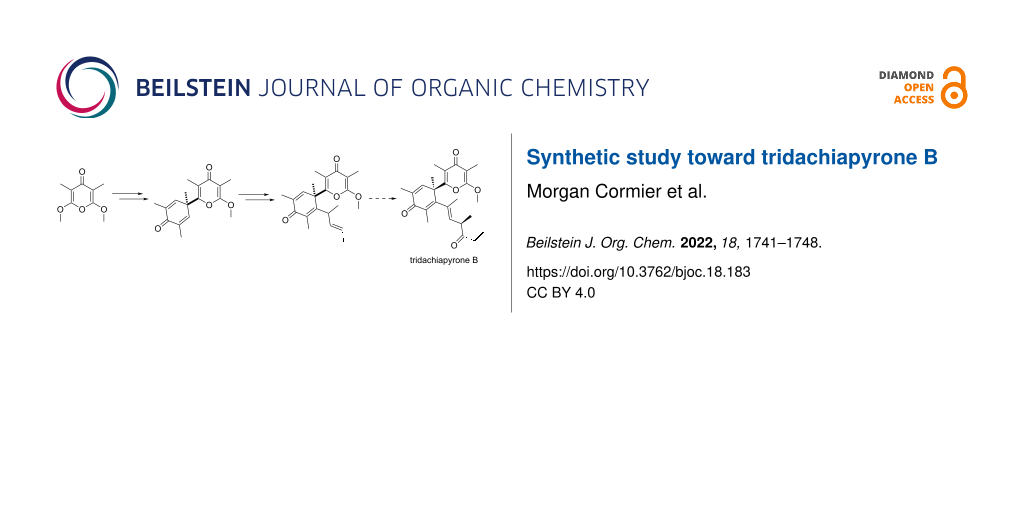
Abstract
A convergent approach to the skeleton of tridachiapyrone B is described taking advantage of the desymmetrization of α,α’-dimethoxy-γ-pyrone leading to α-crotyl-α’-methoxy-γ-pyrone in one step. To construct the quaternary carbon of the 2,5-cyclohexadienone of the target, a strategy based on the Robinson-type annulation of an aldehyde derived from α-crotyl-α’-methoxy-γ-pyrone was applied. The grafting of the simplified target’s side chain was demonstrated through an oxidative anionic oxy-Cope rearrangement of the tertiary alcohol arising from the 1,2-addition of a 1,3-dimethylallyl reagent to 2,5-cyclohexadienone connected to the α’-methoxy-γ-pyrone motif.

Graphical Abstract
Introduction
The α’-methoxy-γ-pyrone motif is present in natural products and bioactive molecules [1-14]. Amidst these targets, a number contains a quaternary carbon vicinal to the scaffold, such as crispatene and photodeoxytridachione (Scheme 1a) [15]. These molecules feature a bicyclo[3.1.0]hexene core which photochemically arises from 1,3-cyclohexadiene precursors, tridachiapyrone A or 9,10-deoxytridachione, as demonstrated by Ireland [16,17]. In turn, the ring system arises from α-tetraenyl-α’-methoxy-γ-pyrone precursor 1a upon heating through 6π-electrocyclization, as illustrated by Baldwin in the synthesis of 9,10-deoxytridachione [18]. In a further demonstration of the versatility of tetraenes connected to α’-methoxy-γ-pyrone, the synthesis of both crispatene and photodeoxytridachione was accomplished by Trauner through the Lewis acid-catalyzed 6π-disrotatory electrocyclization of compounds 1a and b [19-22]. Interestingly, Baldwin and Moses demonstrated the irradiation or sunlight-promoted cycloisomerization of a similar tetraenyl framework into the bicyclo[3.1.0]hexane core through a 6π-conrotatory stereocontrol [23,24].
![[1860-5397-18-183-i1]](/bjoc/content/inline/1860-5397-18-183-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Routes to crispatene, photodeoxytridachione, aureothin, and tridachiapyrone B.
Scheme 1: Routes to crispatene, photodeoxytridachione, aureothin, and tridachiapyrone B.
To date, the known strategies to install a quaternary carbon center connected to α’-methoxy-γ-pyrone therefore rely exclusively on the electrocyclization of tetraenes. With recently demonstrated potent antitumoral [25] and anti-HIV properties [26], aureothin is a natural product featuring the α’-methoxy-γ-pyrone motif connected to a chiral tetrahydrofuran (Scheme 1b). To assemble the skeleton of the natural product, we developed a new strategy in which the α,α’-dimethoxy-γ-pyrone motif 2 was first desymmetrized by a sequence encompassing the conjugate addition of 2-lithio-1,3-dithiane, elimination of methoxide lithium, and deprotonation of 2-(α’-methoxy-γ-pyrone)-1,3-dithiane. The resulting vinylogous enolate intermediate was trapped with the electrophile 3, amounting to the one-pot preparation of compound 4, having a masked carbonyl function connecting both key fragments [27,28].
Isolated and characterized by Schmitz [17], the metabolite tridachiapyrone B is related to tridachiapyrone A (Scheme 1c). As the 1,3-cyclohexadiene motif of the latest is oxidized into 2,5-cyclohexadienone, it is assumed that tridachiapyrone B arises from the ring opening of the epoxide (tridachiapyrone C) of tridachiapyrone A. To our knowledge, the synthesis of tridachiapyrone B was not investigated, even though it seems conceivable that the compound’s stability to light, air or cytochrome would be higher than its 1,3-cyclohexadiene counterpart. As the molecule combines electron acceptor functions such as α’-methoxy-γ-pyrone and 2,5-cyclohexadienone, these considerations are advantageous with a view to evaluate the potential activity of tridachiapyrone B and analogues in biological electron transport processes.
With this context in mind, we sought to establish a straightforward access to the key 2,5-cyclohexadienone core connected to α’-methoxy-γ-pyrone by desymmetrization of α,α’-dimethoxy-γ-pyrone 2 through the addition of hindered nucleophiles to construct the vicinal quaternary carbon. In a subsequent and potentially enantioselective desymmetrization step, compound 5 would be converted into trichiachiapyrone B by 1,4-addition of the side chain to the 2,5-cyclohexadienone scaffold. Avoiding heat and light sensitive tetraenes, the convergent plan would also give the opportunity to assess an enantioselective synthesis of the targets, noting that the C14 epimeric product, isotridachiapyrone B, has also been isolated by Schmitz.
Results and Discussion
At first glance, the structure of 2,5-cyclohexadienone 5 suggests a disconnection involving the dearomative addition of 2,4,6-trimethylphenol to α,α’-dimethoxy-γ-pyrone 2 that we eagerly sought to establish under basic activation of the nucleophile or by protonation of 2 (Scheme 2a). This ambitious coupling, however, met a dead-end and a less direct approach was explored. With a more reactive and less hindered nucleophile, we explored the coupling of lithiocyclopentadiene to compound 2. After conjugate addition and elimination of lithium methoxide, the resulting 6a would be deprotonated by lithiocyclopentadiene and the enolate intercepted with an alkylating reagent to build the quaternary carbon of 6b (Scheme 2b). This one-pot procedure was reminiscent of our previous study describing the addition of an allylic carbanion, generated from allylstannane with n-BuLi, to 2 which was followed by the addition of an aldehyde resulting in a regioselective aldolization [29]. Thereafter, we hypothesized converting cyclopentadiene 6b into 2,5-cyclohexadienone 5 by a sequence involving the oxidation into dialdehyde 7 and treatment with pentan-3-one to enable sequential steps of aldolization and crotonization. As an aromatic carbanion, it was unclear whether the addition of lithiocyclopentadiene to 2 would succeed [30].
![[1860-5397-18-183-i2]](/bjoc/content/inline/1860-5397-18-183-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Pleasingly though, the coupling was successful and moreover simply implemented by reacting lithiocyclopentadiene (2 equiv) with pyrone 2 at room temperature, no reaction occurring at lower temperature in contrast with the nucleophile 2-lithio-1,3-dithiane, and with acetic acid as electrophile (Scheme 3). Among the possible isomers that can be expected, a single one 6a’ was isolated in 49% yield after trituration, as it was found rather unstable on silica gel. While the addition of more reactive carbanions of 1,3-dithiane [27], allyl [27], and methyldienylbenzene groups [31] to compound 2 were demonstrated, this result amounts to the first grafting of an aromatic nucleophile to the motif.
![[1860-5397-18-183-i3]](/bjoc/content/inline/1860-5397-18-183-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Addition of lithiocyclopentadiene to pyrone 2.
Scheme 3: Addition of lithiocyclopentadiene to pyrone 2.
The construction of the vicinal quaternary carbon of 6b was next investigated by methylation of the highly delocalized enolate intermediate 6a·Li. To that end, the addition of lithiocyclopentadiene to 2 was followed by the attendant quenching with a methylating reagent (MeI, Me2SO4, MeOTf) but a complex mixture of products was consistently obtained. To gather information on the reactivity of 6a·Li, the stabilized enolate was treated with 4-nitrobenzaldehyde to promote the aldolization reaction but the corresponding alcohol was not observed, which confirmed the reluctance of 6a·Li to react with other electrophiles than protons.
Unable to form the quaternary carbon of the target from this intermediate, the pathway to 2,5-cyclohexanedione 5 was accordingly updated and an approach to make use of the Robinson-type annulation was devised from aldehyde 9, prepared by oxidation of α-crotyl-α’-methoxy-γ-pyrone 8 (Scheme 4). While its synthesis was initially investigated by the coupling of tri(n-butyl)crotylstannane to 2 in the presence of n-BuLi, the direct transfer of the crotyl appendage failed, likely compromised by the steric hindrance of the nucleophile [31].
![[1860-5397-18-183-i4]](/bjoc/content/inline/1860-5397-18-183-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Plan to reach 2,5-cyclohexadienone 5.
Scheme 4: Plan to reach 2,5-cyclohexadienone 5.
To alleviate this shortcoming, the crotyl appendage of 8 was assembled by methylation of the allylic pyrone counterpart (Scheme 5). Implemented by exposing 2 to the combination of reagents tri(n-butyl)allylstannane/n-BuLi (2 equiv) followed by the treatment with MeI, the crotyl derivative 8 was directly isolated in 55–60% yield (5 g scale) via the regioselective methylation of the intermediate vinylogous enolate 10·Li. The preparation of aldehyde 9 was carried out through a two-step sequence including dihydroxylation (K2OsO4·H2O, 90% yield) of 8 and oxidative cleavage (NaIO4, 91% yield) of the diol intermediate. Note that both ozonolysis and the one-pot Lemieux–Johnson oxidative cleavage process of 8 led instead to methyl ketone 11 in a significant amount (ca. 75% yield), probably by oxidation of the enol form of 9 [32]. The sensitive aldehyde was thus used without purification to investigate the Robinson-type annulation and a protocol was identified allowing the preparation of cyclohexenone 12 in 40% yield (2-step). Accordingly, the Michael addition of ethyl vinyl ketone (EVK), promoted by K2CO3 in a biphasic media (PhMe/H2O), was followed by basic treatment (LiOH) of the keto aldehyde. Since compound 12 bears the desired quaternary carbon of this family of natural products, it was pleasing to reach this milestone, keeping in mind that the fair yield of this two-step transformation may be the consequence of the aldehyde’s low stability.
![[1860-5397-18-183-i5]](/bjoc/content/inline/1860-5397-18-183-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of 2,5-cyclohexadienone 5.
Scheme 5: Preparation of 2,5-cyclohexadienone 5.
Note that the Kotsuki method to perform the Robinson-type annulation of 9 with EVK, catalyzed by a combination of 1,2-cyclohexanediamine and 1,2-cyclohexanedicarboxylic acid, led to product 12 with higher yield but the scale-up of the reaction was less efficient [33]. It has to be underlined that all attempts, using either methods, to couple aldehyde 9 with 2-methylpent-1-en-3-one and directly produce 13 were unsuccessful. To achieve this goal a methylation step of 12 (LiN(iPr)2 (LDA); MeI) was thus necessary (70% yield, 2:1 dr).
The desaturation of the enone compound was next examined and while exposure of 13 to oxidant (o-iodoxybenzoic acid (IBX) or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)) left the starting materials unchanged, treatment with NaH in the presence of oxygen to induce the aerobic oxidation caused instead the degradation of 13. An indirect approach was more rewarding as treatment of the enolate of 13 with PhSeBr led to 2,5-cyclohexadienone 5 in 51% yield after oxidation of the selenoether intermediate with m-chloroperoxybenzoic acid (m-CPBA). An appreciable increase of the yield was actually noted with (PhSe)2 as electrophile, 5 being obtained in 62% yield, enabling thus an evaluation of the next desymmetrization step.
An overview of the scientific literature revealed that, while the asymmetric desymmetrization of prochiral 2,5-cyclohexadienones is a rich topic of investigation, it is mostly restricted to substrates bearing a tertiary alkoxy group [34]. Few examples of this intermolecular strategy actually involve substrates containing a quaternary carbon and the formation of a C–C bond [35,36]. Of note was the report of Takemoto and Iwata describing the 1,4-addition of AlMe3 to 4,4-dimethyl-2,5-cyclohexadienone in the presence of a copper salt/chiral ligand and silylating reagent [37,38]. The racemic conjugate addition of nucleophiles to 5 was first investigated, starting with the Gilman reagent which was used in Takemoto and Iwata study (Scheme 6). In addition, a screening of various organocopper reagents (prepared from MeLi, EtMgBr, ZnEt2 or AlMe3 and copper halide or thiophene-2-carboxylate (CuTC)) was conducted, to no avail. In most cases, the starting material was recovered without indication that the pyrone ring interacted instead with the reagent. To decrease the steric hindrance of the electrophile and potentially facilitate the 1,4-addition, 2,5-cyclohexadienone 14, deprived of a methyl group, was prepared and tested but the reactivity was not significantly improved compared to 5. Both compounds appeared thus mostly reluctant to 1,4-addition and, when it was not the case, the process was poorly reproducible.
![[1860-5397-18-183-i6]](/bjoc/content/inline/1860-5397-18-183-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Attempts to perform the conjugate addition.
Scheme 6: Attempts to perform the conjugate addition.
On the other hand, the 1,2-addition of Grignard reagents to 5 was observed, providing thus an alternative way of grafting a side chain. As summarized in Scheme 7a, this was envisaged through a sequence encompassing the 1,2-addition of the 1,3-dimethylallyl motif to 5 giving 17, followed by the anionic oxy-Cope rearrangement of the dienol into cyclohexenone 18. After desaturation, the resulting 2,5-cyclohexadienone 19 would provide a modular platform to construct the side chain of the target and analogues. Note that this updated route required the 1,2-addition of a rather hindered nucleophile to a carbonyl electrophile with low reactivity. Precedents were noted though, as Carreira used this strategy for the synthesis of indoxamycine B exploiting the reactivity of 1,3-dimethylallyltitanocene species [39]. The 1,2-addition of various allyltitanocene reagents to carbonyl compounds was described by Sato which were prepared from the corresponding allyl carbonates exposed to the combination of [Cp2TiCl2]/n-BuLi [40]. Pleasingly, conducting the coupling of carbonate 20 to 5 in conditions inspired from Carreira’s study (Scheme 7b) led to the desired 1,2-adduct 17 in 50% yield [41].
![[1860-5397-18-183-i7]](/bjoc/content/inline/1860-5397-18-183-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Updated route to tridachiapyrone B.
Scheme 7: Updated route to tridachiapyrone B.
To perform the anionic oxy-Cope rearrangement, alcohol 17 was exposed to t-BuOK, in the presence of 18-crown-6 ether (−78 °C to rt) [42].
However, these conditions did not trigger the rearrangement and the starting material was recovered. On the other hand, treatment with KH in DMSO at room temperature caused the degradation of 17. Scarcely examined for this purpose, hydroxide of quaternary ammonium salt was next evaluated to promote the anionic oxy-Cope rearrangement with the prospect that non-coordinating organic cations could facilitate the transformation by destabilizing the negative charge of the anion (Scheme 7c). Simply generated with Triton B (benzyltrimethylammonium hydroxide), the alcoholate of 17 smoothly (−20 °C to rt) underwent the [3,3]-sigmatropic rearrangement (75% conversion) directly affording 2,5-cyclohexadienone E-19 which was isolated in 41% yield [43] while cyclohexenone 18 was not observed in the crude reaction mixture (as analyzed by 1H NMR spectroscopy). Even though the mechanism was not investigated, the presence of oxygen during the rearrangement step was suspected to account for the oxidation of the enolate intermediate, enabling thus a practical one-pot preparation of 2,5-cyclohexadienone 19 from 17.
Conclusion
While the construction of the side chain’s target remains to be completed, we developed a convergent access to a highly substituted 2,5-cyclohexadienone motif bearing a quaternary carbon connected to α’-methoxy-γ-pyrone, simplified analogues of tridachiapyrone B such as 19. Without requiring the construction of polyenes, the route is enabled by key transformations such as the formal crotylation of α,α’-dimethoxy-γ-pyrone, the Robinson-type annulation of a sensitive aldehyde and 1,2-addition to 2,5-cyclohexadienone followed by an oxidative anionic oxy-Cope rearrangement promoted by hydroxide of quaternary ammonium salt. Moreover, the coupling of lithiocyclopentadiene to α,α’-dimethoxy-γ-pyrone was demonstrated, enlarging the scope of nucleophiles grafted to the pharmaceutically relevant α’-methoxy-γ-pyrone motif. Drawing from this work, future studies will be focused on an enantioselective access to the target.
Supporting Information
| Supporting Information File 1: Experimental details, 1H and 13C spectra of new compounds. | ||
| Format: PDF | Size: 3.4 MB | Download |
References
-
Sharma, P.; Powell, K.; Burnley, J.; Awaad, A. S.; Moses, J. E. Synthesis 2011, 2865–2892. doi:10.1055/s-0030-1260168
Return to citation in text: [1] -
Miller, A. K.; Trauner, D. Synlett 2006, 2295–2316. doi:10.1055/s-2006-949627
Return to citation in text: [1] -
Ui, H.; Shiomi, K.; Suzuki, H.; Hatano, H.; Morimoto, H.; Yamaguchi, Y.; Masuma, R.; Sunazuka, T.; Shimamura, H.; Sakamoto, K.; Kita, K.; Miyoshi, H.; Tomoda, H.; Ōmura, S. J. Antibiot. 2006, 59, 785–790. doi:10.1038/ja.2006.103
Return to citation in text: [1] -
Wilk, W.; Waldmann, H.; Kaiser, M. Bioorg. Med. Chem. 2009, 17, 2304–2309. doi:10.1016/j.bmc.2008.11.001
Return to citation in text: [1] -
Ishibashi, Y.; Ohba, S.; Nishiyama, S.; Yamamura, S. Tetrahedron Lett. 1996, 37, 2997–3000. doi:10.1016/0040-4039(96)00483-2
Return to citation in text: [1] -
Garey, D.; Ramirez, M.-l.; Gonzales, S.; Wertsching, A.; Tith, S.; Keefe, K.; Peña, M. R. J. Org. Chem. 1996, 61, 4853–4856. doi:10.1021/jo960221g
Return to citation in text: [1] -
Manzo, E.; Ciavatta, M. L.; Gavagnin, M.; Mollo, E.; Wahidulla, S.; Cimino, G. Tetrahedron Lett. 2005, 46, 465–468. doi:10.1016/j.tetlet.2004.11.085
Return to citation in text: [1] -
Kim, Y.; Ogura, H.; Akasaka, K.; Oikawa, T.; Matsuura, N.; Imada, C.; Yasuda, H.; Igarashi, Y. Mar. Drugs 2014, 12, 4110–4125. doi:10.3390/md12074110
Return to citation in text: [1] -
Inuzuka, T.; Yamamoto, K.; Iwasaki, A.; Ohno, O.; Suenaga, K.; Kawazoe, Y.; Uemura, D. Tetrahedron Lett. 2014, 55, 6711–6714. doi:10.1016/j.tetlet.2014.10.032
Return to citation in text: [1] -
Fujimaki, T.; Saito, S.; Imoto, M. J. Antibiot. 2017, 70, 328–330. doi:10.1038/ja.2016.162
Return to citation in text: [1] -
Wu, T.; Salim, A. A.; Bernhardt, P. V.; Capon, R. J. J. Nat. Prod. 2021, 84, 474–482. doi:10.1021/acs.jnatprod.0c01343
Return to citation in text: [1] -
Leiris, S. J.; Khdour, O. M.; Segerman, Z. J.; Tsosie, K. S.; Chapuis, J.-C.; Hecht, S. M. Bioorg. Med. Chem. 2010, 18, 3481–3493. doi:10.1016/j.bmc.2010.03.070
Return to citation in text: [1] -
Lin, Z.; Torres, J. P.; Ammon, M. A.; Marett, L.; Teichert, R. W.; Reilly, C. A.; Kwan, J. C.; Hughen, R. W.; Flores, M.; Tianero, M. D.; Peraud, O.; Cox, J. E.; Light, A. R.; Villaraza, A. J. L.; Haygood, M. G.; Concepcion, G. P.; Olivera, B. M.; Schmidt, E. W. Chem. Biol. 2013, 20, 73–81. doi:10.1016/j.chembiol.2012.10.019
Return to citation in text: [1] -
De Paolis, M. Targets Heterocycl. Syst. 2016, 20, 63–84.
Return to citation in text: [1] -
Ireland, C.; Faulkner, J. Tetrahedron 1981, 37, 233–240. doi:10.1016/0040-4020(81)85059-4
Return to citation in text: [1] -
Ireland, C.; Scheuer, P. J. Science 1979, 205, 922–923. doi:10.1126/science.205.4409.922
Return to citation in text: [1] -
Ksebati, M. B.; Schmitz, F. J. J. Org. Chem. 1985, 50, 5637–5642. doi:10.1021/jo00350a042
Return to citation in text: [1] [2] -
Moses, J. E.; Adlington, R. M.; Rodriguez, R.; Eade, S. J.; Baldwin, J. E. Chem. Commun. 2005, 1687–1689. doi:10.1039/b418988d
Return to citation in text: [1] -
Miller, A. K.; Trauner, D. Angew. Chem., Int. Ed. 2003, 42, 549–552. doi:10.1002/anie.200390158
Return to citation in text: [1] -
Miller, A. K.; Byun, D. H.; Beaudry, C. M.; Trauner, D. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12019–12023. doi:10.1073/pnas.0401787101
Return to citation in text: [1] -
Beaudry, C. M.; Malerich, J. P.; Trauner, D. Chem. Rev. 2005, 105, 4757–4778. doi:10.1021/cr0406110
Return to citation in text: [1] -
Zuidema, D. R.; Miller, A. K.; Trauner, D.; Jones, P. B. Org. Lett. 2005, 7, 4959–4962. doi:10.1021/ol051887c
Return to citation in text: [1] -
Brückner, S.; Baldwin, J. E.; Moses, J.; Adlington, R. M.; Cowley, A. R. Tetrahedron Lett. 2003, 44, 7471–7473. doi:10.1016/j.tetlet.2003.08.021
Return to citation in text: [1] -
Eade, S. J.; Walter, M. W.; Byrne, C.; Odell, B.; Rodriguez, R.; Baldwin, J. E.; Adlington, R. M.; Moses, J. E. J. Org. Chem. 2008, 73, 4830–4839. doi:10.1021/jo800220w
Return to citation in text: [1] -
Henrot, M.; Jean, A.; Peixoto, P. A.; Maddaluno, J.; De Paolis, M. J. Org. Chem. 2016, 81, 5190–5201. doi:10.1021/acs.joc.6b00878
Return to citation in text: [1] -
Herrmann, A.; Roesner, M.; Werner, T.; Hauck, S. M.; Koch, A.; Bauer, A.; Schneider, M.; Brack-Werner, R. Sci. Rep. 2020, 10, 1326. doi:10.1038/s41598-020-57843-9
Return to citation in text: [1] -
Henrot, M.; Richter, M. E. A.; Maddaluno, J.; Hertweck, C.; De Paolis, M. Angew. Chem., Int. Ed. 2012, 51, 9587–9591. doi:10.1002/anie.201204259
Return to citation in text: [1] [2] [3] -
De Paolis, M.; Rosso, H.; Henrot, M.; Prandi, C.; d'Herouville, F.; Maddaluno, J. Chem. – Eur. J. 2010, 16, 11229–11232. doi:10.1002/chem.201001780
Return to citation in text: [1] -
Rosso, H.; De Paolis, M.; Collin, V. C.; Dey, S.; Hecht, S. M.; Prandi, C.; Richard, V.; Maddaluno, J. J. Org. Chem. 2011, 76, 9429–9437. doi:10.1021/jo201683u
Return to citation in text: [1] -
Erker, G.; Kehr, G.; Fröhlich, R. Organometallics 2008, 27, 3–14. doi:10.1021/om7007666
Return to citation in text: [1] -
Cormier, M.; Ahmad, M.; Maddaluno, J.; De Paolis, M. Organometallics 2017, 36, 4920–4927. doi:10.1021/acs.organomet.7b00765
Return to citation in text: [1] [2] -
Belotti, D.; Andreatta, G.; Pradaux, F.; BouzBouz, S.; Cossy, J. Tetrahedron Lett. 2003, 44, 3613–3615. doi:10.1016/s0040-4039(03)00695-6
Return to citation in text: [1] -
Inokoishi, Y.; Sasakura, N.; Nakano, K.; Ichikawa, Y.; Kotsuki, H. Org. Lett. 2010, 12, 1616–1619. doi:10.1021/ol100350w
Return to citation in text: [1] -
Kalstabakken, K. A.; Harned, A. M. Tetrahedron 2014, 70, 9571–9585. doi:10.1016/j.tet.2014.07.081
Return to citation in text: [1] -
Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330–7396. doi:10.1021/acs.chemrev.6b00094
Return to citation in text: [1] -
Miyamae, N.; Watanabe, N.; Moritaka, M.; Nakano, K.; Ichikawa, Y.; Kotsuki, H. Org. Biomol. Chem. 2014, 12, 5847–5855. doi:10.1039/c4ob00733f
Return to citation in text: [1] -
Takemoto, Y.; Kuraoka, S.; Hamaue, N.; Iwata, C. Tetrahedron: Asymmetry 1996, 7, 993–996. doi:10.1016/0957-4166(96)00100-0
Return to citation in text: [1] -
Takemoto, Y.; Kuraoka, S.; Hamaue, N.; Aoe, K.; Hiramatsu, H.; Iwata, C. Tetrahedron 1996, 52, 14177–14188. doi:10.1016/0040-4020(96)00869-1
Return to citation in text: [1] -
Jeker, O. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2012, 51, 3474–3477. doi:10.1002/anie.201109175
Return to citation in text: [1] -
Kasatkin, A.; Nakagawa, T.; Okamoto, S.; Sato, F. J. Am. Chem. Soc. 1995, 117, 3881–3882. doi:10.1021/ja00118a030
Return to citation in text: [1] -
Isolated as a single stereoisomer.
Return to citation in text: [1] -
Paquette, L. A. Tetrahedron 1997, 53, 13971–14020. doi:10.1016/s0040-4020(97)00679-0
Return to citation in text: [1] -
Isolated as a single stereoisomer. The dr of the reaction and the relative configuration of the product were not determined.
Return to citation in text: [1]
| 1. | Sharma, P.; Powell, K.; Burnley, J.; Awaad, A. S.; Moses, J. E. Synthesis 2011, 2865–2892. doi:10.1055/s-0030-1260168 |
| 2. | Miller, A. K.; Trauner, D. Synlett 2006, 2295–2316. doi:10.1055/s-2006-949627 |
| 3. | Ui, H.; Shiomi, K.; Suzuki, H.; Hatano, H.; Morimoto, H.; Yamaguchi, Y.; Masuma, R.; Sunazuka, T.; Shimamura, H.; Sakamoto, K.; Kita, K.; Miyoshi, H.; Tomoda, H.; Ōmura, S. J. Antibiot. 2006, 59, 785–790. doi:10.1038/ja.2006.103 |
| 4. | Wilk, W.; Waldmann, H.; Kaiser, M. Bioorg. Med. Chem. 2009, 17, 2304–2309. doi:10.1016/j.bmc.2008.11.001 |
| 5. | Ishibashi, Y.; Ohba, S.; Nishiyama, S.; Yamamura, S. Tetrahedron Lett. 1996, 37, 2997–3000. doi:10.1016/0040-4039(96)00483-2 |
| 6. | Garey, D.; Ramirez, M.-l.; Gonzales, S.; Wertsching, A.; Tith, S.; Keefe, K.; Peña, M. R. J. Org. Chem. 1996, 61, 4853–4856. doi:10.1021/jo960221g |
| 7. | Manzo, E.; Ciavatta, M. L.; Gavagnin, M.; Mollo, E.; Wahidulla, S.; Cimino, G. Tetrahedron Lett. 2005, 46, 465–468. doi:10.1016/j.tetlet.2004.11.085 |
| 8. | Kim, Y.; Ogura, H.; Akasaka, K.; Oikawa, T.; Matsuura, N.; Imada, C.; Yasuda, H.; Igarashi, Y. Mar. Drugs 2014, 12, 4110–4125. doi:10.3390/md12074110 |
| 9. | Inuzuka, T.; Yamamoto, K.; Iwasaki, A.; Ohno, O.; Suenaga, K.; Kawazoe, Y.; Uemura, D. Tetrahedron Lett. 2014, 55, 6711–6714. doi:10.1016/j.tetlet.2014.10.032 |
| 10. | Fujimaki, T.; Saito, S.; Imoto, M. J. Antibiot. 2017, 70, 328–330. doi:10.1038/ja.2016.162 |
| 11. | Wu, T.; Salim, A. A.; Bernhardt, P. V.; Capon, R. J. J. Nat. Prod. 2021, 84, 474–482. doi:10.1021/acs.jnatprod.0c01343 |
| 12. | Leiris, S. J.; Khdour, O. M.; Segerman, Z. J.; Tsosie, K. S.; Chapuis, J.-C.; Hecht, S. M. Bioorg. Med. Chem. 2010, 18, 3481–3493. doi:10.1016/j.bmc.2010.03.070 |
| 13. | Lin, Z.; Torres, J. P.; Ammon, M. A.; Marett, L.; Teichert, R. W.; Reilly, C. A.; Kwan, J. C.; Hughen, R. W.; Flores, M.; Tianero, M. D.; Peraud, O.; Cox, J. E.; Light, A. R.; Villaraza, A. J. L.; Haygood, M. G.; Concepcion, G. P.; Olivera, B. M.; Schmidt, E. W. Chem. Biol. 2013, 20, 73–81. doi:10.1016/j.chembiol.2012.10.019 |
| 14. | De Paolis, M. Targets Heterocycl. Syst. 2016, 20, 63–84. |
| 19. | Miller, A. K.; Trauner, D. Angew. Chem., Int. Ed. 2003, 42, 549–552. doi:10.1002/anie.200390158 |
| 20. | Miller, A. K.; Byun, D. H.; Beaudry, C. M.; Trauner, D. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12019–12023. doi:10.1073/pnas.0401787101 |
| 21. | Beaudry, C. M.; Malerich, J. P.; Trauner, D. Chem. Rev. 2005, 105, 4757–4778. doi:10.1021/cr0406110 |
| 22. | Zuidema, D. R.; Miller, A. K.; Trauner, D.; Jones, P. B. Org. Lett. 2005, 7, 4959–4962. doi:10.1021/ol051887c |
| 31. | Cormier, M.; Ahmad, M.; Maddaluno, J.; De Paolis, M. Organometallics 2017, 36, 4920–4927. doi:10.1021/acs.organomet.7b00765 |
| 18. | Moses, J. E.; Adlington, R. M.; Rodriguez, R.; Eade, S. J.; Baldwin, J. E. Chem. Commun. 2005, 1687–1689. doi:10.1039/b418988d |
| 31. | Cormier, M.; Ahmad, M.; Maddaluno, J.; De Paolis, M. Organometallics 2017, 36, 4920–4927. doi:10.1021/acs.organomet.7b00765 |
| 16. | Ireland, C.; Scheuer, P. J. Science 1979, 205, 922–923. doi:10.1126/science.205.4409.922 |
| 17. | Ksebati, M. B.; Schmitz, F. J. J. Org. Chem. 1985, 50, 5637–5642. doi:10.1021/jo00350a042 |
| 27. | Henrot, M.; Richter, M. E. A.; Maddaluno, J.; Hertweck, C.; De Paolis, M. Angew. Chem., Int. Ed. 2012, 51, 9587–9591. doi:10.1002/anie.201204259 |
| 15. | Ireland, C.; Faulkner, J. Tetrahedron 1981, 37, 233–240. doi:10.1016/0040-4020(81)85059-4 |
| 27. | Henrot, M.; Richter, M. E. A.; Maddaluno, J.; Hertweck, C.; De Paolis, M. Angew. Chem., Int. Ed. 2012, 51, 9587–9591. doi:10.1002/anie.201204259 |
| 27. | Henrot, M.; Richter, M. E. A.; Maddaluno, J.; Hertweck, C.; De Paolis, M. Angew. Chem., Int. Ed. 2012, 51, 9587–9591. doi:10.1002/anie.201204259 |
| 28. | De Paolis, M.; Rosso, H.; Henrot, M.; Prandi, C.; d'Herouville, F.; Maddaluno, J. Chem. – Eur. J. 2010, 16, 11229–11232. doi:10.1002/chem.201001780 |
| 29. | Rosso, H.; De Paolis, M.; Collin, V. C.; Dey, S.; Hecht, S. M.; Prandi, C.; Richard, V.; Maddaluno, J. J. Org. Chem. 2011, 76, 9429–9437. doi:10.1021/jo201683u |
| 26. | Herrmann, A.; Roesner, M.; Werner, T.; Hauck, S. M.; Koch, A.; Bauer, A.; Schneider, M.; Brack-Werner, R. Sci. Rep. 2020, 10, 1326. doi:10.1038/s41598-020-57843-9 |
| 30. | Erker, G.; Kehr, G.; Fröhlich, R. Organometallics 2008, 27, 3–14. doi:10.1021/om7007666 |
| 25. | Henrot, M.; Jean, A.; Peixoto, P. A.; Maddaluno, J.; De Paolis, M. J. Org. Chem. 2016, 81, 5190–5201. doi:10.1021/acs.joc.6b00878 |
| 23. | Brückner, S.; Baldwin, J. E.; Moses, J.; Adlington, R. M.; Cowley, A. R. Tetrahedron Lett. 2003, 44, 7471–7473. doi:10.1016/j.tetlet.2003.08.021 |
| 24. | Eade, S. J.; Walter, M. W.; Byrne, C.; Odell, B.; Rodriguez, R.; Baldwin, J. E.; Adlington, R. M.; Moses, J. E. J. Org. Chem. 2008, 73, 4830–4839. doi:10.1021/jo800220w |
| 17. | Ksebati, M. B.; Schmitz, F. J. J. Org. Chem. 1985, 50, 5637–5642. doi:10.1021/jo00350a042 |
| 34. | Kalstabakken, K. A.; Harned, A. M. Tetrahedron 2014, 70, 9571–9585. doi:10.1016/j.tet.2014.07.081 |
| 32. | Belotti, D.; Andreatta, G.; Pradaux, F.; BouzBouz, S.; Cossy, J. Tetrahedron Lett. 2003, 44, 3613–3615. doi:10.1016/s0040-4039(03)00695-6 |
| 33. | Inokoishi, Y.; Sasakura, N.; Nakano, K.; Ichikawa, Y.; Kotsuki, H. Org. Lett. 2010, 12, 1616–1619. doi:10.1021/ol100350w |
| 43. | Isolated as a single stereoisomer. The dr of the reaction and the relative configuration of the product were not determined. |
| 42. | Paquette, L. A. Tetrahedron 1997, 53, 13971–14020. doi:10.1016/s0040-4020(97)00679-0 |
| 39. | Jeker, O. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2012, 51, 3474–3477. doi:10.1002/anie.201109175 |
| 40. | Kasatkin, A.; Nakagawa, T.; Okamoto, S.; Sato, F. J. Am. Chem. Soc. 1995, 117, 3881–3882. doi:10.1021/ja00118a030 |
| 35. | Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330–7396. doi:10.1021/acs.chemrev.6b00094 |
| 36. | Miyamae, N.; Watanabe, N.; Moritaka, M.; Nakano, K.; Ichikawa, Y.; Kotsuki, H. Org. Biomol. Chem. 2014, 12, 5847–5855. doi:10.1039/c4ob00733f |
| 37. | Takemoto, Y.; Kuraoka, S.; Hamaue, N.; Iwata, C. Tetrahedron: Asymmetry 1996, 7, 993–996. doi:10.1016/0957-4166(96)00100-0 |
| 38. | Takemoto, Y.; Kuraoka, S.; Hamaue, N.; Aoe, K.; Hiramatsu, H.; Iwata, C. Tetrahedron 1996, 52, 14177–14188. doi:10.1016/0040-4020(96)00869-1 |
© 2022 Cormier et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.