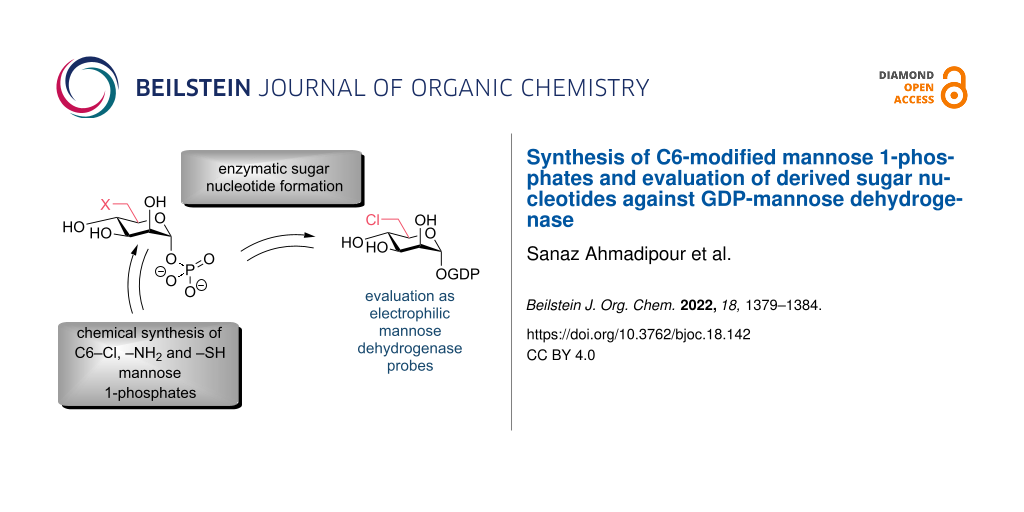
Abstract
Sufferers of cystic fibrosis are at significant risk of contracting chronic bacterial lung infections. The dominant pathogen in these cases is mucoid Pseudomonas aeruginosa. Such infections are characterised by overproduction of the exopolysaccharide alginate. We present herein the design and chemoenzymatic synthesis of sugar nucleotide tools to probe a critical enzyme within alginate biosynthesis, GDP-mannose dehydrogenase (GMD). We first synthesise C6-modified glycosyl 1-phosphates, incorporating 6-amino, 6-chloro and 6-sulfhydryl groups, followed by their evaluation as substrates for enzymatic pyrophosphorylative coupling. The development of this methodology enables access to GDP 6-chloro-6-deoxy-ᴅ-mannose and its evaluation against GMD.

Graphical Abstract
Introduction
The opportunistic Gram-negative pathogen, Pseudomonas aeruginosa (PA), becomes the dominant pathogen in patients suffering from cystic fibrosis (CF) and causes a chronic respiratory infection that infects more than 80% of those affected [1]. PA strains colonising the respiratory tract in CF undergo a phenomenon known as mucoid conversion, a phenotype characterised by the secretion of copious amounts of the carbohydrate exopolysaccharide alginate. The manifestation of alginate overproduction contributes to the PA biofilm environment and the resultant and deleterious bacterial resistance to current antibiotic treatments [2]. Alginate production is therefore established as a major virulence factor within PA respiratory tract infections for CF sufferers, and interventions that could halt its production would be of significant value.
Within the biosynthetic assembly of alginate there is a critical dependence upon the provision of one sugar nucleotide building block, GDP-mannuronic acid (GDP-ManA, 5). This material is sourced from the cytosolic metabolic pool, starting from fructose 6-phosphate and its synthesis ultimately arrives at the limiting step in alginate precursor biosynthesis, the action of GDP-mannose dehydrogenase (GMD), which oxidises GDP-mannose 1 to 5 (Figure 1a). GMD oxidation is suggested to have four discrete steps, first oxidising C6 to an aldehyde 2 followed by substrate attachment through Cys268 to form thiohemiacetal 3. A second oxidation reveals thioester 4 which, following hydrolysis, releases the product 5. As GMD does not exist in humans, strategies that could prevent its mechanism of action could open a pathway for new and selective inhibitors to disrupt bacterial alginate production [3].
![[1860-5397-18-142-1]](/bjoc/content/figures/1860-5397-18-142-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Proposed oxidative pathway for provision of GDP-ManA 5 from GDP-Man 1, C6 stereochemistry of 3 is unknown; b) previously disclosed C6–Me and C6–amido structure-to-function tools for GMD; c) C6-modified GDP-Mans of type 8 and 9, targeted in this work.
Figure 1: a) Proposed oxidative pathway for provision of GDP-ManA 5 from GDP-Man 1, C6 stereochemistry of 3 i...
We recently disclosed the first series of targeted sugar nucleotide probes for GMD (Figure 1b) [4-6]. A C6-methyl analogue 6 was oxidised by GMD with direct evidence for a ketone product obtained. Most recently, C6-amide sugar nucleotide 7 was shown to be a micromolar inhibitor of GMD (IC50 = 112 μM). Access to these chemical tools was established using a chemoenzymatic approach, whereby bespoke structural modifications were made to the mannose component, delivering an appropriate glycosyl 1-phosphate, followed by pyrophosphorylative enzymatic coupling to complete the sugar nucleotide [7,8].
With this capability in place, we wished to explore the synthesis of further tools, targeting the active site cysteine residue (Figure 1c). We envisaged that access to C6–amino or C6–sulfhydryl species of type 8 would offer prospect to establish active site thiohemiaminal (amine to imine oxidation) or disulfide formation, respectively. Additionally, C6–Cl derivative 9 could probe cysteine alkylation. Reported herein is our exploration of this synthesis and the evaluation of GDP 6-chloro-6-deoxy-ᴅ-mannose 18 against GMD.
Results and Discussion
Chemical synthesis of 6-chloro-6-deoxy- and 6-amino-6-deoxy-mannose 1-phosphates
We first required access to appropriate glycosyl 1-phosphates (as putative substrates for enzymatic pyrophosphorylative coupling) to then access the sugar nucleotide GMD probes of type 8 and 9 (Figure 1c). Accordingly, syntheses of C6-modified mannose 1-phosphates 13 and 17 were developed (Scheme 1).
![[1860-5397-18-142-i1]](/bjoc/content/inline/1860-5397-18-142-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Syntheses of C6-modified mannose 1-phosphates 13 and 17. Conditions a) PPh3, CBr4, DCM, rt, 75%; b) NaN3, DMF, 75 °C, 64%; c) HO(O)P(OBn)2, NIS, AgOTf, DCM, rt, 65%; d) Pd/C, Pd(OH)2, H2, HCl, EtOH, THF, 24 h, 90%; e) NH4OAc, DMF, 30 °C, 80%; f) n-BuLi, Cl(O)P(OPh)2, THF, −78 °C to rt, 58%; g) i) PtO2, H2, EtOH, ii) Et3N, H2O, MeOH, 99%, 2 steps.
Scheme 1: Syntheses of C6-modified mannose 1-phosphates 13 and 17. Conditions a) PPh3, CBr4, DCM, rt, 75%; b)...
The synthesis of 6-amino-6-deoxymannose 1-phosphate 13 started from protected thioglycoside 10 [6]. A two-step modification using Appel halogenation followed by nucleophilic substitution with azide furnished 11. Conversion of the intermediate C6–bromide to 11 was confirmed by 13C NMR, with C6 shifting downfield from δC 33.4 ppm to 51.6 ppm. Following this, dibenzyl phosphate was glycosylated with 11 using NIS/AgOTf activation of the thioglycoside, which proceeded in good yield (65%) to deliver 12. A final global deprotection with concomitant azide reduction was completed using hydrogen and Pd/C and Pd(OH)2/C catalysts, affording 13 as the disodium salt in 90% yield, after anion exchange chromatography.
Synthesis of 6-deoxy-6-thio-ᴅ-mannose 1-phosphate (18) was completed from 14 via C6 substitution with thioacetate and MacDonald phosphorylation [9]. Details of this synthesis were reported by us previously [10]. In this work, we attempted to use these optimised MacDonald conditions to directly access 17 from 14, but the reaction exhibited extensive degradation upon multiple attempts.
Instead we started from 14 and completed a selective anomeric deacetylation on a gram-scale using ammonium acetate in DMF, to afford hemi-acetal 15 in good yield (80%) [11]. This was followed by phosphorylation of the anomeric position using diphenylphosphoryl chloride as the phosphorous electrophile, following deprotonation of 15 using n-BuLi at −78 °C. This afforded protected anomeric phosphate 16, confirmed by 1H and 13C NMR data (H1 δH 6.01 ppm, 3JH1–P = 6.7 Hz, 3JH1–H2 = 1.8 Hz; C1 δC 96.1 ppm, 2JC1–P = 5.9 Hz). Following an initial purification of 16 using silica gel flash chromatography, the material was crystallised using a minimum volume of hot EtOH, enabling solid state analysis (Figure 2).
![[1860-5397-18-142-2]](/bjoc/content/figures/1860-5397-18-142-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of 16 with ADPs rendered at the 50% probability level. Acetyl group disorder is omitted for clarity. Atom colour scheme: carbon = black, oxygen = red, phosphorus = purple, chlorine = green, hydrogen = pink. C6–Cl is the gg rotamer (from the Cl6–C6–C5–O5 torsion angle).
Figure 2: Structure of 16 with ADPs rendered at the 50% probability level. Acetyl group disorder is omitted f...
Crystals of 16 were analysed by single crystal X-ray diffraction and the data were solved in the monoclinic space group P21. Although the crystals suffered from intrinsic non-merohedral twinning through a non-crystallographic rotation, the structural model allowed unambiguous assignment of the solid state molecular structure and absolute configuration. As shown in Figure 2, the desired axial anomeric phosphate was clearly visible alongside an interesting gg rotameric form for the C6–chloro side chain substituent (pyranose side chain conformation has recently been shown to be an important factor contributing to anomeric reactivity of canonical pyranoses [12]). One phenyl ring of the phosphate ester group occupied a position folded underneath the pyranose ring and engaged in an intramolecular C–H···π interaction with the axial C5 proton (Figure 2, dotted line). The C···π (mean plane) distance of 3.60 Å corresponded to an H···π distance of ca. 2.6 Å, consistent with the expected values for similar interactions among aryl-substituted monosaccharides [13,14]. The remaining weak interactions in the extended structure largely consist of diffuse intermolecular C–H···O contacts involving the phosphate and acetyl oxygen atoms.
Deprotection of 16 was completed in two steps, first using hydrogenolysis with Adam’s catalyst (PtO2), followed by removal of the acetate protecting groups with Et3N/H2O/MeOH, and furnished the target glycosyl 1-phosphate 17 as the sodium triethylamine salt. Overall, the synthesis of 17 was developed in 4 steps and 46% overall yield from 14. With the required C6-modified glycosyl 1-phosphates in hand, we next evaluated them as substrates for enzymatic sugar nucleotide synthesis.
Enzymatic synthesis of C6-modified sugar nucleotides
To evaluate pyrophosphorylative coupling using the C6-modified mannose 1-phosphates we selected the recombinant GDP-mannose pyrophosphorylase (GDP-Man PP) from Salmonella enterica, previously established to have a broad promiscuity for GDP-Man synthesis [4,6,15]. Glycosyl 1-phosphates 13, 17, and 18 were individually incubated with GDP-Man PP at 37 °C (Scheme 2) and the reactions monitored by TLC (iPrOH/NH4OH/H2O 6:3:1).
![[1860-5397-18-142-i2]](/bjoc/content/inline/1860-5397-18-142-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Evaluation of enzymatic GDP-Man synthesis using C6-modified mannose 1-phosphates 13, 17, and 18; Y+ = appropriate counterion.
Scheme 2: Evaluation of enzymatic GDP-Man synthesis using C6-modified mannose 1-phosphates 13, 17, and 18; Y+...
Monitoring of analytical scale reactions showed an indicative conversion for substrate 17 after 16 h; no conversion was observed after this time for 13 or 18 (see Figure S3 in Supporting Information File 1). Further analytical HPLC analysis for the formation of 19 from 17 after 16 h indicated a product had formed with a similar retention time to the control synthesis of 1; this was tentatively assigned as 19. Given this, we completed further enzymatic synthesis on a preparative scale to yield 19 in milligram quantities and in an isolated yield of 59%.
We previously demonstrated the GDP-Man-PP from S. enterica exhibited relaxed specificity towards C4 & C5 pyranose modification, although there is a fine balance to be struck between size and charge when modifying C6 [4]. Substrate 18 was known to form a disulfide in solution [10], presumably resulting in the glycosyl-1-phosphate being unable to access the enzyme active site; unfortunately, the addition of higher concentrations of reducing agent (DTT) and solid-supported PPh3 to access the reduced form for pyrophosphorylation had no positive effect. In the case of no observable reactivity for substrate 13, likely protonation of the amine would result in unfavourable positive charge within the active site and the absence of vital H-bond contacts. X-ray crystallographic data of a related Man-PP from T. maritima show Asp260 & Asp109 in coordination with Mg2+ and two water molecules in close proximity to C6 [16].
Evaluation of GDP 6-chloro-6-deoxy-ᴅ-mannose against GMD
GMD from P. aeruginosa was co-incubated with GDP-mannose 1 and sugar nucleotide 19, and the production of NADH monitored by fluorescence (excitation 355 nm; emission 460 nm). Figure 3 illustrates a reduced rate of NADH production by GMD upon incubation with both probe 19 and the native substrate 1 (blue line). No significant increase in fluorescence was observed upon incubation of probe 19 and GMD (green line). However, a moderate level of NADH production was restored upon spiking the incubation with GDP-Man 1 after 85 minutes. The rate of fluorescence increase was reduced compared to both the positive control and incubation with 19 and 1 (Figure 3, blue line). This suggests that 19 may bind to GMD, but not as a substrate. Furthermore, we extended the GMD-19 incubation time to overnight, followed by protein-MS analysis, but found no evidence of sugar nucleotide–protein conjugation; by contrast a positive control treating GMD with iodoacetamide showed multiple alkylation of the protein (see Figure S4 in Supporting Information Information File 1).
![[1860-5397-18-142-3]](/bjoc/content/figures/1860-5397-18-142-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: GMD function with probe 19 over 120 min (GMD (100 µg/mL), GDP sugars (50 µM), NAD+ (200 µM)). Dotted trace dictates expected fluorescence output following spiking with GDP-Man 1 if GMD activity fully restored (or probe 19 was not an inhibitor).
Figure 3: GMD function with probe 19 over 120 min (GMD (100 µg/mL), GDP sugars (50 µM), NAD+ (200 µM)). Dotte...
Conclusion
We have developed a synthetic approach to a small series of C6-modified mannose 1-phosphates (6-amino, 6-chloro and 6-thio) and with these tools further explored the substrate promiscuity of the mannose pyrophosphorylase from S. enterica, observing that larger (than canonical OH) chlorine is accepted. Evaluation of GDP 6-chloro-6-deoxy-ᴅ-mannose suggests that the ligand can bind to GMD, but that targeting inhibitive S-alkylation of an sp3-hybridised C6 electrophilic probe is ineffective here.
Supporting Information
| Supporting Information File 1: Detailed experimental protocols and characterisation data; spectral NMR data (1H, 13C and 31P NMR for compounds 10–17 and 19). | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Li, Z.; Kosorok, M. R.; Farrell, P. M.; Laxova, A.; West, S. E. H.; Green, C. G.; Collins, J.; Rock, M. J.; Splaingard, M. L. J. Am. Med. Assoc. 2005, 293, 581–588. doi:10.1001/jama.293.5.581
Return to citation in text: [1] -
Hodges, N. A.; Gordon, C. A. Antimicrob. Agents Chemother. 1991, 35, 2450–2452. doi:10.1128/aac.35.11.2450
Return to citation in text: [1] -
Ahmadipour, S.; Reynisson, J.; Field, R. A.; Miller, G. J. Future Med. Chem. 2022, 14, 295–298. doi:10.4155/fmc-2021-0286
Return to citation in text: [1] -
Beswick, L.; Dimitriou, E.; Ahmadipour, S.; Zafar, A.; Rejzek, M.; Reynisson, J.; Field, R. A.; Miller, G. J. ACS Chem. Biol. 2020, 15, 3086–3092. doi:10.1021/acschembio.0c00426
Return to citation in text: [1] [2] [3] -
Beswick, L.; Ahmadipour, S.; Dolan, J. P.; Rejzek, M.; Field, R. A.; Miller, G. J. Carbohydr. Res. 2019, 485, 107819. doi:10.1016/j.carres.2019.107819
Return to citation in text: [1] -
Ahmadipour, S.; Pergolizzi, G.; Rejzek, M.; Field, R. A.; Miller, G. J. Org. Lett. 2019, 21, 4415–4419. doi:10.1021/acs.orglett.9b00967
Return to citation in text: [1] [2] [3] -
Ahmadipour, S.; Miller, G. J. Carbohydr. Res. 2017, 451, 95–109. doi:10.1016/j.carres.2017.08.014
Return to citation in text: [1] -
Ahmadipour, S.; Beswick, L.; Miller, G. J. Carbohydr. Res. 2018, 469, 38–47. doi:10.1016/j.carres.2018.09.002
Return to citation in text: [1] -
MacDonald, D. L. J. Org. Chem. 1962, 27, 1107–1109. doi:10.1021/jo01050a552
Return to citation in text: [1] -
Beswick, L.; Ahmadipour, S.; Hofman, G.-J.; Wootton, H.; Dimitriou, E.; Reynisson, J.; Field, R. A.; Linclau, B.; Miller, G. J. Carbohydr. Res. 2020, 488, 107896. doi:10.1016/j.carres.2019.107896
Return to citation in text: [1] [2] -
Zhu, J.-S.; McCormick, N. E.; Timmons, S. C.; Jakeman, D. L. J. Org. Chem. 2016, 81, 8816–8825. doi:10.1021/acs.joc.6b01485
Return to citation in text: [1] -
Quirke, J. C. K.; Crich, D. ACS Catal. 2021, 11, 5069–5078. doi:10.1021/acscatal.1c00896
Return to citation in text: [1] -
Terraneo, G.; Potenza, D.; Canales, A.; Jiménez-Barbero, J.; Baldridge, K. K.; Bernardi, A. J. Am. Chem. Soc. 2007, 129, 2890–2900. doi:10.1021/ja066633g
Return to citation in text: [1] -
Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M. S.; Chakrabarti, P. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. doi:10.1039/c4cp00099d
Return to citation in text: [1] -
Zou, L.; Zheng, R. B.; Lowary, T. L. Beilstein J. Org. Chem. 2012, 8, 1219–1226. doi:10.3762/bjoc.8.136
Return to citation in text: [1] -
Pelissier, M.-C.; Lesley, S. A.; Kuhn, P.; Bourne, Y. J. Biol. Chem. 2010, 285, 27468–27476. doi:10.1074/jbc.m109.095182
Return to citation in text: [1]
| 1. | Li, Z.; Kosorok, M. R.; Farrell, P. M.; Laxova, A.; West, S. E. H.; Green, C. G.; Collins, J.; Rock, M. J.; Splaingard, M. L. J. Am. Med. Assoc. 2005, 293, 581–588. doi:10.1001/jama.293.5.581 |
| 7. | Ahmadipour, S.; Miller, G. J. Carbohydr. Res. 2017, 451, 95–109. doi:10.1016/j.carres.2017.08.014 |
| 8. | Ahmadipour, S.; Beswick, L.; Miller, G. J. Carbohydr. Res. 2018, 469, 38–47. doi:10.1016/j.carres.2018.09.002 |
| 16. | Pelissier, M.-C.; Lesley, S. A.; Kuhn, P.; Bourne, Y. J. Biol. Chem. 2010, 285, 27468–27476. doi:10.1074/jbc.m109.095182 |
| 4. | Beswick, L.; Dimitriou, E.; Ahmadipour, S.; Zafar, A.; Rejzek, M.; Reynisson, J.; Field, R. A.; Miller, G. J. ACS Chem. Biol. 2020, 15, 3086–3092. doi:10.1021/acschembio.0c00426 |
| 5. | Beswick, L.; Ahmadipour, S.; Dolan, J. P.; Rejzek, M.; Field, R. A.; Miller, G. J. Carbohydr. Res. 2019, 485, 107819. doi:10.1016/j.carres.2019.107819 |
| 6. | Ahmadipour, S.; Pergolizzi, G.; Rejzek, M.; Field, R. A.; Miller, G. J. Org. Lett. 2019, 21, 4415–4419. doi:10.1021/acs.orglett.9b00967 |
| 3. | Ahmadipour, S.; Reynisson, J.; Field, R. A.; Miller, G. J. Future Med. Chem. 2022, 14, 295–298. doi:10.4155/fmc-2021-0286 |
| 4. | Beswick, L.; Dimitriou, E.; Ahmadipour, S.; Zafar, A.; Rejzek, M.; Reynisson, J.; Field, R. A.; Miller, G. J. ACS Chem. Biol. 2020, 15, 3086–3092. doi:10.1021/acschembio.0c00426 |
| 2. | Hodges, N. A.; Gordon, C. A. Antimicrob. Agents Chemother. 1991, 35, 2450–2452. doi:10.1128/aac.35.11.2450 |
| 10. | Beswick, L.; Ahmadipour, S.; Hofman, G.-J.; Wootton, H.; Dimitriou, E.; Reynisson, J.; Field, R. A.; Linclau, B.; Miller, G. J. Carbohydr. Res. 2020, 488, 107896. doi:10.1016/j.carres.2019.107896 |
| 11. | Zhu, J.-S.; McCormick, N. E.; Timmons, S. C.; Jakeman, D. L. J. Org. Chem. 2016, 81, 8816–8825. doi:10.1021/acs.joc.6b01485 |
| 13. | Terraneo, G.; Potenza, D.; Canales, A.; Jiménez-Barbero, J.; Baldridge, K. K.; Bernardi, A. J. Am. Chem. Soc. 2007, 129, 2890–2900. doi:10.1021/ja066633g |
| 14. | Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M. S.; Chakrabarti, P. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. doi:10.1039/c4cp00099d |
| 10. | Beswick, L.; Ahmadipour, S.; Hofman, G.-J.; Wootton, H.; Dimitriou, E.; Reynisson, J.; Field, R. A.; Linclau, B.; Miller, G. J. Carbohydr. Res. 2020, 488, 107896. doi:10.1016/j.carres.2019.107896 |
| 4. | Beswick, L.; Dimitriou, E.; Ahmadipour, S.; Zafar, A.; Rejzek, M.; Reynisson, J.; Field, R. A.; Miller, G. J. ACS Chem. Biol. 2020, 15, 3086–3092. doi:10.1021/acschembio.0c00426 |
| 6. | Ahmadipour, S.; Pergolizzi, G.; Rejzek, M.; Field, R. A.; Miller, G. J. Org. Lett. 2019, 21, 4415–4419. doi:10.1021/acs.orglett.9b00967 |
| 15. | Zou, L.; Zheng, R. B.; Lowary, T. L. Beilstein J. Org. Chem. 2012, 8, 1219–1226. doi:10.3762/bjoc.8.136 |
| 6. | Ahmadipour, S.; Pergolizzi, G.; Rejzek, M.; Field, R. A.; Miller, G. J. Org. Lett. 2019, 21, 4415–4419. doi:10.1021/acs.orglett.9b00967 |
| 12. | Quirke, J. C. K.; Crich, D. ACS Catal. 2021, 11, 5069–5078. doi:10.1021/acscatal.1c00896 |
© 2022 Ahmadipour et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.