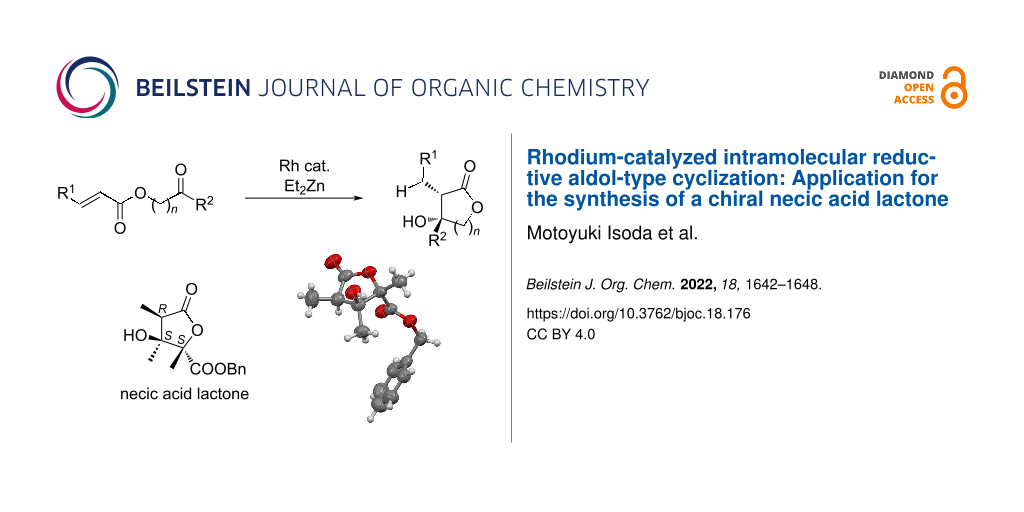
Abstract
A rhodium-catalyzed intramolecular reductive aldol-type cyclization is described to give β-hydroxylactones with high diastereoselectivities. The stereoselectivity of this cyclization is highly solvent dependent and can give syn- or anti-β-hydroxylactones with high diastereoselectivity. This methodology was also applied to the synthesis of a chiral necic acid lactone which is a structural component of the pyrrolizidine alkaloid monocrotaline.

Graphical Abstract
Introduction
Carbon–carbon bond-forming reactions are among the most important reactions in the synthetic chemistry toolbox and the aldol reaction is one of the most powerful tools to achieve this transformation [1-8]. In particular, the intramolecular aldol condensation is an important approach to the formation of ring systems such as cyclic β-hydroxy carbonyl products or cyclic α,β-unsaturated carbonyl products. Therefore, various types of intramolecular aldol-type reactions have been developed and widely applied to the total synthesis of diverse natural products [9-18]. The reductive aldol-type reaction is another important variation that has been reported using metal catalysts such as Co [19-21], Cu [22-25], and others [26-32] with hydrosilanes (R3Si-H) or hydrogen as the reductant. In this area, rhodium catalysis has received significant attention [33-40], and we have also reported reductive α-acylations, reductive aldol-type reactions, and reductive Mannich-type reactions using RhCl(PPh3)3 with Et2Zn [41-47]. The rhodium-catalyzed reductive aldol reaction of α,β-unsaturated esters with aldehydes or ketones gives aldol-type products in good to excellent yields (Scheme 1) [43,44]. In addition, the reductive aldol-type reaction could also be applied to an asymmetric system, although the diastereoselectivity was poor. On the other hand, reductive Mannich-type reactions were achieved in good to excellent yields with high diastereoselectivity [45,46]. As part of a wider program of C–C bond formation systems, we herein report a rhodium-catalyzed intramolecular reductive aldol-type cyclization and its application for the synthesis of a chiral necic acid lactone.
![[1860-5397-18-176-i1]](/bjoc/content/inline/1860-5397-18-176-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Rh-catalyzed intramolecular cyclization
When applying our previously reported conditions [43], the intramolecular reductive aldol-type cyclization of 1a proceeded smoothly and gave the desired product 2a in a good yield, but the diastereomeric ratio was not sufficient as shown in entry 1 (Table 1). To improve the diastereoselectivity of the reaction, we optimized the conditions for the reductive aldol-type reaction by intramolecular cyclization of 1, and the results are summarized in Table 1. The use of [RhCl(cod)]2 in dichloromethane gave the best result with high diastereoselectivity, and the stereochemistry of the major product 2a was found to be the syn-form with regard to the CH3 (Cα) and OH (Cβ) moieties (Table 1, entry 7). Interestingly, using the higher coordinating solvents, DMF or DMPU, preferentially gave the opposite diastereomer, i.e., the major product being the anti-form with regard to CH3 (Cα) and OH (Cβ) moieties (anti-2a, Table 1, entries 10 and 11). For stereochemistry assignment, the relative configurations of syn-2a and anti-2a were confirmed by X-ray crystallography. In addition, a NOESY experiment of the product syn-2a showed an nOe correlation between the methine proton on Cα and one of the protons of the benzene ring on Cβ, but not in anti-2a.
Table 1: Optimization of the reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-176-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Rh cat. | Solvent | Time (h) | Yielda | dr [syn:anti]b,c |
|---|---|---|---|---|---|
| 1 | RhCl(PPh3)3 | THF | 1 | 64 | [3:1] |
| 2 | [RhCl(cod)]2 | THF | 1.5 | 72 | [30:1] |
| 3 | RhClCO(PPh3)2 | THF | 1 | 85 | [9:1] |
| 4 | Rh(acac)(CO)2 | THF | 1 | trace | – |
| 5 | [RhCl(cod)]2 | toluene | 1 | 68 | [14:1] |
| 6 | [RhCl(cod)]2 | AcOEt | 1 | 79 | [25:1] |
| 7 | [RhCl(cod)]2 | CH2Cl2 | 1 | 77 | [31:1] |
| 8 | [RhCl(cod)]2 | DME | 2 | 17d | – |
| 9 | [RhCl(cod)]2 | CH3CN | 1 | 71 | [2:1] |
| 10 | [RhCl(cod)]2 | DMF | 3 | 42 | [1:27] |
| 11 | [RhCl(cod)]2 | DMPU | 1 | 52 | [1:50] |
aIsolated yield; bthe stereochemistry between CH3 (Cα) and OH (Cβ) moieties; cdiastereomeric ratio was determined after purification; ddiastereomeric mixture.
Next, various substrates were investigated and the results are summarized in Scheme 2. The synthesis of products 2a–c proceeded smoothly to give the corresponding β-hydroxylactones 2 in moderate to good yields with high diastereoselectivities, although 2d was obtained in low yield. It may suggest that the existence of substituent(s) in γ- and/or δ-position of 2 help the formation of the intermediate structure which works in favor of the intramolecular cyclization. β-Substituted substrates on α,β-unsaturated ester moiety of 1 also gave the products (2g and 2h) in low yields, but the formation of the 7-membered ring (2f) was not achieved. On the other hand, when the previous conditions using the RhCl(PPh3)3 catalyst was applied to the aldol-type cyclization, 5- and 6-membered products were obtained in good yields (see the yields and dr in parentheses in Scheme 2). However, the yields were also greatly affected by the substituents on the β-position of the α,β-unsaturated ester moiety, and all diastereomeric ratios were inferior in the case of the RhCl(PPh3)3 catalyst. The relative configurations of 2b were confirmed by X-ray crystallography, and the relative configurations of 2c, 2g, and 2h were confirmed by NOESY experiments.
![[1860-5397-18-176-i2]](/bjoc/content/inline/1860-5397-18-176-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope and limitation of the rhodium-catalyzed reductive aldol-type cyclization. aIsolated yield. bDiastereomeric ratio was determined after purification. cDiastereomeric mixture. dDiastereomeric ratio was determined by 1H NMR. eThe reaction was carried out using RhCl(PPh3)3 in THF at rt.
Scheme 2: Scope and limitation of the rhodium-catalyzed reductive aldol-type cyclization. aIsolated yield. bD...
Mechanistic investigation of the intramolecular cyclization
The reaction mechanism of the intramolecular cyclization can only be speculative at this stage. We have already reported the generation of a rhodium hydride (Rh–H) complex from RhCl(PPh3)3 and Et2Zn, in which the reaction with tert-butyl acrylate formed the corresponding E-silylenolate via 1,4-reduction at 0 °C [46], even if the reaction was performed at −45 °C (Scheme 3). Also in relation to this result, Mikami and his group reported a rhodium-catalyzed carboxylation of alkenes or activated alkenes by using CO2 with Et2Zn, and a similar Rh–H complex derived from [RhCl(cod)]2 and Et2Zn played an important role in this reaction [48]. Furthermore, Hopmann et al. detected the Rh–H complex derived from [RhCl(cod)]2 and Et2Zn by 1H NMR, and the detailed mechanism disclosed that the Rh–H complex did not interact with CO2 but with the benzene ring in the substrates through an η6 binding intermediate by DFT calculation [49].
![[1860-5397-18-176-i3]](/bjoc/content/inline/1860-5397-18-176-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Detection of metal-enolate and proposed mechanism of intramolecular cyclization.
Scheme 3: Detection of metal-enolate and proposed mechanism of intramolecular cyclization.
Although another mechanism could not be denied in which a Z-enolate intermediate changes to an E-enolate under thermodynamic control, we propose the following mechanism on the basis of the above results (Scheme 3). The Rh–H complex 5 from [RhCl(cod)]2 and Et2Zn would generate predominantly the corresponding E-enolate 6 via 1,4-reduction, which is stabilized through η6 binding with benzene ring of the substrate. Subsequent transmetalation with zinc species 4 readily reacts with the carbonyl group to form the intramolecular C−C bonds at the α-position, then providing the product syn-2a with high regioselectivity. On the other hand, the use of higher coordinating solvents such as DMF or DMPU might break the weak η6 binding of rhodium complex to give anti-2a, predominantly.
Synthesis of a chiral necic acid lactone of monocrotaline
There are several reports of bioactive natural products that have a 3-hydroxy-2-methyllactone scaffold in the molecular structure. For example, cytospolide K2 [50] containing a 10-membered lactone and feigrisolide [51] containing a 7-membered lactone are known to exhibit cytotoxicity and antimicrobial activity. Moreover, antiviral activity was also confirmed for aggregatin B [52] containing a 7-membered lactone ring, in which the β-position hydroxy group was dehydrated (Figure 1). Monocrotaline is a kind of pyrrolizidine alkaloid and was isolated from seeds of Crotalaria spectabilis in 1935 [53]. Monocrotaline is used as compound for pulmonary hypertension model in rats. To date, some groups have reported synthetic methods and its synthetic supply will potentially contribute to hypertension treatment [54-57]. Although there have been a lot of reports of pyrrolizidine scaffolds or necine base, the synthesis of necic acid lactones such as monocrotalic acid is rare (Figure 2). Consequently, we attempted to apply the rhodium-catalyzed intramolecular reductive aldol-type reaction to the synthesis of a chiral necic acid lactone that is a part of structural component of monocrotaline.
![[1860-5397-18-176-1]](/bjoc/content/figures/1860-5397-18-176-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bioactive natural products bearing a 3-hydroxy-2-methyllactone scaffold.
Figure 1: Bioactive natural products bearing a 3-hydroxy-2-methyllactone scaffold.
![[1860-5397-18-176-2]](/bjoc/content/figures/1860-5397-18-176-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Monocrotaline and its structural components.
Figure 2: Monocrotaline and its structural components.
According to the literature, a Sharpless dihydroxylation of benzyl tiglate (8) to form a chiral diol 9 was followed by a Parikh–Doering oxidation to give the corresponding product 10 in 62% yield (Scheme 4) [58,59]. Subsequent acryloylation in the presence of DMAP and hydroquinone gave the intramolecular cyclization starting material (S)-1j in 61% yield. The transformation of the compound (S)-1j in the rhodium-catalyzed intramolecular reductive aldol-type cyclization proceeded smoothly and gave the chiral necic acid lactone (2S,3S,4R)-2j in 32% yield (Figure 3).
![[1860-5397-18-176-i4]](/bjoc/content/inline/1860-5397-18-176-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthetic route towards chiral necic acid lactone (2S,3S,4R)-2j. Conditions: a) CH3SO2NH2, AD-mix-β, t-BuOH, H2O. b) SO3·Py, Et3N, DMSO, CH2Cl2. c) DMAP, CH2Cl2, Et3N, acryloyl chloride, hydroquinone. d) [RhCl(cod)]2, THF, Et2Zn.
Scheme 4: Synthetic route towards chiral necic acid lactone (2S,3S,4R)-2j. Conditions: a) CH3SO2NH2, AD-mix-β...
![[1860-5397-18-176-3]](/bjoc/content/figures/1860-5397-18-176-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure of necic acid lactone (2S,3S,4R)-2j in the crystal.
Figure 3: Molecular structure of necic acid lactone (2S,3S,4R)-2j in the crystal.
Conclusion
In conclusion, during the development of a rhodium-catalyzed intramolecular reductive cyclization, we found that using [RhCl(cod)]2 improved the diastereomeric ratio of the products compared with other Rh catalysts. It seems that using [RhCl(cod)]2 leads to milder reaction conditions that lead to highly improved diastereomeric ratios. In addition, we demonstrated a new approach to a necic acid lactone 2j that is a diastereomer of monocrotalic acid, a key intermediate of monocrotalin.
Supporting Information
| Supporting Information File 1: General procedures and analytical data, including copies of 1H NMR, 13C NMR, and X-ray crystallography. | ||
| Format: PDF | Size: 4.1 MB | Download |
References
-
Volla, C. M. R.; Atodiresei, I.; Rueping, M. Chem. Rev. 2014, 114, 2390–2431. doi:10.1021/cr400215u
Return to citation in text: [1] -
Roscales, S.; Csákÿ, A. G. Chem. Soc. Rev. 2014, 43, 8215–8225. doi:10.1039/c4cs00195h
Return to citation in text: [1] -
Thankachan, A. P.; Asha, S.; Sindhu, K. S.; Anilkumar, G. RSC Adv. 2015, 5, 62179–62193. doi:10.1039/c5ra10102f
Return to citation in text: [1] -
Brahmachari, G. RSC Adv. 2016, 6, 64676–64725. doi:10.1039/c6ra14399g
Return to citation in text: [1] -
Becker, M. R.; Watson, R. B.; Schindler, C. S. Chem. Soc. Rev. 2018, 47, 7867–7881. doi:10.1039/c8cs00391b
Return to citation in text: [1] -
Kwiatkowski, M. R.; Alexanian, E. J. Acc. Chem. Res. 2019, 52, 1134–1144. doi:10.1021/acs.accounts.9b00044
Return to citation in text: [1] -
Ye, S.; Xiang, T.; Li, X.; Wu, J. Org. Chem. Front. 2019, 6, 2183–2199. doi:10.1039/c9qo00272c
Return to citation in text: [1] -
Rej, S.; Ano, Y.; Chatani, N. Chem. Rev. 2020, 120, 1788–1887. doi:10.1021/acs.chemrev.9b00495
Return to citation in text: [1] -
Ochi, Y.; Yokoshima, S.; Fukuyama, F. Synthesis 2017, 49, 96–114. doi:10.1055/s-0036-1588878
Return to citation in text: [1] -
Xiao, Z.; Li, Y.; Gao, S. Org. Lett. 2017, 19, 1834–1837. doi:10.1021/acs.orglett.7b00592
Return to citation in text: [1] -
Yuan, P.; Liu, X.; Yang, X.; Zhang, Y.; Chen, X. J. Org. Chem. 2017, 82, 3692–3701. doi:10.1021/acs.joc.7b00181
Return to citation in text: [1] -
Wolleb, H.; Carreira, E. M. Angew. Chem., Int. Ed. 2017, 56, 10890–10893. doi:10.1002/anie.201705809
Return to citation in text: [1] -
Zhang, Q.; Zhang, Z.; Huang, Z.; Zhang, C.; Xi, S.; Zhang, M. Angew. Chem., Int. Ed. 2018, 57, 937–941. doi:10.1002/anie.201711414
Return to citation in text: [1] -
Abe, H.; Fujimaki, M.; Nakagawa, E.; Kobayashi, T.; Ito, H. Chem. Commun. 2018, 54, 6165–6168. doi:10.1039/c8cc03438a
Return to citation in text: [1] -
Kalmode, H. P.; Handore, K. L.; Rajput, R.; Shaikh, S. R.; Gonnade, R. G.; Kulkarni, K. A.; Reddy, D. S. Org. Lett. 2018, 20, 7003–7006. doi:10.1021/acs.orglett.8b02838
Return to citation in text: [1] -
Kawamoto, Y.; Ozone, D.; Kobayashi, T.; Ito, H. Org. Biomol. Chem. 2018, 16, 8477–8480. doi:10.1039/c8ob02557f
Return to citation in text: [1] -
Tsukamoto, H.; Hanada, S.; Nomura, Y.; Doi, T. J. Org. Chem. 2018, 83, 9430–9441. doi:10.1021/acs.joc.8b01075
Return to citation in text: [1] -
Kalmode, H. P.; Patil, S. S.; Handore, K. L.; Athawale, P. R.; Dandela, R.; Verma, A. K.; Basu, A.; Reddy, D. S. Eur. J. Org. Chem. 2019, 2376–2381. doi:10.1002/ejoc.201900048
Return to citation in text: [1] -
Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 2005–2008. doi:10.1246/cl.1989.2005
Return to citation in text: [1] -
Lam, H. W.; Joensuu, P. M.; Murray, G. J.; Fordyce, E. A. F.; Prieto, O.; Luebbers, T. Org. Lett. 2006, 8, 3729–3732. doi:10.1021/ol061329d
Return to citation in text: [1] -
Lumby, R. J. R.; Joensuu, P. M.; Lam, H. W. Org. Lett. 2007, 9, 4367–4370. doi:10.1021/ol701980e
Return to citation in text: [1] -
Chiu, P.; Chen, B.; Cheng, K. F. Tetrahedron Lett. 1998, 39, 9229–9232. doi:10.1016/s0040-4039(98)02130-3
Return to citation in text: [1] -
Ooi, T.; Doda, K.; Sakai, D.; Maruoka, K. Tetrahedron Lett. 1999, 40, 2133–2136. doi:10.1016/s0040-4039(99)00130-6
Return to citation in text: [1] -
Deschamp, J.; Chuzel, O.; Hannedouche, J.; Riant, O. Angew. Chem., Int. Ed. 2006, 45, 1292–1297. doi:10.1002/anie.200503791
Return to citation in text: [1] -
Kato, M.; Oki, H.; Ogata, K.; Fukuzawa, S. Synlett 2009, 1299–1302. doi:10.1055/s-0029-1216724
Return to citation in text: [1] -
Chrovian, C. C.; Montgomery, J. Org. Lett. 2007, 9, 537–540. doi:10.1021/ol063028+
Return to citation in text: [1] -
Kiyooka, S.-i.; Shimizu, A.; Torii, S. Tetrahedron Lett. 1998, 39, 5237–5238. doi:10.1016/s0040-4039(98)01030-2
Return to citation in text: [1] -
Zhao, C.-X.; Duffey, M. O.; Taylor, S. J.; Morken, J. P. Org. Lett. 2001, 3, 1829–1831. doi:10.1021/ol015859f
Return to citation in text: [1] -
Shibahara, F.; Krische, M. J. Chem. Lett. 2008, 37, 1102–1107. doi:10.1246/cl.2008.1102
Return to citation in text: [1] -
Kawakami, T.; Miyatake, M.; Shibata, I.; Baba, A. J. Org. Chem. 1996, 61, 376–379. doi:10.1021/jo951500r
Return to citation in text: [1] -
Węglarz, I.; Szewczyk, M.; Mlynarski, J. Adv. Synth. Catal. 2020, 362, 1532–1536. doi:10.1002/adsc.201901457
Return to citation in text: [1] -
Meyer, C. C.; Ortiz, E.; Krische, M. J. Chem. Rev. 2020, 120, 3721–3748. doi:10.1021/acs.chemrev.0c00053
Return to citation in text: [1] -
Revis, A.; Hilty, T. K. Tetrahedron Lett. 1987, 28, 4809–4812. doi:10.1016/s0040-4039(00)96631-0
Return to citation in text: [1] -
Taylor, S. J.; Morken, J. P. J. Am. Chem. Soc. 1999, 121, 12202–12203. doi:10.1021/ja992952e
Return to citation in text: [1] -
Emiabata-Smith, D.; McKillop, A.; Mills, C.; Motherwell, W. B.; Whitehead, A. J. Synlett 2001, 1302–1304. doi:10.1055/s-2001-16044
Return to citation in text: [1] -
Jang, H.-Y.; Huddleston, R. R.; Krische, M. J. J. Am. Chem. Soc. 2002, 124, 15156–15157. doi:10.1021/ja021163l
Return to citation in text: [1] -
Nishiyama, H.; Shiomi, T.; Tsuchiya, Y.; Matsuda, I. J. Am. Chem. Soc. 2005, 127, 6972–6973. doi:10.1021/ja050698m
Return to citation in text: [1] -
Shiomi, T.; Nishiyama, H. Org. Lett. 2007, 9, 1651–1654. doi:10.1021/ol070251d
Return to citation in text: [1] -
Bee, C.; Han, S. B.; Hassan, A.; Iida, H.; Krische, M. J. J. Am. Chem. Soc. 2008, 130, 2746–2747. doi:10.1021/ja710862u
Return to citation in text: [1] -
Shiomi, T.; Adachi, T.; Ito, J.-i.; Nishiyama, H. Org. Lett. 2009, 11, 1011–1014. doi:10.1021/ol802939u
Return to citation in text: [1] -
Sato, K.; Yamazoe, S.; Yamamoto, R.; Ohata, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Org. Lett. 2008, 10, 2405–2408. doi:10.1021/ol800660y
Return to citation in text: [1] -
Sato, K.; Isoda, M.; Ohata, S.; Morita, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Adv. Synth. Catal. 2012, 354, 510–514. doi:10.1002/adsc.201100463
Return to citation in text: [1] -
Sato, K.; Isoda, M.; Tokura, Y.; Omura, K.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Tetrahedron Lett. 2013, 54, 5913–5915. doi:10.1016/j.tetlet.2013.08.109
Return to citation in text: [1] [2] [3] -
Isoda, M.; Sato, K.; Tokura, Y.; Tarui, A.; Omote, M.; Ando, A. Chem. Pharm. Bull. 2014, 62, 956–961. doi:10.1248/cpb.c14-00223
Return to citation in text: [1] [2] -
Isoda, M.; Sato, K.; Funakoshi, M.; Omura, K.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233
Return to citation in text: [1] [2] -
Isoda, M.; Sato, K.; Kunugi, Y.; Tokonishi, S.; Tarui, A.; Omote, M.; Minami, H.; Ando, A. Beilstein J. Org. Chem. 2016, 12, 1608–1615. doi:10.3762/bjoc.12.157
Return to citation in text: [1] [2] [3] -
Sato, K.; Isoda, M.; Tarui, A.; Omote, M. Eur. J. Org. Chem. 2020, 6503–6511. doi:10.1002/ejoc.202001041
Return to citation in text: [1] -
Kawashima, S.; Aikawa, K.; Mikami, K. Eur. J. Org. Chem. 2016, 3166–3170. doi:10.1002/ejoc.201600338
Return to citation in text: [1] -
Pavlovic, L.; Vaitla, J.; Bayer, A.; Hopmann, K. H. Organometallics 2018, 37, 941–948. doi:10.1021/acs.organomet.7b00899
Return to citation in text: [1] -
Lu, S.; Sun, P.; Li, T.; Kurtán, T.; Mándi, A.; Antus, S.; Krohn, K.; Draeger, S.; Schulz, B.; Yi, Y.; Li, L.; Zhang, W. J. Org. Chem. 2011, 76, 9699–9710. doi:10.1021/jo201755v
Return to citation in text: [1] -
Tang, Y.-Q.; Sattler, I.; Thiericke, R.; Grabley, S. J. Antibiot. 2000, 53, 934–943. doi:10.7164/antibiotics.53.934
Return to citation in text: [1] -
Verdan, M. H.; Barison, A.; Lemos de Sá, E.; Salvador, M. J.; Poliquesi, C. B.; Eberlin, M. N.; Stefanello, M. É. A. J. Nat. Prod. 2010, 73, 1434–1437. doi:10.1021/np1002466
Return to citation in text: [1] -
Neal, W. M.; Rusoff, L. L.; Ahmann, C. F. J. Am. Chem. Soc. 1935, 57, 2560–2561. doi:10.1021/ja01315a073
Return to citation in text: [1] -
Vedejs, E.; Ahmad, S.; Larsen, S. D.; Westwood, S. J. Org. Chem. 1987, 52, 3937–3938. doi:10.1021/jo00226a045
Return to citation in text: [1] -
Niwa, H.; Okamoto, O.; Yamada, K. Tetrahedron Lett. 1988, 29, 5139–5142. doi:10.1016/s0040-4039(00)80702-9
Return to citation in text: [1] -
Niwa, H.; Ogawa, T.; Okamoto, O.; Yamada, K. Tetrahedron 1992, 48, 10531–10548. doi:10.1016/s0040-4020(01)88350-2
Return to citation in text: [1] -
Honda, T.; Tomitsuka, K.; Tsubuki, M. J. Org. Chem. 1993, 58, 4274–4279. doi:10.1021/jo00068a022
Return to citation in text: [1] -
Shao, H.; Rueter, J. K.; Goodman, M. J. Org. Chem. 1998, 63, 5240–5244. doi:10.1021/jo971983u
Return to citation in text: [1] -
Weber, F.; Brückner, R. Org. Lett. 2014, 16, 6428–6431. doi:10.1021/ol5032602
Return to citation in text: [1]
| 58. | Shao, H.; Rueter, J. K.; Goodman, M. J. Org. Chem. 1998, 63, 5240–5244. doi:10.1021/jo971983u |
| 59. | Weber, F.; Brückner, R. Org. Lett. 2014, 16, 6428–6431. doi:10.1021/ol5032602 |
| 53. | Neal, W. M.; Rusoff, L. L.; Ahmann, C. F. J. Am. Chem. Soc. 1935, 57, 2560–2561. doi:10.1021/ja01315a073 |
| 54. | Vedejs, E.; Ahmad, S.; Larsen, S. D.; Westwood, S. J. Org. Chem. 1987, 52, 3937–3938. doi:10.1021/jo00226a045 |
| 55. | Niwa, H.; Okamoto, O.; Yamada, K. Tetrahedron Lett. 1988, 29, 5139–5142. doi:10.1016/s0040-4039(00)80702-9 |
| 56. | Niwa, H.; Ogawa, T.; Okamoto, O.; Yamada, K. Tetrahedron 1992, 48, 10531–10548. doi:10.1016/s0040-4020(01)88350-2 |
| 57. | Honda, T.; Tomitsuka, K.; Tsubuki, M. J. Org. Chem. 1993, 58, 4274–4279. doi:10.1021/jo00068a022 |
| 1. | Volla, C. M. R.; Atodiresei, I.; Rueping, M. Chem. Rev. 2014, 114, 2390–2431. doi:10.1021/cr400215u |
| 2. | Roscales, S.; Csákÿ, A. G. Chem. Soc. Rev. 2014, 43, 8215–8225. doi:10.1039/c4cs00195h |
| 3. | Thankachan, A. P.; Asha, S.; Sindhu, K. S.; Anilkumar, G. RSC Adv. 2015, 5, 62179–62193. doi:10.1039/c5ra10102f |
| 4. | Brahmachari, G. RSC Adv. 2016, 6, 64676–64725. doi:10.1039/c6ra14399g |
| 5. | Becker, M. R.; Watson, R. B.; Schindler, C. S. Chem. Soc. Rev. 2018, 47, 7867–7881. doi:10.1039/c8cs00391b |
| 6. | Kwiatkowski, M. R.; Alexanian, E. J. Acc. Chem. Res. 2019, 52, 1134–1144. doi:10.1021/acs.accounts.9b00044 |
| 7. | Ye, S.; Xiang, T.; Li, X.; Wu, J. Org. Chem. Front. 2019, 6, 2183–2199. doi:10.1039/c9qo00272c |
| 8. | Rej, S.; Ano, Y.; Chatani, N. Chem. Rev. 2020, 120, 1788–1887. doi:10.1021/acs.chemrev.9b00495 |
| 26. | Chrovian, C. C.; Montgomery, J. Org. Lett. 2007, 9, 537–540. doi:10.1021/ol063028+ |
| 27. | Kiyooka, S.-i.; Shimizu, A.; Torii, S. Tetrahedron Lett. 1998, 39, 5237–5238. doi:10.1016/s0040-4039(98)01030-2 |
| 28. | Zhao, C.-X.; Duffey, M. O.; Taylor, S. J.; Morken, J. P. Org. Lett. 2001, 3, 1829–1831. doi:10.1021/ol015859f |
| 29. | Shibahara, F.; Krische, M. J. Chem. Lett. 2008, 37, 1102–1107. doi:10.1246/cl.2008.1102 |
| 30. | Kawakami, T.; Miyatake, M.; Shibata, I.; Baba, A. J. Org. Chem. 1996, 61, 376–379. doi:10.1021/jo951500r |
| 31. | Węglarz, I.; Szewczyk, M.; Mlynarski, J. Adv. Synth. Catal. 2020, 362, 1532–1536. doi:10.1002/adsc.201901457 |
| 32. | Meyer, C. C.; Ortiz, E.; Krische, M. J. Chem. Rev. 2020, 120, 3721–3748. doi:10.1021/acs.chemrev.0c00053 |
| 51. | Tang, Y.-Q.; Sattler, I.; Thiericke, R.; Grabley, S. J. Antibiot. 2000, 53, 934–943. doi:10.7164/antibiotics.53.934 |
| 22. | Chiu, P.; Chen, B.; Cheng, K. F. Tetrahedron Lett. 1998, 39, 9229–9232. doi:10.1016/s0040-4039(98)02130-3 |
| 23. | Ooi, T.; Doda, K.; Sakai, D.; Maruoka, K. Tetrahedron Lett. 1999, 40, 2133–2136. doi:10.1016/s0040-4039(99)00130-6 |
| 24. | Deschamp, J.; Chuzel, O.; Hannedouche, J.; Riant, O. Angew. Chem., Int. Ed. 2006, 45, 1292–1297. doi:10.1002/anie.200503791 |
| 25. | Kato, M.; Oki, H.; Ogata, K.; Fukuzawa, S. Synlett 2009, 1299–1302. doi:10.1055/s-0029-1216724 |
| 52. | Verdan, M. H.; Barison, A.; Lemos de Sá, E.; Salvador, M. J.; Poliquesi, C. B.; Eberlin, M. N.; Stefanello, M. É. A. J. Nat. Prod. 2010, 73, 1434–1437. doi:10.1021/np1002466 |
| 19. | Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 2005–2008. doi:10.1246/cl.1989.2005 |
| 20. | Lam, H. W.; Joensuu, P. M.; Murray, G. J.; Fordyce, E. A. F.; Prieto, O.; Luebbers, T. Org. Lett. 2006, 8, 3729–3732. doi:10.1021/ol061329d |
| 21. | Lumby, R. J. R.; Joensuu, P. M.; Lam, H. W. Org. Lett. 2007, 9, 4367–4370. doi:10.1021/ol701980e |
| 49. | Pavlovic, L.; Vaitla, J.; Bayer, A.; Hopmann, K. H. Organometallics 2018, 37, 941–948. doi:10.1021/acs.organomet.7b00899 |
| 9. | Ochi, Y.; Yokoshima, S.; Fukuyama, F. Synthesis 2017, 49, 96–114. doi:10.1055/s-0036-1588878 |
| 10. | Xiao, Z.; Li, Y.; Gao, S. Org. Lett. 2017, 19, 1834–1837. doi:10.1021/acs.orglett.7b00592 |
| 11. | Yuan, P.; Liu, X.; Yang, X.; Zhang, Y.; Chen, X. J. Org. Chem. 2017, 82, 3692–3701. doi:10.1021/acs.joc.7b00181 |
| 12. | Wolleb, H.; Carreira, E. M. Angew. Chem., Int. Ed. 2017, 56, 10890–10893. doi:10.1002/anie.201705809 |
| 13. | Zhang, Q.; Zhang, Z.; Huang, Z.; Zhang, C.; Xi, S.; Zhang, M. Angew. Chem., Int. Ed. 2018, 57, 937–941. doi:10.1002/anie.201711414 |
| 14. | Abe, H.; Fujimaki, M.; Nakagawa, E.; Kobayashi, T.; Ito, H. Chem. Commun. 2018, 54, 6165–6168. doi:10.1039/c8cc03438a |
| 15. | Kalmode, H. P.; Handore, K. L.; Rajput, R.; Shaikh, S. R.; Gonnade, R. G.; Kulkarni, K. A.; Reddy, D. S. Org. Lett. 2018, 20, 7003–7006. doi:10.1021/acs.orglett.8b02838 |
| 16. | Kawamoto, Y.; Ozone, D.; Kobayashi, T.; Ito, H. Org. Biomol. Chem. 2018, 16, 8477–8480. doi:10.1039/c8ob02557f |
| 17. | Tsukamoto, H.; Hanada, S.; Nomura, Y.; Doi, T. J. Org. Chem. 2018, 83, 9430–9441. doi:10.1021/acs.joc.8b01075 |
| 18. | Kalmode, H. P.; Patil, S. S.; Handore, K. L.; Athawale, P. R.; Dandela, R.; Verma, A. K.; Basu, A.; Reddy, D. S. Eur. J. Org. Chem. 2019, 2376–2381. doi:10.1002/ejoc.201900048 |
| 50. | Lu, S.; Sun, P.; Li, T.; Kurtán, T.; Mándi, A.; Antus, S.; Krohn, K.; Draeger, S.; Schulz, B.; Yi, Y.; Li, L.; Zhang, W. J. Org. Chem. 2011, 76, 9699–9710. doi:10.1021/jo201755v |
| 45. | Isoda, M.; Sato, K.; Funakoshi, M.; Omura, K.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233 |
| 46. | Isoda, M.; Sato, K.; Kunugi, Y.; Tokonishi, S.; Tarui, A.; Omote, M.; Minami, H.; Ando, A. Beilstein J. Org. Chem. 2016, 12, 1608–1615. doi:10.3762/bjoc.12.157 |
| 46. | Isoda, M.; Sato, K.; Kunugi, Y.; Tokonishi, S.; Tarui, A.; Omote, M.; Minami, H.; Ando, A. Beilstein J. Org. Chem. 2016, 12, 1608–1615. doi:10.3762/bjoc.12.157 |
| 43. | Sato, K.; Isoda, M.; Tokura, Y.; Omura, K.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Tetrahedron Lett. 2013, 54, 5913–5915. doi:10.1016/j.tetlet.2013.08.109 |
| 44. | Isoda, M.; Sato, K.; Tokura, Y.; Tarui, A.; Omote, M.; Ando, A. Chem. Pharm. Bull. 2014, 62, 956–961. doi:10.1248/cpb.c14-00223 |
| 48. | Kawashima, S.; Aikawa, K.; Mikami, K. Eur. J. Org. Chem. 2016, 3166–3170. doi:10.1002/ejoc.201600338 |
| 41. | Sato, K.; Yamazoe, S.; Yamamoto, R.; Ohata, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Org. Lett. 2008, 10, 2405–2408. doi:10.1021/ol800660y |
| 42. | Sato, K.; Isoda, M.; Ohata, S.; Morita, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Adv. Synth. Catal. 2012, 354, 510–514. doi:10.1002/adsc.201100463 |
| 43. | Sato, K.; Isoda, M.; Tokura, Y.; Omura, K.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Tetrahedron Lett. 2013, 54, 5913–5915. doi:10.1016/j.tetlet.2013.08.109 |
| 44. | Isoda, M.; Sato, K.; Tokura, Y.; Tarui, A.; Omote, M.; Ando, A. Chem. Pharm. Bull. 2014, 62, 956–961. doi:10.1248/cpb.c14-00223 |
| 45. | Isoda, M.; Sato, K.; Funakoshi, M.; Omura, K.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233 |
| 46. | Isoda, M.; Sato, K.; Kunugi, Y.; Tokonishi, S.; Tarui, A.; Omote, M.; Minami, H.; Ando, A. Beilstein J. Org. Chem. 2016, 12, 1608–1615. doi:10.3762/bjoc.12.157 |
| 47. | Sato, K.; Isoda, M.; Tarui, A.; Omote, M. Eur. J. Org. Chem. 2020, 6503–6511. doi:10.1002/ejoc.202001041 |
| 33. | Revis, A.; Hilty, T. K. Tetrahedron Lett. 1987, 28, 4809–4812. doi:10.1016/s0040-4039(00)96631-0 |
| 34. | Taylor, S. J.; Morken, J. P. J. Am. Chem. Soc. 1999, 121, 12202–12203. doi:10.1021/ja992952e |
| 35. | Emiabata-Smith, D.; McKillop, A.; Mills, C.; Motherwell, W. B.; Whitehead, A. J. Synlett 2001, 1302–1304. doi:10.1055/s-2001-16044 |
| 36. | Jang, H.-Y.; Huddleston, R. R.; Krische, M. J. J. Am. Chem. Soc. 2002, 124, 15156–15157. doi:10.1021/ja021163l |
| 37. | Nishiyama, H.; Shiomi, T.; Tsuchiya, Y.; Matsuda, I. J. Am. Chem. Soc. 2005, 127, 6972–6973. doi:10.1021/ja050698m |
| 38. | Shiomi, T.; Nishiyama, H. Org. Lett. 2007, 9, 1651–1654. doi:10.1021/ol070251d |
| 39. | Bee, C.; Han, S. B.; Hassan, A.; Iida, H.; Krische, M. J. J. Am. Chem. Soc. 2008, 130, 2746–2747. doi:10.1021/ja710862u |
| 40. | Shiomi, T.; Adachi, T.; Ito, J.-i.; Nishiyama, H. Org. Lett. 2009, 11, 1011–1014. doi:10.1021/ol802939u |
| 43. | Sato, K.; Isoda, M.; Tokura, Y.; Omura, K.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Tetrahedron Lett. 2013, 54, 5913–5915. doi:10.1016/j.tetlet.2013.08.109 |
© 2022 Isoda et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.