [38-40]. The synthesis of corresponding 1,2,3-cyclopropenium bromides was realized by a classical approach: combination of C1 and C2 building blocks, i.e., the addition of a carbene species to a triple bond of diarylacetylene, followed by treatment of the produced cyclopropene with HBr to convert it to the corresponding cyclopropenylium cation. Using this approach, tris(4-chlorophenyl)- and tris(3-chlorophenyl)cyclopropenyl bromides were prepared for the first time. The advantage of this approach is the possibility of synthesis of substituted diarylacetylenes, the corresponding substituted benzal chlorides, and triarylcyclopropenyl bromides from one starting aryl aldehyde.
Diethyl phosphite was allowed to react with appropriately substituted benzaldehydes in THF for 48 hours at 25 °C to afford diethyl (hydroxy(aryl)methyl)phosphonates 1, which were detected by 31P NMR spectroscopy in THF (21.4 ppm for 1a, 21.0 ppm for 1b, and 21.5 ppm for 1c). Further, reaction mixtures with compounds 1 were treated with SOCl2 for 3–4 h at 0 °C and converted to chloro derivatives 2. In the next step, compounds 2 and starting substituted benzaldehydes were subsequently treated with 2 equiv of potassium tert-butoxide in THF for 18 hours at room temperature to afford substituted diarylacetylenes 3. Based on this reaction, the desired compounds 3 were prepared from 2-chloro-, 3-chloro-, and 4-chlorophenyl aldehydes, respectively, in 3 steps in 10–53% yield (10% for 3a, 48% for 3b, 53% for 3c, Scheme 1). This method is an alternative way to different transition metal-catalyzed cross-coupling reactions broadly used for the preparation of different diarylacetylenes and, rarely, bis(chlorophenyl)acetylenes.
![[1860-5397-18-139-i1]](/bjoc/content/inline/1860-5397-18-139-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of bis(chlorophenyl)acetylenes 3.
Scheme 1: Synthesis of bis(chlorophenyl)acetylenes 3.
Next, starting substituted benzaldehydes were treated with an excess of SOCl2 for 24 h at 25 °C. Corresponding substituted benzal chlorides 4 were distilled at reduced pressure to give pure compounds. In a final step, we used the above mentioned approach of combining the C1 and C2 building blocks and found that chloroarylcarbenes, generated from the corresponding benzal chlorides 4b,c under the action of potassium tert-butoxide, reacted with 1,2-bis(chlorophenyl)ethynes 3b,c to form triarylcyclopropenylium salts 5b,c in 22 and 15% yield (Scheme 2). Unfortunately, it was not possible to synthesize tris(2-chlorophenyl)cyclopropenylium bromide 5a using this method.
![[1860-5397-18-139-i2]](/bjoc/content/inline/1860-5397-18-139-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1,2,3-tris(chlorophenyl)cyclopropenylium bromides 5 and tributyl(1,2,3-tris(chlorophenyl)cyclopropenyl)phosphonium bromides 6.
Scheme 2: Synthesis of 1,2,3-tris(chlorophenyl)cyclopropenylium bromides 5 and tributyl(1,2,3-tris(chlorophen...
The structures of 3–5 were confirmed by 1H and 13C NMR as well as IR spectroscopic methods and, for 5c, single-crystal X-ray crystallography (Figure 1a). The 13C NMR signals of the cationic carbon atoms of the three-membered ring appeared at about 145 ppm. Besides, the 1H NMR spectra of 5 were unremarkable and consistent with the suggested formulas.
![[1860-5397-18-139-1]](/bjoc/content/figures/1860-5397-18-139-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP representations for cations 5c (a) and 6c (b) at the 50% probability level. Bromide anion and co-crystallized solvent molecules are omitted for clarity. For 6c, only one of two symmetry-independent molecules is shown. Selected interatomic distances (Å): C1–C2 1.387(7), C1–C3 1.372(7), C2–C3 1.380(7) for 5c; C1–C2 1.521(5), C1–C3 1.521(5), C2–C3 1.298(5), P1–C1 1.837(3) for 6c. Deposition numbers 2176393 for 5c and 2176394 for 6c contain the supplementary crystallographic data for this paper [41].
Figure 1: ORTEP representations for cations 5c (a) and 6c (b) at the 50% probability level. Bromide anion and...
As a next step, we synthesized a series of tributyl(1,2,3-triarylcyclopropenyl)phosphonium bromides 6 containing a Cl substituent in the meta- or para-position of each aryl group. This was done by reaction of appropriate 1,2,3-triarylcyclopropenylium bromides 5 with PBu3 at 25 °C in THF in 34 and 39% yield (Scheme 2). The structures of 6 were confirmed by 31P, 1H, and 13C NMR spectroscopy. The 31P{1H} NMR spectra of phosphonium bromides 6 showed a singlet at about 40 ppm, which is typical for phosphonium salts. The 13C{1H} NMR spectra consisted of a doublet at about 20 ppm, corresponding to the carbon atom C1, which is characteristic for the sp3-hybridized carbon atom, with a coupling constant of 1JCP ≈ 45 Hz. Additionally, the structure of 6c in the crystal was confirmed by X-ray crystallography (Figure 1b).
Synthesis, structure, and electrochemical properties of 3,4,5-tris(chlorophenyl)-1,2-diphosphaferrocenes
The obtained phosphonium salts 6 were treated with a mixture of sodium polyphosphides of the type NaxPy (obtained in situ from sodium metal and white phosphorus P4), containing mainly NaP5 and Na3P7 [42]), resulting in sodium 3,4,5-tris(chlorophenyl)-1,2-diphosphacyclopentadienides 7 in good yields (60 and 63%, Scheme 3). This reaction allowed a selective and controllable conversion of NaxPy to the 1,2-diphospholide anion, in which two new C−P bonds could selectively be formed [43,44]. The obtained sodium 3,4,5-triaryl-1,2-diphospholides 7 were isolated in good purity from the reaction mixture by filtration and further washing with a mixture of THF/n-hexane. The 31P{1H} NMR spectra of 7 showed a singlet at about 200 ppm, which is typical for sodium 1,2-diphospholides (31P{1H} in THF: 201 ppm for 7b and 198 ppm for 7c). Further, the 13C{1H} NMR spectra of 7 showed two multiplets at about 147 and 160 ppm for the heteroaromatic P2C3 ring backbone.
![[1860-5397-18-139-i3]](/bjoc/content/inline/1860-5397-18-139-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 3,4,5-tris(chlorophenyl)-1,2-diphosphacyclopentadienides 7 and 3,4,5-tris(chlorophenyl)-1,2-diphosphaferrocenes 8.
Scheme 3: Synthesis of 3,4,5-tris(chlorophenyl)-1,2-diphosphacyclopentadienides 7 and 3,4,5-tris(chlorophenyl...
Recently, we have reported a convenient method for the preparation of 1,2-diphosphaferrocenes [37] and 1,2,3-triphosphaferrocenes [25] with various substituents at para-positions of aryl groups. Using this approach, sodium bis(diglyme) 3,4,5-tris(3-chlorophenyl)-1,2-diphosphacyclopentadienide (7b) was treated in a 1:1 ratio with [FeCp(η6-C6H5CH3)][PF6] at 160 °C in diglyme. Evaporation of diglyme at reduced pressure and extraction of the product with toluene, followed by filtration through silica, resulted in 3,4,5-tris(3-chlorophenyl)-1,2-diphosphaferrocene (8b) in 68% yield and high purity (Scheme 3).
The structure of 3,4,5-triaryl-1,2-diphosphaferrocene 8b was confirmed by 31P, 1H, and 13C NMR spectroscopy. The 31P{1H} NMR spectrum of 8b showed a singlet at about −10 ppm, shifted upfield by about 210 ppm in comparison to the starting 1,2-diphospholide anion 7b. In the 1H NMR spectra, characteristic signals of the cyclopentadienyl ring (4.61 ppm) and ClC6H4 substituents (6.88–7.42 ppm) were observed. The 13C{1H} NMR spectrum of 8b showed pseudotriplets at about 106 ppm and 117 ppm for the carbon atoms of the P2C3 ring and a singlet at about 75 ppm for the cyclopentadienyl ring.
Quantum chemically, two possible conformations of 8b were considered, 8b-I and 8b-II (Figure 2). Similar to a previous report on 8c [37], both 8b-I and 8b-II adopted an almost eclipsed conformation during optimization. Computations predicted slightly lower energy (1 kcal⋅mol−1) for conformation 8b-I, with Cl oriented towards the Fe atom. According to the computations, 8b-I is also slightly more advantageous compared to 8c with the same energy difference.
![[1860-5397-18-139-2]](/bjoc/content/figures/1860-5397-18-139-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Considered conformations of 8b-I and 8b-II.
Figure 2: Considered conformations of 8b-I and 8b-II.
The experimental UV–vis spectra of 8b and 8c in CH2Cl2 were similar and contained bands at about 280, 320, and 380 nm. The bands at 280 and 320 nm were more intense in the spectrum of 8c, which is in full agreement with quantum chemical predictions (Figure 3). According to the computations, 8b-I and 8b-II demonstrated almost the same absorption. The bands at about 250 and 280 nm were caused by π–π* transitions. The dominating transition contributing to the lowest-energy absorption (380 nm) was the one corresponding to a transition between HOMO−1 and LUMO+1 (Figure 4). Both orbitals were localized mostly at the P2C3–Fe–Cp moiety, and the former was also contributed to by atomic orbitals of the aryl ring in the 4-position. Similar to 8c, the atomic orbitals of the Cl atoms in 8b practically did not participate in the frontier orbitals, which explained the similarity of the low-energy range of the experimental spectra.
![[1860-5397-18-139-3]](/bjoc/content/figures/1860-5397-18-139-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Top: experimental UV–vis spectra of 8с (black) and 8b (red). Bottom: broadened calculated UV–vis spectra of 8c (black), 8b-I (red), and 8b-II (blue). Experimental UV–vis spectra of 8с republished with permission of Royal Society of Chemistry from [37] (“Synthesis, structure and electrochemical properties of 3,4,5-triaryl-1,2-diphosphaferocenes” by I. A. Bezkishko et al., Inorg. Chem. Front., vol. 9, Issue 11, © 2022); permission conveyed through Copyright Clearance Center, Inc. This content is not subject to CC BY 4.0.
Figure 3: Top: experimental UV–vis spectra of 8с (black) and 8b (red). Bottom: broadened calculated UV–vis sp...
![[1860-5397-18-139-4]](/bjoc/content/figures/1860-5397-18-139-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Frontier orbitals of 8b-II contributing to absorption bands at about 380 nm.
Figure 4: Frontier orbitals of 8b-II contributing to absorption bands at about 380 nm.
The electrochemical properties of 1,2-diphosphaferrocene 8b were studied by cyclic voltammetry and compared to data for 8c (Table 1 and Figure 5). During oxidation, there were no noticeable differences between 8b and 8c. The oxidation potentials of 8b and 8c were shifted to the positive region relative to pure ferrocene by 0.48–0.53 V. This, in turn, indicated that the chlorine atoms in the η5-P2C3R3 fragment did not significantly affect the HOMO energy of 1,2-diphosphaferrocenes 8. The number of phosphorus atoms in cyclopentadiene had a much greater effect on the shifts of the FeII/FeIII oxidation potential. As shown earlier, an increase in the number of phosphorus atoms led to the irreversible oxidation of phosphaferrocenes containing an unsubstituted Cp ring. It was shown that Cp*Fe(η5-P5) was irreversibly oxidized at a potential of 0.57 V relative to Fс/Fс+, and the presence of even five phosphorus atoms makes an insignificant contribution to the HOMO energy level [45]. The situation changed fundamentally when both rings were replaced with phosphacyclopentadienyl ligands. Related diphosphacyclobutadiene complexes Fe(η4-P2C2R2)2 were oxidized much more cathodically (negative by 1.7–2.0 V) [46,47], which indicated a significant contribution of the phosphacyclopentadienyl ligands to the iron atomic orbitals. Of course the structures of 8 and diphosphacyclobutadiene complexes are not isolobal, but it would be interesting to study electrochemical properties of Fe(η5-P2C3R3)2 complexes in the future.
Table 1: Electrochemical data for the redox properties of 3,4,5-triaryl-1,2-diphosphaferrocenes 8b and 8c.
| compound | Eox1 (V) vs Ag/AgCl | Ered1 (V) | 1EHOMO (eV) | 1ELUMO (eV) | gap (eV) |
| ferrocene [48] | 0.48 | −3.19a | −4.79a | −1.61a | 3.18a |
| FeCp(η5-P2C3R3) (R = 3-Cl-C6H4, 8b) | 0.96 | −2.15 | −5.28 | −2.2 | 2.84 |
| FeCp(η5-P2C3R3) (R = 4-Cl-C6H4, 8c) [37] | 1.01 | −1.83 | −5.36 | −2.48 | 2.88 |
| Cp*Fe(η5-P5) [45] | 1.12b | −1.55b | −5.47b | −2.80b | 2.57b |
aConditions: −50 °C, glassy carbon working electrode, Ag/AgCl reference electrode, c 0.5 mM, Bu4NBF4, DMF, 100 mV⋅s−1. bConditions: −13 °C, Pt working electrode, Ag/AgCl reference electrode (recalculated from Fc/Fc+), c 0.5 mM, Bu4NPF6, CH2Cl2, 500 mV⋅s−1.
![[1860-5397-18-139-5]](/bjoc/content/figures/1860-5397-18-139-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Cyclic voltammograms of 3,4,5-triaryl-1,2-diphosphaferrocenes 8b and 8c in CH3CN on glassy carbon electrode (0.5 mM complex). Potentials vs Ag/AgCl. Scan rate = 100 mV⋅s−1, room temperature. Cyclic voltammogram of 8c republished with permission of Royal Society of Chemistry from [37] (“Synthesis, structure and electrochemical properties of 3,4,5-triaryl-1,2-diphosphaferocenes” by I. A. Bezkishko et al., Inorg. Chem. Front., vol. 9, Issue 11, © 2022); permission conveyed through Copyright Clearance Center, Inc. This content is not subject to CC BY 4.0.
Figure 5: Cyclic voltammograms of 3,4,5-triaryl-1,2-diphosphaferrocenes 8b and 8c in CH3CN on glassy carbon e...
For reduction, the electrochemical properties changed more noticeable since the contribution to the LUMO came from the cyclopentadiene fragments. For 1,2-diphosphaferrocene 8b, the reduction potential was positively shifted by 0.32 V as compared to 8c. It should be noted that an increase of phosphorus atoms' number in phosphaferrocenes leads to a greater positive potential, which in turn leads to the formation of dimers, which was shown for pentaphosphaferrocene Cp*Fe(η5-P5) [49] and the corresponding Sm complexes [50].
Conclusion
In summary, a series of bis(сhlorophenyl)acetylenes 3, substituted benzal chlorides 4, and tris(chlorophenyl)cyclopropenylium bromides 5 were synthesized starting from corresponding chloro-substituted benzaldehydes. We found that the reaction of tributyl(1,2,3-tris(chlorophenyl)cyclopropenyl)phosphonium bromides 6 with sodium polyphosphides can be successfully used for the preparation of sodium 3,4,5-tris(chlorophenyl)-1,2-diphosphacyclopentadienides 7. A facile synthesis of 3,4,5-tris(3-chlorophenyl)-1,2-diphosphaferrocene (8b) from sodium bis(diglyme) 3,4,5-tris(3-chlorophenyl)-1,2-diphosphacyclopentadienide (7b) and [FeCp(η6-C6H5CH3)][PF6] is described. The structure of 8b was studied using experimental NMR, UV–vis, and electrochemical analyses as well as theoretical studies. The meta- and para-substitution of the Cl atoms in the aryl fragments did not significantly effect the oxidation potentials of 1,2-diphosphaferrocenes 8, while the reduction potential of 8b was shifted by 0.33 V to a more negative region as compared to 8c.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data of synthesized compounds. | ||
| Format: PDF | Size: 413.5 KB | Download |
References
-
Le Floch, P. Coord. Chem. Rev. 2006, 250, 627–681. doi:10.1016/j.ccr.2005.04.032
Return to citation in text: [1] -
Carmichael, D.; Mathey, F. Top. Curr. Chem. 2002, 220, 27–51. doi:10.1007/3-540-45731-3_2
Return to citation in text: [1] -
Bezkishko, I. A.; Zagidullin, A. A.; Milyukov, V. A.; Sinyashin, O. G. Russ. Chem. Rev. 2014, 83, 555–574. doi:10.1070/rc2014v083n06abeh004442
Return to citation in text: [1] -
Bai, J.; Virovets, A. V.; Scheer, M. Science 2003, 300, 781–783. doi:10.1126/science.1081119
Return to citation in text: [1] -
Peresypkina, E.; Virovets, A.; Scheer, M. Coord. Chem. Rev. 2021, 446, 213995. doi:10.1016/j.ccr.2021.213995
Return to citation in text: [1] -
Fu, G. C. Acc. Chem. Res. 2006, 39, 853–860. doi:10.1021/ar068115g
Return to citation in text: [1] -
Ogasawara, M.; Watanabe, S.; Nakajima, K.; Takahashi, T. J. Am. Chem. Soc. 2010, 132, 2136–2137. doi:10.1021/ja910348z
Return to citation in text: [1] -
Frison, G.; Mathey, F.; Sevin, A. J. Phys. Chem. A 2002, 106, 5653–5659. doi:10.1021/jp0258816
Return to citation in text: [1] -
Kostic, N. M.; Fenske, R. F. Organometallics 1983, 2, 1008–1013. doi:10.1021/om50002a012
Return to citation in text: [1] -
Willms, H.; Frank, W.; Ganter, C. Organometallics 2009, 28, 3049–3058. doi:10.1021/om8012025
Return to citation in text: [1] -
Carmichael, D.; Goldet, G.; Klankermayer, J.; Ricard, L.; Seeboth, N.; Stankevič, M. Chem. – Eur. J. 2007, 13, 5492–5502. doi:10.1002/chem.200601715
Return to citation in text: [1] -
Ogasawara, M.; Ito, A.; Yoshida, K.; Hayashi, T. Organometallics 2006, 25, 2715–2718. doi:10.1021/om060138b
Return to citation in text: [1] -
Wang, L.-S.; Hollis, T. K. Org. Lett. 2003, 5, 2543–2545. doi:10.1021/ol034816r
Return to citation in text: [1] -
Garrett, C. E.; Fu, G. C. J. Org. Chem. 1997, 62, 4534–4535. doi:10.1021/jo970419g
Return to citation in text: [1] -
Reichl, S.; Mädl, E.; Riedlberger, F.; Piesch, M.; Balázs, G.; Seidl, M.; Scheer, M. Nat. Commun. 2021, 12, 5774. doi:10.1038/s41467-021-26002-7
Return to citation in text: [1] -
Liebscher, M.; Bruhn, C.; Siemeling, U.; Baio, J.; Lu, H.; Weidner, T. Eur. J. Inorg. Chem. 2017, 351–359. doi:10.1002/ejic.201600892
Return to citation in text: [1] -
Piesch, M.; Dielmann, F.; Reichl, S.; Scheer, M. Chem. – Eur. J. 2020, 26, 1518–1524. doi:10.1002/chem.201905240
Return to citation in text: [1] -
Elsayed Moussa, M.; Welsch, S.; Dütsch, L.; Piesch, M.; Reichl, S.; Seidl, M.; Scheer, M. Molecules 2019, 24, 325. doi:10.3390/molecules24020325
Return to citation in text: [1] -
Peresypkina, E.; Heindl, C.; Virovets, A.; Brake, H.; Mädl, E.; Scheer, M. Chem. – Eur. J. 2018, 24, 2503–2508. doi:10.1002/chem.201705883
Return to citation in text: [1] -
Brake, H.; Peresypkina, E.; Heindl, C.; Virovets, A. V.; Kremer, W.; Scheer, M. Chem. Sci. 2019, 10, 2940–2944. doi:10.1039/c8sc05471a
Return to citation in text: [1] -
Schiller, J.; Peresypkina, E.; Virovets, A. V.; Scheer, M. Angew. Chem., Int. Ed. 2020, 59, 13647–13650. doi:10.1002/anie.202004988
Return to citation in text: [1] -
Ogasawara, M.; Arae, S.; Watanabe, S.; Subbarayan, V.; Sato, H.; Takahashi, T. Organometallics 2013, 32, 4997–5000. doi:10.1021/om400496b
Return to citation in text: [1] -
Escobar, A.; Mathey, F. Organometallics 2010, 29, 1053–1056. doi:10.1021/om9010164
Return to citation in text: [1] -
Masaoka, K.; Ohkubo, M.; Taue, H.; Wakioka, M.; Ohki, Y.; Ogasawara, M. ChemistrySelect 2022, 7, e202104472. doi:10.1002/slct.202104472
Return to citation in text: [1] -
Petrov, A. V.; Zagidullin, A. A.; Bezkishko, I. A.; Khrizanforov, M. N.; Kholin, K. V.; Gerasimova, T. P.; Ivshin, K. A.; Shekurov, R. P.; Katsyuba, S. A.; Kataeva, O. N.; Budnikova, Y. H.; Miluykov, V. A. Dalton Trans. 2020, 49, 17252–17262. doi:10.1039/d0dt03281f
Return to citation in text: [1] [2] -
Scherer, O. J.; Hilt, T.; Wolmershäuser, G. Angew. Chem., Int. Ed. 2000, 39, 1425–1427. doi:10.1002/(sici)1521-3773(20000417)39:8<1425::aid-anie1425>3.0.co;2-1
Return to citation in text: [1] -
Deng, S.; Schwarzmaier, C.; Zabel, M.; Nixon, J. F.; Timoshkin, A. Y.; Scheer, M. Organometallics 2009, 28, 1075–1081. doi:10.1021/om801118k
Return to citation in text: [1] -
Heindl, C.; Schindler, A.; Bodensteiner, M.; Peresypkina, E. V.; Virovets, A. V.; Scheer, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 397–403. doi:10.1080/10426507.2014.979985
Return to citation in text: [1] -
Schindler, A.; Zabel, M.; Nixon, J. F.; Scheer, M. Z. Naturforsch., B: J. Chem. Sci. 2009, 64, 1429–1437. doi:10.1515/znb-2009-11-1225
Return to citation in text: [1] -
Schindler, A.; Balázs, G.; Zabel, M.; Gröger, C.; Kalbitzer, R.; Scheer, M. C. R. Chim. 2010, 13, 1241–1248. doi:10.1016/j.crci.2010.05.003
Return to citation in text: [1] -
Milyukov, V. A.; Hey-Hawkins, E.; Sinyashin, O. G. Russ. Chem. Bull. 2007, 56, 549–551. doi:10.1007/s11172-007-0089-x
Return to citation in text: [1] -
Dielmann, F.; Merkle, R.; Heinl, S.; Scheer, M. Z. Naturforsch., B: J. Chem. Sci. 2009, 64, 3–10. doi:10.1515/znb-2009-0102
Return to citation in text: [1] -
Heinl, C.; Peresypkina, E.; Balázs, G.; Mädl, E.; Virovets, A. V.; Scheer, M. Chem. – Eur. J. 2021, 27, 7542–7548. doi:10.1002/chem.202100203
Return to citation in text: [1] -
Weber, L.; Sommer, O.; Stammler, H.-G.; Neumann, B.; Kölle, U. Chem. Ber. 1995, 128, 665–671. doi:10.1002/cber.19951280703
Return to citation in text: [1] -
Weber, L.; Kirchhoff, R.; Boese, R.; Stammler, H.-G.; Neumann, B. Organometallics 1993, 12, 731–737. doi:10.1021/om00027a024
Return to citation in text: [1] -
Maigrot, N.; Avarvari, N.; Charrier, C.; Mathey, F. Angew. Chem., Int. Ed. Engl. 1995, 34, 590–592. doi:10.1002/anie.199505901
Return to citation in text: [1] -
Bezkishko, I. A.; Zagidullin, A. A.; Khrizanforov, M. N.; Gerasimova, T. P.; Ivshin, K. A.; Kataeva, O. N.; Ganushevich, Y. S.; Miluykov, V. A.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. Front. 2022, 9, 2608–2616. doi:10.1039/d2qi00446a
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Komatsu, K.; Kitagawa, T. Chem. Rev. 2003, 103, 1371–1428. doi:10.1021/cr010011q
Return to citation in text: [1] -
Lyons, D. J. M.; Crocker, R. D.; Blümel, M.; Nguyen, T. V. Angew. Chem., Int. Ed. 2017, 56, 1466–1484. doi:10.1002/anie.201605979
Return to citation in text: [1] -
Ranga, P. K.; Ahmad, F.; Singh, G.; Tyagi, A.; Vijaya Anand, R. Org. Biomol. Chem. 2021, 19, 9541–9564. doi:10.1039/d1ob01549d
Return to citation in text: [1] -
These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service http://www.ccdc.cam.ac.uk/structures.
Return to citation in text: [1] -
Zagidullin, A. A.; Petrov, A. V.; Bezkishko, I. A.; Miluykov, V. A. Russ. Chem. Bull. 2021, 70, 1260–1268. doi:10.1007/s11172-021-3209-0
Return to citation in text: [1] -
Bezkishko, I. A.; Zagidullin, A. A.; Milyukov, V. A. Russ. Chem. Bull. 2020, 69, 435–448. doi:10.1007/s11172-020-2782-y
Return to citation in text: [1] -
Zagidullin, A. A.; Khrizanforov, M. N.; Bezkishko, I. A.; Lönnecke, P.; Hey-Hawkins, E.; Miluykov, V. A. J. Organomet. Chem. 2021, 956, 122122. doi:10.1016/j.jorganchem.2021.122122
Return to citation in text: [1] -
Winter, R. F.; Geiger, W. E. Organometallics 1999, 18, 1827–1833. doi:10.1021/om9809724
Return to citation in text: [1] [2] -
Wolf, R.; Ehlers, A. W.; Khusniyarov, M. M.; Hartl, F.; de Bruin, B.; Long, G. J.; Grandjean, F.; Schappacher, F. M.; Pöttgen, R.; Slootweg, J. C.; Lutz, M.; Spek, A. L.; Lammertsma, K. Chem. – Eur. J. 2010, 16, 14322–14334. doi:10.1002/chem.201001913
Return to citation in text: [1] -
Hierlmeier, G.; Coburger, P.; Scott, D. J.; Maier, T. M.; Pelties, S.; Wolf, R.; Pividori, D. M.; Meyer, K.; Leest, N. P.; Bruin, B. Chem. – Eur. J. 2021, 27, 14936–14946. doi:10.1002/chem.202102335
Return to citation in text: [1] -
Khrizanforov, M.; Strekalova, S.; Kholin, K.; Khrizanforova, V.; Grinenko, V.; Gryaznova, T.; Budnikova, Y. RSC Adv. 2016, 6, 42701–42707. doi:10.1039/c6ra04480h
Return to citation in text: [1] -
Butovskiy, M. V.; Balázs, G.; Bodensteiner, M.; Peresypkina, E. V.; Virovets, A. V.; Sutter, J.; Scheer, M. Angew. Chem., Int. Ed. 2013, 52, 2972–2976. doi:10.1002/anie.201209329
Return to citation in text: [1] -
Li, T.; Gamer, M. T.; Scheer, M.; Konchenko, S. N.; Roesky, P. W. Chem. Commun. 2013, 49, 2183–2185. doi:10.1039/c3cc38841g
Return to citation in text: [1]
| 45. | Winter, R. F.; Geiger, W. E. Organometallics 1999, 18, 1827–1833. doi:10.1021/om9809724 |
| 37. | Bezkishko, I. A.; Zagidullin, A. A.; Khrizanforov, M. N.; Gerasimova, T. P.; Ivshin, K. A.; Kataeva, O. N.; Ganushevich, Y. S.; Miluykov, V. A.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. Front. 2022, 9, 2608–2616. doi:10.1039/d2qi00446a |
| 49. | Butovskiy, M. V.; Balázs, G.; Bodensteiner, M.; Peresypkina, E. V.; Virovets, A. V.; Sutter, J.; Scheer, M. Angew. Chem., Int. Ed. 2013, 52, 2972–2976. doi:10.1002/anie.201209329 |
| 1. | Le Floch, P. Coord. Chem. Rev. 2006, 250, 627–681. doi:10.1016/j.ccr.2005.04.032 |
| 2. | Carmichael, D.; Mathey, F. Top. Curr. Chem. 2002, 220, 27–51. doi:10.1007/3-540-45731-3_2 |
| 10. | Willms, H.; Frank, W.; Ganter, C. Organometallics 2009, 28, 3049–3058. doi:10.1021/om8012025 |
| 11. | Carmichael, D.; Goldet, G.; Klankermayer, J.; Ricard, L.; Seeboth, N.; Stankevič, M. Chem. – Eur. J. 2007, 13, 5492–5502. doi:10.1002/chem.200601715 |
| 12. | Ogasawara, M.; Ito, A.; Yoshida, K.; Hayashi, T. Organometallics 2006, 25, 2715–2718. doi:10.1021/om060138b |
| 37. | Bezkishko, I. A.; Zagidullin, A. A.; Khrizanforov, M. N.; Gerasimova, T. P.; Ivshin, K. A.; Kataeva, O. N.; Ganushevich, Y. S.; Miluykov, V. A.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. Front. 2022, 9, 2608–2616. doi:10.1039/d2qi00446a |
| 8. | Frison, G.; Mathey, F.; Sevin, A. J. Phys. Chem. A 2002, 106, 5653–5659. doi:10.1021/jp0258816 |
| 9. | Kostic, N. M.; Fenske, R. F. Organometallics 1983, 2, 1008–1013. doi:10.1021/om50002a012 |
| 38. | Komatsu, K.; Kitagawa, T. Chem. Rev. 2003, 103, 1371–1428. doi:10.1021/cr010011q |
| 39. | Lyons, D. J. M.; Crocker, R. D.; Blümel, M.; Nguyen, T. V. Angew. Chem., Int. Ed. 2017, 56, 1466–1484. doi:10.1002/anie.201605979 |
| 40. | Ranga, P. K.; Ahmad, F.; Singh, G.; Tyagi, A.; Vijaya Anand, R. Org. Biomol. Chem. 2021, 19, 9541–9564. doi:10.1039/d1ob01549d |
| 6. | Fu, G. C. Acc. Chem. Res. 2006, 39, 853–860. doi:10.1021/ar068115g |
| 7. | Ogasawara, M.; Watanabe, S.; Nakajima, K.; Takahashi, T. J. Am. Chem. Soc. 2010, 132, 2136–2137. doi:10.1021/ja910348z |
| 31. | Milyukov, V. A.; Hey-Hawkins, E.; Sinyashin, O. G. Russ. Chem. Bull. 2007, 56, 549–551. doi:10.1007/s11172-007-0089-x |
| 32. | Dielmann, F.; Merkle, R.; Heinl, S.; Scheer, M. Z. Naturforsch., B: J. Chem. Sci. 2009, 64, 3–10. doi:10.1515/znb-2009-0102 |
| 33. | Heinl, C.; Peresypkina, E.; Balázs, G.; Mädl, E.; Virovets, A. V.; Scheer, M. Chem. – Eur. J. 2021, 27, 7542–7548. doi:10.1002/chem.202100203 |
| 3. | Bezkishko, I. A.; Zagidullin, A. A.; Milyukov, V. A.; Sinyashin, O. G. Russ. Chem. Rev. 2014, 83, 555–574. doi:10.1070/rc2014v083n06abeh004442 |
| 4. | Bai, J.; Virovets, A. V.; Scheer, M. Science 2003, 300, 781–783. doi:10.1126/science.1081119 |
| 5. | Peresypkina, E.; Virovets, A.; Scheer, M. Coord. Chem. Rev. 2021, 446, 213995. doi:10.1016/j.ccr.2021.213995 |
| 34. | Weber, L.; Sommer, O.; Stammler, H.-G.; Neumann, B.; Kölle, U. Chem. Ber. 1995, 128, 665–671. doi:10.1002/cber.19951280703 |
| 35. | Weber, L.; Kirchhoff, R.; Boese, R.; Stammler, H.-G.; Neumann, B. Organometallics 1993, 12, 731–737. doi:10.1021/om00027a024 |
| 36. | Maigrot, N.; Avarvari, N.; Charrier, C.; Mathey, F. Angew. Chem., Int. Ed. Engl. 1995, 34, 590–592. doi:10.1002/anie.199505901 |
| 19. | Peresypkina, E.; Heindl, C.; Virovets, A.; Brake, H.; Mädl, E.; Scheer, M. Chem. – Eur. J. 2018, 24, 2503–2508. doi:10.1002/chem.201705883 |
| 20. | Brake, H.; Peresypkina, E.; Heindl, C.; Virovets, A. V.; Kremer, W.; Scheer, M. Chem. Sci. 2019, 10, 2940–2944. doi:10.1039/c8sc05471a |
| 21. | Schiller, J.; Peresypkina, E.; Virovets, A. V.; Scheer, M. Angew. Chem., Int. Ed. 2020, 59, 13647–13650. doi:10.1002/anie.202004988 |
| 25. | Petrov, A. V.; Zagidullin, A. A.; Bezkishko, I. A.; Khrizanforov, M. N.; Kholin, K. V.; Gerasimova, T. P.; Ivshin, K. A.; Shekurov, R. P.; Katsyuba, S. A.; Kataeva, O. N.; Budnikova, Y. H.; Miluykov, V. A. Dalton Trans. 2020, 49, 17252–17262. doi:10.1039/d0dt03281f |
| 26. | Scherer, O. J.; Hilt, T.; Wolmershäuser, G. Angew. Chem., Int. Ed. 2000, 39, 1425–1427. doi:10.1002/(sici)1521-3773(20000417)39:8<1425::aid-anie1425>3.0.co;2-1 |
| 27. | Deng, S.; Schwarzmaier, C.; Zabel, M.; Nixon, J. F.; Timoshkin, A. Y.; Scheer, M. Organometallics 2009, 28, 1075–1081. doi:10.1021/om801118k |
| 16. | Liebscher, M.; Bruhn, C.; Siemeling, U.; Baio, J.; Lu, H.; Weidner, T. Eur. J. Inorg. Chem. 2017, 351–359. doi:10.1002/ejic.201600892 |
| 17. | Piesch, M.; Dielmann, F.; Reichl, S.; Scheer, M. Chem. – Eur. J. 2020, 26, 1518–1524. doi:10.1002/chem.201905240 |
| 18. | Elsayed Moussa, M.; Welsch, S.; Dütsch, L.; Piesch, M.; Reichl, S.; Seidl, M.; Scheer, M. Molecules 2019, 24, 325. doi:10.3390/molecules24020325 |
| 28. | Heindl, C.; Schindler, A.; Bodensteiner, M.; Peresypkina, E. V.; Virovets, A. V.; Scheer, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 397–403. doi:10.1080/10426507.2014.979985 |
| 29. | Schindler, A.; Zabel, M.; Nixon, J. F.; Scheer, M. Z. Naturforsch., B: J. Chem. Sci. 2009, 64, 1429–1437. doi:10.1515/znb-2009-11-1225 |
| 30. | Schindler, A.; Balázs, G.; Zabel, M.; Gröger, C.; Kalbitzer, R.; Scheer, M. C. R. Chim. 2010, 13, 1241–1248. doi:10.1016/j.crci.2010.05.003 |
| 15. | Reichl, S.; Mädl, E.; Riedlberger, F.; Piesch, M.; Balázs, G.; Seidl, M.; Scheer, M. Nat. Commun. 2021, 12, 5774. doi:10.1038/s41467-021-26002-7 |
| 50. | Li, T.; Gamer, M. T.; Scheer, M.; Konchenko, S. N.; Roesky, P. W. Chem. Commun. 2013, 49, 2183–2185. doi:10.1039/c3cc38841g |
| 13. | Wang, L.-S.; Hollis, T. K. Org. Lett. 2003, 5, 2543–2545. doi:10.1021/ol034816r |
| 14. | Garrett, C. E.; Fu, G. C. J. Org. Chem. 1997, 62, 4534–4535. doi:10.1021/jo970419g |
| 22. | Ogasawara, M.; Arae, S.; Watanabe, S.; Subbarayan, V.; Sato, H.; Takahashi, T. Organometallics 2013, 32, 4997–5000. doi:10.1021/om400496b |
| 23. | Escobar, A.; Mathey, F. Organometallics 2010, 29, 1053–1056. doi:10.1021/om9010164 |
| 24. | Masaoka, K.; Ohkubo, M.; Taue, H.; Wakioka, M.; Ohki, Y.; Ogasawara, M. ChemistrySelect 2022, 7, e202104472. doi:10.1002/slct.202104472 |
| 43. | Bezkishko, I. A.; Zagidullin, A. A.; Milyukov, V. A. Russ. Chem. Bull. 2020, 69, 435–448. doi:10.1007/s11172-020-2782-y |
| 44. | Zagidullin, A. A.; Khrizanforov, M. N.; Bezkishko, I. A.; Lönnecke, P.; Hey-Hawkins, E.; Miluykov, V. A. J. Organomet. Chem. 2021, 956, 122122. doi:10.1016/j.jorganchem.2021.122122 |
| 41. | These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service http://www.ccdc.cam.ac.uk/structures. |
| 42. | Zagidullin, A. A.; Petrov, A. V.; Bezkishko, I. A.; Miluykov, V. A. Russ. Chem. Bull. 2021, 70, 1260–1268. doi:10.1007/s11172-021-3209-0 |
| 48. | Khrizanforov, M.; Strekalova, S.; Kholin, K.; Khrizanforova, V.; Grinenko, V.; Gryaznova, T.; Budnikova, Y. RSC Adv. 2016, 6, 42701–42707. doi:10.1039/c6ra04480h |
| 37. | Bezkishko, I. A.; Zagidullin, A. A.; Khrizanforov, M. N.; Gerasimova, T. P.; Ivshin, K. A.; Kataeva, O. N.; Ganushevich, Y. S.; Miluykov, V. A.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. Front. 2022, 9, 2608–2616. doi:10.1039/d2qi00446a |
| 45. | Winter, R. F.; Geiger, W. E. Organometallics 1999, 18, 1827–1833. doi:10.1021/om9809724 |
| 46. | Wolf, R.; Ehlers, A. W.; Khusniyarov, M. M.; Hartl, F.; de Bruin, B.; Long, G. J.; Grandjean, F.; Schappacher, F. M.; Pöttgen, R.; Slootweg, J. C.; Lutz, M.; Spek, A. L.; Lammertsma, K. Chem. – Eur. J. 2010, 16, 14322–14334. doi:10.1002/chem.201001913 |
| 47. | Hierlmeier, G.; Coburger, P.; Scott, D. J.; Maier, T. M.; Pelties, S.; Wolf, R.; Pividori, D. M.; Meyer, K.; Leest, N. P.; Bruin, B. Chem. – Eur. J. 2021, 27, 14936–14946. doi:10.1002/chem.202102335 |
| 37. | Bezkishko, I. A.; Zagidullin, A. A.; Khrizanforov, M. N.; Gerasimova, T. P.; Ivshin, K. A.; Kataeva, O. N.; Ganushevich, Y. S.; Miluykov, V. A.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. Front. 2022, 9, 2608–2616. doi:10.1039/d2qi00446a |
| 37. | Bezkishko, I. A.; Zagidullin, A. A.; Khrizanforov, M. N.; Gerasimova, T. P.; Ivshin, K. A.; Kataeva, O. N.; Ganushevich, Y. S.; Miluykov, V. A.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. Front. 2022, 9, 2608–2616. doi:10.1039/d2qi00446a |
| 37. | Bezkishko, I. A.; Zagidullin, A. A.; Khrizanforov, M. N.; Gerasimova, T. P.; Ivshin, K. A.; Kataeva, O. N.; Ganushevich, Y. S.; Miluykov, V. A.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. Front. 2022, 9, 2608–2616. doi:10.1039/d2qi00446a |
| 25. | Petrov, A. V.; Zagidullin, A. A.; Bezkishko, I. A.; Khrizanforov, M. N.; Kholin, K. V.; Gerasimova, T. P.; Ivshin, K. A.; Shekurov, R. P.; Katsyuba, S. A.; Kataeva, O. N.; Budnikova, Y. H.; Miluykov, V. A. Dalton Trans. 2020, 49, 17252–17262. doi:10.1039/d0dt03281f |
© 2022 Zagidullin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.