Abstract
The cyanation reaction has achieved rapid progress in recent times. The ability to exhibit multiple oxidation states increased the demand of ruthenium in the field of catalysis. These cyanation reactions have wide application in pharmacological and biological fields. This review gives an overview of the ruthenium-catalyzed cyanation reactions covering literature up to 2021.

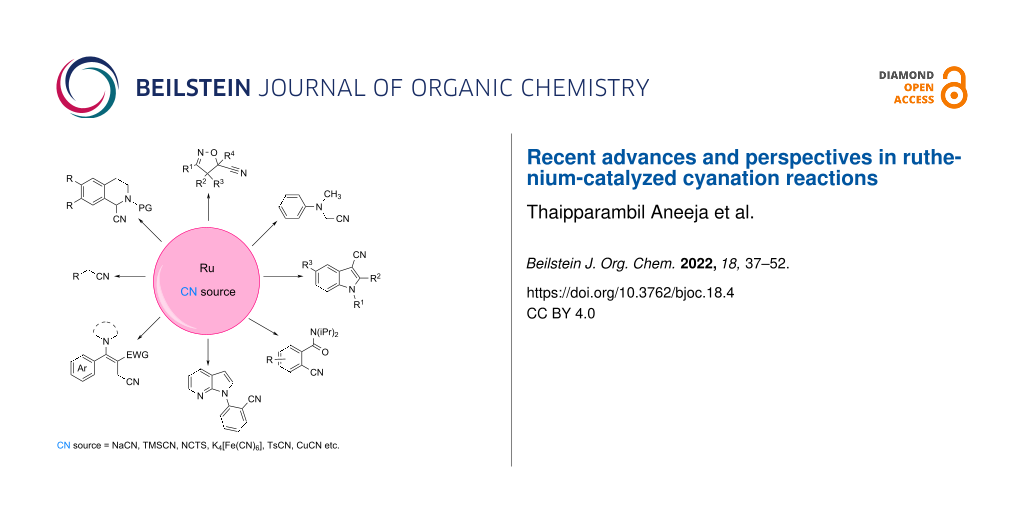
Graphical Abstract
Introduction
Nitriles are a major class of organic compounds having wide significance in materials science, agrochemical and pharmaceutical industry [1]. They are the privileged compounds finding broad applications in natural product synthesis, pigments and dyes.
Different variety of α-amino carbonyls [2], α-amino acids [3] and 1,2-diamines were prepared from the nitriles using homogeneous and heterogeneous catalysis [4-6]. One of the astonishing aspects of nitriles is that it can be easily converted to amines, carboxylic acids, tetrazoles, aldehydes, amidines, and amides [7-11]. This has been suitably transformed into structurally diverse and complex molecules.
In 1927, Pongratz reported a method towards cyanation reactions [12]. From then, onwards, cyanation gained prime focus and achieved much acceptance. The conventional approaches towards the synthesis of nitriles include Rosenmund–von Braun reactions, Sandmeyer reactions and industrial ammoxidation reactions [13,14]. But these strategies involve harsh reaction conditions and generation of large amount of heavy metal waste. These drawbacks demanded the need of an alternative method for the synthesis of nitriles.
Nowadays transition-metal-catalyzed reactions have received tremendous interest. Various transition metals such as Fe [15], Co [16], Ni [17], Pd [18], Cu [19], Rh [20] etc. were well explored in cyanation owing to its cost-effective and earth abundant characteristics. Moreover, much greener methodologies like microwave-assisted cyanation reactions also received much attention in recent times [21].
The cyanation can be carried out using electrophilic and nucleophilic cyanating agents [22]. Usually a cyanation is accomplished via the nucleophilic attack of a CN− at an electrophilic carbon center. But there are some reagents that react as CN+ and thus attack the nucleophilic carbon center. Tosyl cyanide [23], 2-chlorobenzylthiocyanate [24], and cyanogen chloride [25] are some of the examples for electrophilic cyanating agents. Commonly used metallic cyanating agents include K4Fe(CN)6, CuCN, KCN, NaCN, TMSCN etc.
Ruthenium-catalyzed reactions have gained significant attention in recent times [26]. Ruthenium has the ability to show a large number of oxidation states, and thus a large number of complexes can be prepared using this metal. Ruthenium complexes have astonishing characteristics such as high electron transfer ability, low redox potentials, high Lewis acidity, and greater stabilities of the reactive metallic species like oxometals, metallacycles, and metal carbene complexes [27]. The wide availability of highly reactive ruthenium complexes which are efficient as catalysts elevated the scope of this metal in synthetic organic chemistry.
For clarity and ease of understanding of the topic, this review is categorized into four sections: cyanation of amines, cyanation of arenes and heteroarenes, photocatalyzed cyanation, and miscellaneous. To the best of our knowledge, this is the first review summarizing an overview of ruthenium-catalyzed cyanation reactions covering literature up to 2021.
Review
1 Cyanation of amines
1.1 Cyanation of amines using heterogeneous catalysts
Sain and co-workers investigated the ability of starch-immobilized ruthenium trichloride to catalyze the oxidative cyanation of tertiary amines [28]. The reaction was performed using 2 mol % of catalyst, 2.5 mmol H2O2, 1.2 mmol NaCN, and 1 mL CH3COOH in methanol at room temperature (Scheme 1). Better yields of the α-aminonitrile products were obtained for aromatic amines with electron-donating substituents than electron-withdrawing ones. Under the optimized conditions, tributylamine was found inactive to afford the desired product. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis confirmed the purity of the prepared compounds without any ruthenium contamination. The attractive features of this reaction include environment-friendly conditions, effective recycling and activity of the catalyst, and high product selectivity. A plausible mechanism of this transformation is depicted in Scheme 2.
![[1860-5397-18-4-i1]](/bjoc/content/inline/1860-5397-18-4-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Starch-immobilized ruthenium trichloride-catalyzed cyanation of tertiary amines.
Scheme 1: Starch-immobilized ruthenium trichloride-catalyzed cyanation of tertiary amines.
![[1860-5397-18-4-i2]](/bjoc/content/inline/1860-5397-18-4-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the cyanation of tertiary amines using starch-immobilized ruthenium trichloride as the catalyst.
Scheme 2: Proposed mechanism for the cyanation of tertiary amines using starch-immobilized ruthenium trichlor...
Later in 2012, Nageswar et al. have established a methodology for the oxidative cyanation of tertiary amines using a heterogeneous Ru/C catalyst [29]. Most of the reported works involve the use of toxic cyanating agents like NaCN or TMSCN. In this protocol, the authors have exploited the cost-effective, less toxic, and easily available ethyl cyanoformate as the cyanide source. They carried out the optimization studies using N,N-dimethylaniline (1.0 mmol) and ethyl cyanoformate (2.0 mmol) as the model substrates and the optimized conditions include 5 wt % Ru/C (20 mg) as catalyst and 2.5 equiv of TBHP (in decane) as oxidant in methanol at 60 °C for 6 h (Scheme 3). Differently substituted tertiary amines with electron-rich and electron-deficient substituents afforded the required products in good yields. This reaction was found to be suitable for N-aryl-substituted cyclic amines such as N-arylpiperidine, N-arylpyrrolidine, and N-aryltetrahydroisoquinoline. However, aliphatic tertiary amines failed to achieve the desired product by this method. The proposed mechanism is initiated with the formation of a Ru–oxo species by the reaction between Ru/C and TBHP. The next step involves the formation of an iminium ion intermediate through reaction of the Ru–oxo species with the tertiary amine. The subsequent reaction between this iminium ion intermediate and CN− furnishes the required product (Scheme 4).
![[1860-5397-18-4-i3]](/bjoc/content/inline/1860-5397-18-4-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyanation of tertiary amines using heterogeneous Ru/C catalyst.
Scheme 3: Cyanation of tertiary amines using heterogeneous Ru/C catalyst.
![[1860-5397-18-4-i4]](/bjoc/content/inline/1860-5397-18-4-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for cyanation of tertiary amines using a heterogeneous Ru/C catalyst.
Scheme 4: Proposed mechanism for cyanation of tertiary amines using a heterogeneous Ru/C catalyst.
In 2013, Jain and co-workers described a novel strategy for the synthesis of a ruthenium-carbamato complex and its promising catalytic application in the oxidative cyanation reaction [30]. The prepared catalyst was found highly active in the oxidative cyanation of tertiary amines to the corresponding α-aminonitriles in excellent yields. The cyanation reaction of N,N-dimethylaniline using NaCN catalyzed by ruthenium-carbamato complex in AcOH/MeOH furnished the desired product in 92% yield within 2.5 h (Scheme 5). Meanwhile, the ruthenium chloride-catalyzed cyanation reaction required 3.5 h to complete the reaction and achieved the product in 90% yield. Thus, the studies revealed the higher efficiency of the ruthenium-carbamato complex in catalyzing the cyanation reaction. This strategy utilized eco-friendly hydrogen peroxide and molecular oxygen as the oxidant system. This method was found highly favorable to tertiary amines with electron-donating substituents.
![[1860-5397-18-4-i5]](/bjoc/content/inline/1860-5397-18-4-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Ruthenium-carbamato complex-catalyzed oxidative cyanation of tertiary amines.
Scheme 5: Ruthenium-carbamato complex-catalyzed oxidative cyanation of tertiary amines.
The first report on an MCM-41-immobilized N-alkylethylenediamine Ru(III) complex (MCM-41-2N-RuCl3) catalyzed oxidative cyanation of tertiary amines was achieved by Cai et al. [31]. The optimized conditions were MCM-41-2N-RuCl3 (5 mol %), NaCN (1.2 mmol), AcOH (6.0 mmol), H2O2 (2.5 mmol) in methanol at 60 °C under Ar atmosphere for 4 h (Scheme 6). The reaction proceeded smoothly for N-aryl-substituted cyclic tertiary amines including N-arylpiperidines and N-aryltetrahydroisoquinolines. The high reusability of the catalyst at least seven times further enhanced the importance of this strategy in the field of organic synthesis.
![[1860-5397-18-4-i6]](/bjoc/content/inline/1860-5397-18-4-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Cyanation of tertiary amines using immobilized MCM-41-2N-RuCl3 as the catalyst.
Scheme 6: Cyanation of tertiary amines using immobilized MCM-41-2N-RuCl3 as the catalyst.
1.2 Cyanation of amines using homogeneous catalysts
An interesting ruthenium-catalyzed oxidative cyanation of tertiary amines using molecular oxygen was reported by Murahashi and co-workers [32]. This RuCl3·nH2O-catalyzed protocol used NaCN in acetic acid as the cyano source, methanol as the solvent under molecular oxygen at 60 °C for 1–2 h (Scheme 7). The extensive substrate scope studies under optimized conditions disclosed that differently substituted N,N-dimethylanilines gave the desired products in excellent yields. The reaction was also found suitable for N-phenyltetrahydroisoquinoline and delivered the expected product in good yields.
![[1860-5397-18-4-i7]](/bjoc/content/inline/1860-5397-18-4-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Cyanation of tertiary amines using RuCl3·nH2O as the catalyst and molecular oxygen as oxidant.
Scheme 7: Cyanation of tertiary amines using RuCl3·nH2O as the catalyst and molecular oxygen as oxidant.
Later, the same group reported a novel synthetic pathway for the oxidative cyanation of tertiary amines using sodium cyanide [33]. The optimized conditions for this reaction included the use of 0.05 mmol of RuCl3 as the catalyst, 1.2 mmol of NaCN in acetic acid as the cyanide source and 2.5 mmol of H2O2 as the oxidant in methanol (Scheme 8). Both, substituted N,N-dimethylanilines with electron-donating and electron-withdrawing groups were well tolerated in this reaction. The N-aryl-substituted cyclic amines such as N-arylpiperidine, N-aryltetrahydroisoquinoline and N-arylpyrrolidine derivatives also reacted well to furnish the desired products in good yields.
![[1860-5397-18-4-i8]](/bjoc/content/inline/1860-5397-18-4-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: RuCl3-catalyzed cyanation of tertiary amines using NaCN/HCN and H2O2 as oxidant.
Scheme 8: RuCl3-catalyzed cyanation of tertiary amines using NaCN/HCN and H2O2 as oxidant.
In 2008, they described the scope and mechanism of the oxidative cyanation of tertiary amines using H2O2 and O2 [34]. They pointed out the fact that the hydrogen peroxide system was found to be more efficient in catalyzing the cyanation reaction of cyclic amines than the aerobic oxidation system. The catalytic cycle for the hydrogen peroxide system involves the formation of the oxoruthenium species (A) and the low-valent ruthenium species (B), whereas the aerobic oxidation system includes C–H activation and a subsequent reaction with molecular oxygen (Scheme 9 and Scheme 10). Thus, the authors came to the conclusion that the higher yields of cyanation product obtained in the case of the hydrogen peroxide system was due to the highly reactive oxoruthenium species and low-valence ruthenium species.
![[1860-5397-18-4-i9]](/bjoc/content/inline/1860-5397-18-4-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Proposed mechanism for the ruthenium-catalyzed oxidative cyanation using H2O2.
Scheme 9: Proposed mechanism for the ruthenium-catalyzed oxidative cyanation using H2O2.
![[1860-5397-18-4-i10]](/bjoc/content/inline/1860-5397-18-4-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Proposed mechanism for the ruthenium-catalyzed aerobic oxidative cyanation.
Scheme 10: Proposed mechanism for the ruthenium-catalyzed aerobic oxidative cyanation.
Sain et al. disclosed a new methodology towards the oxidative cyanation of tertiary amines using Ru as the catalyst (Scheme 11) [35]. In this reaction, the comparatively safer acetone cyanohydrin was utilized as the cyanating agent. Better yields of products were obtained for both electron-rich and electron-deficient tertiary amines. Cyclic amines such as piperidine, tetrahydroisoquinoline derivatives, and pyrrolidine were also tolerated well in this reaction. The use of the non-toxic and inexpensive acetone cyanohydrin makes this method more advantageous compared to the known methods.
![[1860-5397-18-4-i11]](/bjoc/content/inline/1860-5397-18-4-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: RuCl3-catalyzed oxidative cyanation of tertiary amines using acetone cyanohydrin as the cyanating agent.
Scheme 11: RuCl3-catalyzed oxidative cyanation of tertiary amines using acetone cyanohydrin as the cyanating a...
2 Cyanation of arenes and heteroarenes
2.1 Cyanation of arenes and heteroarenes using heterogeneous catalysts
In 2012, a novel strategy for the 3-cyanation of indole in the presence of Ru(III)-exchanged NaY zeolite (RuY) was reported [36]. In this reaction K4[Fe(CN)6] was utilized as the cyano source in DMF at 110 °C. The Cu(OAc)2 and O2 (1 atm) was found essential for promoting this reaction (Scheme 12). Both electron-rich and electron-deficient indoles afforded the desired products in good yields. The major advantages of this method include high regioselectivity, mild reaction conditions, and reusability of the catalyst.
![[1860-5397-18-4-i12]](/bjoc/content/inline/1860-5397-18-4-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Cyanation of indoles using K4[Fe(CN)6] as cyano source and Ru(III)-exchanged NaY zeolite (RuY) as catalyst.
Scheme 12: Cyanation of indoles using K4[Fe(CN)6] as cyano source and Ru(III)-exchanged NaY zeolite (RuY) as c...
Two years later, in 2014, Ackermann and co-workers disclosed an astonishing protocol towards the cyanation of arenes and heteroarenes using a ruthenium(II) catalyst [37]. This was the first report on a C(sp2)–H cyanation reaction using ruthenium as the catalyst. In this reaction the authors utilized the less toxic, environment-friendly, and easily available N-cyano-N-phenyl-p-toluenesulfonamide (NCTS) as the cyanating reagent. The reaction exhibited high chemoselectivity and good functional group tolerance. The optimized conditions for the reaction were [RuCl2(p-cymene)]2 (5 mol %), AgSbF6 (20 mol %), NaOAc (20 mol %) in DCE at 120 °C for 24 h (Scheme 13). Differently substituted aromatic amides with a range of functional groups such as fluoro, chloro, bromo, ester etc. were tolerated well in this method. The authors also performed the cyanation of heteroarenes at C-2 and C-3 positions and obtained excellent results. Various heteroarenes such as thiophenes, benzofurans, furans, and indoles were found suitable substrates and afforded the desired products with high chemo- and site-selectivity. A possible mechanism for the reaction was also described. The first step of the catalytic cycle involves the formation of a cationic complex I which after coordination and subsequent NCTS insertion is transformed to intermadiate II. β-Elimination finally delivers the required product (Scheme 14).
![[1860-5397-18-4-i13]](/bjoc/content/inline/1860-5397-18-4-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Cyanation of arenes and heteroarenes using a ruthenium(II) catalyst and N-cyano-N-phenyl-p-toluenesulfonamide (NCTS) as cyanating reagent.
Scheme 13: Cyanation of arenes and heteroarenes using a ruthenium(II) catalyst and N-cyano-N-phenyl-p-toluenes...
![[1860-5397-18-4-i14]](/bjoc/content/inline/1860-5397-18-4-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Proposed mechanism for the cyanation of arenes and heteroarenes using ruthenium(II) as catalyst and NCTS as cyanation reagent.
Scheme 14: Proposed mechanism for the cyanation of arenes and heteroarenes using ruthenium(II) as catalyst and...
Later, Deb and co-workers developed a methodology towards the synthesis of N-(2-cyanoaryl)-7-azaindoles using [RuCl2(p-cymene)]2/AgOTf/NaOAc as the catalyst system (Scheme 15) [38]. They carried out extensive substrate scope studies and it was pointed out that N-aryl-7-azaindoles substituted with electron-donating groups gave better yields of products. Heteroarenes also performed well in this reaction with high reactivity. The authors also investigated the cyanation of differently substituted 7-azaindoles and achieved excellent results. They also succeeded in applying this strategy to N-aryl-α-carbolines to synthesize the respective cyanated N-aryl-α-carboline products.
![[1860-5397-18-4-i15]](/bjoc/content/inline/1860-5397-18-4-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of N-(2-cyanoaryl)-7-azaindoles.
Scheme 15: Synthesis of N-(2-cyanoaryl)-7-azaindoles.
3 Photocatalyzed cyanation reactions
Nowadays photocatalysis has emerged as an efficient tool for the synthesis of organic compounds. Jain et al. put forward an oxidative cyanation of tertiary amines using an immobilized heterogeneous ruthenium catalyst (Figure 1) [39]. The optimized conditions comprised sodium cyanide (NaCN) (1.2 mmol) in acetic acid as cyanide source, molecular oxygen as the oxidant, and TiO2-immobilized ruthenium(II) polyazine complex as the heterogeneous photoredox catalyst in methanol at room temperature (Table 1). The substrate scope studies revealed a better reactivity of aromatic tertiary amines substituted with electron-donating groups compared to the electron-withdrawing ones. The N-aryl cyclic amines including N-phenylpiperidine, N-phenyltetrahydroisoquinoline, and N-phenylpyrrolidine smoothly reacted to deliver the required α-aminonitriles in good yields. Under the optimized conditions, aliphatic tributylamine failed to achieve the desired product (Table 1, entry 5). However, tertiary amines with a benzyl group reacted very slowly and afforded the expected product in moderate yield (Table 1, entry 6). The reason for the incompatibility of tributylamine towards this method has yet to be explored.
![[1860-5397-18-4-1]](/bjoc/content/figures/1860-5397-18-4-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the TiO2-immobilized ruthenium polyazine complex.
Figure 1: Structure of the TiO2-immobilized ruthenium polyazine complex.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-4-i28.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-4-i29.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-4-i30.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-4-i31.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-18-4-i32.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-18-4-i33.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-18-4-i34.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-18-4-i35.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-18-4-i36.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-18-4-i37.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-18-4-i38.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-18-4-i39.svg?max-width=637&scale=1.0)
A synthesis of allylic cyanides via visible-light-mediated oxidative cyanation of aza-Baylis–Hillman adducts was developed by Yadav and co-workers [40]. The cyanating agent employed was TMSCN in acetonitrile (Scheme 16). The aza-BH adducts with electron-donating groups gave higher yields in comparison with electron-deficient ones. Mild reaction conditions, readily available reagents, lower catalyst loading, and the use of cost effective atmospheric oxygen and visible light are the most attractive characteristics of this reaction.
![[1860-5397-18-4-i16]](/bjoc/content/inline/1860-5397-18-4-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Visible-light-induced oxidative cyanation of aza-Baylis–Hillman adducts.
Scheme 16: Visible-light-induced oxidative cyanation of aza-Baylis–Hillman adducts.
Xu et al. in 2016 discussed the deboronative cyanation of alkyltrifluoroborates using [Ru(bpy)3](PF6)2 as the photoredox catalyst [41]. This method provides an efficient pathway to 1°, 2°, and 3° alkyl nitriles using p‐toluenesulfonyl cyanide (TsCN) in CH2Cl2/H2O under visible-light irradiation (Scheme 17). 1‐Acetoxy‐1,2‐benziodoxol‐3‐(1H)‐one (BI‐OAc) was chosen as the oxidant and TFA as the additive in this method. A lower reactivity was observed for 2° and 3° alkyltrifluoroborates under the optimized conditions. The authors were able to improve the product yield by increasing the amount of TsCN and avoiding the use of additive (Scheme 18). A wide variety of functional groups such as esters, cyano, amides, ethers, ketones, alkynes, and halides were compatible with this strategy.
![[1860-5397-18-4-i17]](/bjoc/content/inline/1860-5397-18-4-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of 1° alkyl nitriles using [Ru(bpy)3](PF6)2 as the photocatalyst.
Scheme 17: Synthesis of 1° alkyl nitriles using [Ru(bpy)3](PF6)2 as the photocatalyst.
![[1860-5397-18-4-i18]](/bjoc/content/inline/1860-5397-18-4-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of 2° and 3° alkyl nitriles using [Ru(bpy)3](PF6)2 as the photocatalyst.
Scheme 18: Synthesis of 2° and 3° alkyl nitriles using [Ru(bpy)3](PF6)2 as the photocatalyst.
A photoredox-catalyzed oxidative coupling of 4-alkyl-3,4-dihydroquinoxalin-2(1H)-ones with nucleophiles was reported by Hong and co-workers [42]. The reaction was performed using 20 mol % of Ru(bpy)3Cl2.6H2O in methanol under CFL light irradiation at room temperature (Scheme 19). They were able to synthesize the cyano derivative by utilizing TMSCN as the cyanating medium, and obtained the desired product in 74% yield.
![[1860-5397-18-4-i19]](/bjoc/content/inline/1860-5397-18-4-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Photoredox cross coupling reaction.
Scheme 19: Photoredox cross coupling reaction.
Espino and co-workers developed and characterized a variety of dicationic Ru(II) polypyridyl complexes with 2-(pyridyl)benzimidazole or its N-alkylated derivatives as the ancillary ligands (N^N) [43]. The prepared Ru(II) derivatives were found efficient in the synthesis of α-amino nitriles from amines via a one-pot strategy. This synthetic pathway comprises two consecutive reactions including photooxidation of the amine and the cyanation of resultant aldimine intermediate to afford the α-amino nitriles (Scheme 20). This reaction worked well under eco-friendly conditions with low catalyst loading, and utilizing O2 as green oxidant to give excellent yields of the products. They also disclosed the efficiency of these Ru(II) complexes in the photooxidation of primary and secondary amines. Moreover, 1-hexylamine worked well in this reaction affording the desired product in moderate to good yields. The efficient transformation of 1-hexylamine proved that this reaction was even applicable to amines with non-activated α-H atoms. The proposed mechanism for the cyanation step is depicted in Scheme 21.
![[1860-5397-18-4-i20]](/bjoc/content/inline/1860-5397-18-4-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of α-amino nitriles from amines via a one-pot strategy.
Scheme 20: Synthesis of α-amino nitriles from amines via a one-pot strategy.
![[1860-5397-18-4-i21]](/bjoc/content/inline/1860-5397-18-4-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Proposed mechanistic pathway for the cyanation of the aldimine intermediate.
Scheme 21: Proposed mechanistic pathway for the cyanation of the aldimine intermediate.
Chemical synthesis usually needs labor-intensive and tiresome procedures. One of the approaches to overcome these challenges is to perform the reactions in flow [44]. The major advantages of this approach include predictable reaction scale-up, high reproducibility and yields, lower catalyst loading, high product purity, and excellent selectivity.
A visible-light-promoted Strecker-type functionalization of N-aryl-substituted tetrahydroisoquinolines under flow conditions was reported (Scheme 22) [45]. The cyanating agent utilized was trimethylsilyl cyanide (TMSCN) in acetonitrile solvent under CFL light irradiation.
![[1860-5397-18-4-i22]](/bjoc/content/inline/1860-5397-18-4-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Strecker-type functionalization of N-aryl-substituted tetrahydroisoquinolines under flow conditions.
Scheme 22: Strecker-type functionalization of N-aryl-substituted tetrahydroisoquinolines under flow conditions....
4 Miscellaneous
An efficient methodology for the synthesis of α-aminonitriles via the one-pot coupling of aldehydes, amines and trimethylsilyl cyanide was reported [46]. This reaction was catalyzed by RuCl3 and used acetonitrile as solvent (Scheme 23). Both aromatic and aliphatic aldehydes performed well and gave the desired products in good yields. At the same time ketones were found less active in this strategy. The operational simplicity and high yield of products are the prominent characteristics of this reaction.
![[1860-5397-18-4-i23]](/bjoc/content/inline/1860-5397-18-4-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: One-pot synthesis of α-aminonitriles using RuCl3 as catalyst.
Scheme 23: One-pot synthesis of α-aminonitriles using RuCl3 as catalyst.
In 2008, Bhanage et al. developed a novel methodology for the synthesis of alkyl iodides/nitriles using ruthenium tris(2,2,6,6-tetramethyl-3,5-heptanedionate) (Ru(TMHD)3) as the catalyst (Scheme 24) [47]. This catalyst was found highly efficient in the hydrogenation, iodination, and cyanation reaction of carbonyl compounds under environmentally benign conditions. This method provides an efficient synthetic route towards alkyl iodides and nitriles in one pot. Carbonyl compounds such as cinnamaldehyde, acetophenone, and cyclohexanone etc. were well tolerated in this reaction and afforded the corresponding nitriles in moderate to good yields.
![[1860-5397-18-4-i24]](/bjoc/content/inline/1860-5397-18-4-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Synthesis of alkyl nitriles using (Ru(TMHD)3) as the catalyst.
Scheme 24: Synthesis of alkyl nitriles using (Ru(TMHD)3) as the catalyst.
Zhang and co-workers developed an efficient methodology for the synthesis of cyanated isoxazolines from the corresponding alkenyl oximes under mild reaction conditions (Scheme 25) [48]. The dual role of tert-butyl nitrite as oxidant and as a nitrogen source further enhanced the significance of this method. [RuCl2(p-cymene)]2 was identified as the best choice of catalyst. Differently substituted alkenyl oximes with aryl, heteroaryl, and alkyl substituents performed well in this reaction. The major advantage of this method is the formation of C–O and C≡N bonds in a single-step. The proposed mechanism is depicted in Scheme 26.
![[1860-5397-18-4-i25]](/bjoc/content/inline/1860-5397-18-4-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Synthesis of cyanated isoxazolines from alkenyl oximes catalyzed by [RuCl2(p-cymene)]2 in the presence of tert-butyl nitrite.
Scheme 25: Synthesis of cyanated isoxazolines from alkenyl oximes catalyzed by [RuCl2(p-cymene)]2 in the prese...
![[1860-5397-18-4-i26]](/bjoc/content/inline/1860-5397-18-4-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Proposed mechanism for the synthesis of cyanated isoxazolines from alkenyl oximes.
Scheme 26: Proposed mechanism for the synthesis of cyanated isoxazolines from alkenyl oximes.
Xiao and co-workers developed an environmentally benign strategy for the oxidative cyanation of differently substituted alcohols using a manganese oxide nanorod-supported ruthenium catalyst (Scheme 27) [49]. They also evaluated the efficiency of other metals such as Au, Pt, Pd, Rh, Ag, and Fe and found that they were less efficient compared to Ru. The benzylic alcohols showed higher reactivity than aliphatic alcohols towards this methodology. Moreover, this protocol worked well for differently substituted heterocyclic alcohols and afforded the products in excellent yields. The authors also conducted various experimental and theoretical studies to analyze the reaction mechanism. The proposed mechanism begins with the oxidative dehydrogenation of the alcohol to afford the aldehyde which undergoes condensation with ammonia to give the corresponding imine. Finally, oxidative dehydrogenation results in the formation of the nitrile.
![[1860-5397-18-4-i27]](/bjoc/content/inline/1860-5397-18-4-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Oxidative cyanation of differently substituted alcohols.
Scheme 27: Oxidative cyanation of differently substituted alcohols.
Conclusion
This review summarizes the recent progress in ruthenium-catalyzed cyanation reactions. Due to the wide application of nitrile compounds in pharmaceutical and biological fields, cyanation reactions have achieved significant progress in recent times.
Transition-metal-catalyzed cyanation reactions have emerged as an alternative approach to conventional cyanation strategies. Nowadays ruthenium has gained much acceptance owing to its wide range of oxidation states and ability to form a large number of complexes. From our discussion it is evident that the most commonly used cyanating reagents include highly toxic compounds such as K4[Fe(CN)6], NaCN, CuCN, TMSCN etc. However, some of the reported works involve the use of safer and greener cyanation sources like NCTS, acetone cyanohydrin, ethyl cyanoformate etc.
Our studies disclosed that many of the reports were mainly focusing on the oxidative cyanation of tertiary amines. Extensive research was carried out in the cyanation of tertiary amines using different oxidants such as O2, H2O2, and TBHP. Ruthenium was also found to be effective in the cyanation of arenes and heteroarenes with high functional group tolerance. Some of the suggested methods utilized photoredox catalysts and thus offer new avenues towards greener and economical nitrile synthesis.
In spite of these advancements, the use of toxic cyanating agents like NaCN in almost all the methods remains as a limitation in this area. The use of non-toxic and environment friendly cyanating agents such as NCTS, acetone cyanohydrin, ethyl cyanoformate etc needs more attention in future. Scientists can also focus more on cyanation reactions using photoredox catalyst.
The larger availability of reactive complexes of ruthenium and the high reusability and easy separation procedures of heterogeneous catalysts have proven the fact that this metal serve as highly effective catalyst for a wide range of organic transformations. It will surely motivate the scientific community to develop more environmentally benign cyanation reactions using ruthenium in future.
Funding
TA, CMA, and PVS thank the Council of Scientific and Industrial Research (CSIR New Delhi) for the award of research fellowships. GA thanks the Kerala State Council for Science Technology & Environment (KSCSTE, Trivandrum) for financial support in the form of a research grant (No. 341/2013/KSCSTE dated 15.03.2013).
References
-
Ding, R.; Liu, Y.; Han, M.; Jiao, W.; Li, J.; Tian, H.; Sun, B. J. Org. Chem. 2018, 83, 12939–12944. doi:10.1021/acs.joc.8b02190
Return to citation in text: [1] -
Enders, D.; Shilvock, J. P. Chem. Soc. Rev. 2000, 29, 359–373. doi:10.1039/a908290e
Return to citation in text: [1] -
Strecker, A. Justus Liebigs Ann. Chem. 1850, 75, 27–45. doi:10.1002/jlac.18500750103
Return to citation in text: [1] -
Iyer, M. S.; Gigstad, K. M.; Namdev, N. D.; Lipton, M. J. Am. Chem. Soc. 1996, 118, 4910–4911. doi:10.1021/ja952686e
Return to citation in text: [1] -
Rafiee, E.; Azad, A. Synth. Commun. 2007, 37, 1127–1132. doi:10.1080/00397910701198914
Return to citation in text: [1] -
Rafiee, E.; Rashidzadeh, S.; Azad, A. J. Mol. Catal. A: Chem. 2007, 261, 49–52. doi:10.1016/j.molcata.2006.07.058
Return to citation in text: [1] -
Haddenham, D.; Pasumansky, L.; DeSoto, J.; Eagon, S.; Singaram, B. J. Org. Chem. 2009, 74, 1964–1970. doi:10.1021/jo8023329
Return to citation in text: [1] -
Knight, J. A.; Zook, H. D. J. Am. Chem. Soc. 1952, 74, 4560–4562. doi:10.1021/ja01138a031
Return to citation in text: [1] -
Demko, Z. P.; Sharpless, K. B. J. Org. Chem. 2001, 66, 7945–7950. doi:10.1021/jo010635w
Return to citation in text: [1] -
Garduño, J. A.; Garcı́a, J. J. ACS Catal. 2015, 5, 3470–3477. doi:10.1021/acscatal.5b00348
Return to citation in text: [1] -
Allen, C. L.; Lapkin, A. A.; Williams, J. M. J. Tetrahedron Lett. 2009, 50, 4262–4264. doi:10.1016/j.tetlet.2009.05.021
Return to citation in text: [1] -
Pongratz, A. Monatsh. Chem. 1927, 48, 585–591. doi:10.1007/bf01518076
Return to citation in text: [1] -
Nielsen, M. A.; Nielsen, M. K.; Pittelkow, T. Org. Process Res. Dev. 2004, 8, 1059–1064. doi:10.1021/op0498823
Return to citation in text: [1] -
Koelsch, C. F.; Whitney, A. G. J. Org. Chem. 1941, 6, 795–803. doi:10.1021/jo01206a002
Return to citation in text: [1] -
Saranya, S.; Neetha, M.; Aneeja, T.; Anilkumar, G. Adv. Synth. Catal. 2020, 362, 4543–4551. doi:10.1002/adsc.202000827
Return to citation in text: [1] -
Pawar, A. B.; Chang, S. Org. Lett. 2015, 17, 660–663. doi:10.1021/ol503680d
Return to citation in text: [1] -
Kanchana, U. S.; Mathew, T. V.; Anilkumar, G. J. Organomet. Chem. 2020, 920, 121337. doi:10.1016/j.jorganchem.2020.121337
Return to citation in text: [1] -
Neetha, M.; Afsina, C. M. A.; Aneeja, T.; Anilkumar, G. RSC Adv. 2020, 10, 33683–33699. doi:10.1039/d0ra05960a
Return to citation in text: [1] -
Li, Z.-l.; Sun, K.-k.; Cai, C. Org. Chem. Front. 2018, 5, 1848–1853. doi:10.1039/c8qo00322j
Return to citation in text: [1] -
Soumya, P. K.; Vaishak, T. B.; Saranya, S.; Anilkumar, G. Appl. Organomet. Chem. 2021, 35, e6340. doi:10.1002/aoc.6340
Return to citation in text: [1] -
Fairoosa, J.; Shamna, S.; Neetha, M.; Anilkumar, G. Appl. Organomet. Chem. 2021, 35, e6356. doi:10.1002/aoc.6356
Return to citation in text: [1] -
Hughes, T. V.; Cava, M. P. J. Org. Chem. 1999, 64, 313–315. doi:10.1021/jo981924w
Return to citation in text: [1] -
Kahne, D.; Collum, D. B. Tetrahedron Lett. 1981, 22, 5011–5014. doi:10.1016/s0040-4039(01)92406-2
Return to citation in text: [1] -
Davis, W. A.; Cava, M. P. J. Org. Chem. 1983, 48, 2774–2775. doi:10.1021/jo00164a030
Return to citation in text: [1] -
Wheland, R. C.; Martin, E. L. J. Org. Chem. 1975, 40, 3101–3109. doi:10.1021/jo00909a019
Return to citation in text: [1] -
Maas, G. Chem. Soc. Rev. 2004, 33, 183–190. doi:10.1039/b309046a
Return to citation in text: [1] -
Naota, T.; Takaya, H.; Murahashi, S.-I. Chem. Rev. 1998, 98, 2599–2660. doi:10.1021/cr9403695
Return to citation in text: [1] -
Verma, S.; Jain, S. L.; Sain, B. ChemCatChem 2011, 3, 1329–1332. doi:10.1002/cctc.201100111
Return to citation in text: [1] -
Reddy, K. H. V.; Satish, G.; Reddy, V. P.; Anil Kumar, B. S. P.; Nageswar, Y. V. D. RSC Adv. 2012, 2, 11084–11088. doi:10.1039/c2ra21630b
Return to citation in text: [1] -
Kumar, S.; Kumar, P.; Jain, S. L. RSC Adv. 2013, 3, 24013–24016. doi:10.1039/c3ra44051f
Return to citation in text: [1] -
Wang, X.; Xiao, R.; Ai, J.; Cai, M. J. Chem. Res. 2017, 41, 576–580. doi:10.3184/174751917x15064232103065
Return to citation in text: [1] -
Murahashi, S.-I.; Komiya, N.; Terai, H.; Nakae, T. J. Am. Chem. Soc. 2003, 125, 15312–15313. doi:10.1021/ja0390303
Return to citation in text: [1] -
Murahashi, S.-I.; Komiya, N.; Terai, H. Angew. Chem., Int. Ed. 2005, 44, 6931–6933. doi:10.1002/anie.200501496
Return to citation in text: [1] -
Murahashi, S.-I.; Nakae, T.; Terai, H.; Komiya, N. J. Am. Chem. Soc. 2008, 130, 11005–11012. doi:10.1021/ja8017362
Return to citation in text: [1] -
Verma, S.; Jain, S. L.; Sain, B. Catal. Lett. 2011, 141, 882–885. doi:10.1007/s10562-011-0582-6
Return to citation in text: [1] -
Khorshidi, A. Chin. Chem. Lett. 2012, 23, 903–906. doi:10.1016/j.cclet.2012.06.030
Return to citation in text: [1] -
Liu, W.; Ackermann, L. Chem. Commun. 2014, 50, 1878–1881. doi:10.1039/c3cc49502g
Return to citation in text: [1] -
Mishra, A.; Vats, T. K.; Deb, I. J. Org. Chem. 2016, 81, 6525–6534. doi:10.1021/acs.joc.6b01148
Return to citation in text: [1] -
Kumar, P.; Varma, S.; Jain, S. L. J. Mater. Chem. A 2014, 2, 4514–4519. doi:10.1039/c3ta14783e
Return to citation in text: [1] -
Srivastava, V. P.; Yadav, A. K.; Yadav, L. D. S. Tetrahedron Lett. 2014, 55, 1788–1792. doi:10.1016/j.tetlet.2014.01.121
Return to citation in text: [1] -
Dai, J.-J.; Zhang, W.-M.; Shu, Y.-J.; Sun, Y.-Y.; Xu, J.; Feng, Y.-S.; Xu, H.-J. Chem. Commun. 2016, 52, 6793–6796. doi:10.1039/c6cc01530a
Return to citation in text: [1] -
Akula, P. S.; Hong, B.-C.; Lee, G.-H. RSC Adv. 2018, 8, 19580–19584. doi:10.1039/c8ra03259a
Return to citation in text: [1] -
Echevarría, I.; Vaquero, M.; Quesada, R.; Espino, G. Inorg. Chem. Front. 2020, 7, 3092–3105. doi:10.1039/d0qi00580k
Return to citation in text: [1] -
Bédard, A.-C.; Adamo, A.; Aroh, K. C.; Russell, M. G.; Bedermann, A. A.; Torosian, J.; Yue, B.; Jensen, K. F.; Jamison, T. F. Science 2018, 361, 1220–1225. doi:10.1126/science.aat0650
Return to citation in text: [1] -
Filipović, A.; Džambaski, Z.; Vasiljević-Radović, D.; Bondžić, B. P. Org. Biomol. Chem. 2021, 19, 2668–2675. doi:10.1039/d0ob02582h
Return to citation in text: [1] -
De, S. K. Synth. Commun. 2005, 35, 653–656. doi:10.1081/scc-200050347
Return to citation in text: [1] -
Bhor, M. D.; Panda, A. G.; Nandurkar, N. S.; Bhanage, B. M. Tetrahedron Lett. 2008, 49, 6475–6479. doi:10.1016/j.tetlet.2008.09.002
Return to citation in text: [1] -
Wang, D.-J.; Chen, B.-Y.; Wang, Y.-Q.; Zhang, X.-W. Eur. J. Org. Chem. 2018, 1342–1346. doi:10.1002/ejoc.201701651
Return to citation in text: [1] -
Wang, H.; Xu, D.; Guan, E.; Wang, L.; Zhang, J.; Wang, C.; Wang, S.; Xu, H.; Meng, X.; Yang, B.; Gates, B. C.; Xiao, F.-S. ACS Catal. 2020, 10, 6299–6308. doi:10.1021/acscatal.0c00485
Return to citation in text: [1]
| 35. | Verma, S.; Jain, S. L.; Sain, B. Catal. Lett. 2011, 141, 882–885. doi:10.1007/s10562-011-0582-6 |
| 36. | Khorshidi, A. Chin. Chem. Lett. 2012, 23, 903–906. doi:10.1016/j.cclet.2012.06.030 |
| 37. | Liu, W.; Ackermann, L. Chem. Commun. 2014, 50, 1878–1881. doi:10.1039/c3cc49502g |
| 1. | Ding, R.; Liu, Y.; Han, M.; Jiao, W.; Li, J.; Tian, H.; Sun, B. J. Org. Chem. 2018, 83, 12939–12944. doi:10.1021/acs.joc.8b02190 |
| 7. | Haddenham, D.; Pasumansky, L.; DeSoto, J.; Eagon, S.; Singaram, B. J. Org. Chem. 2009, 74, 1964–1970. doi:10.1021/jo8023329 |
| 8. | Knight, J. A.; Zook, H. D. J. Am. Chem. Soc. 1952, 74, 4560–4562. doi:10.1021/ja01138a031 |
| 9. | Demko, Z. P.; Sharpless, K. B. J. Org. Chem. 2001, 66, 7945–7950. doi:10.1021/jo010635w |
| 10. | Garduño, J. A.; Garcı́a, J. J. ACS Catal. 2015, 5, 3470–3477. doi:10.1021/acscatal.5b00348 |
| 11. | Allen, C. L.; Lapkin, A. A.; Williams, J. M. J. Tetrahedron Lett. 2009, 50, 4262–4264. doi:10.1016/j.tetlet.2009.05.021 |
| 22. | Hughes, T. V.; Cava, M. P. J. Org. Chem. 1999, 64, 313–315. doi:10.1021/jo981924w |
| 44. | Bédard, A.-C.; Adamo, A.; Aroh, K. C.; Russell, M. G.; Bedermann, A. A.; Torosian, J.; Yue, B.; Jensen, K. F.; Jamison, T. F. Science 2018, 361, 1220–1225. doi:10.1126/science.aat0650 |
| 4. | Iyer, M. S.; Gigstad, K. M.; Namdev, N. D.; Lipton, M. J. Am. Chem. Soc. 1996, 118, 4910–4911. doi:10.1021/ja952686e |
| 5. | Rafiee, E.; Azad, A. Synth. Commun. 2007, 37, 1127–1132. doi:10.1080/00397910701198914 |
| 6. | Rafiee, E.; Rashidzadeh, S.; Azad, A. J. Mol. Catal. A: Chem. 2007, 261, 49–52. doi:10.1016/j.molcata.2006.07.058 |
| 23. | Kahne, D.; Collum, D. B. Tetrahedron Lett. 1981, 22, 5011–5014. doi:10.1016/s0040-4039(01)92406-2 |
| 45. | Filipović, A.; Džambaski, Z.; Vasiljević-Radović, D.; Bondžić, B. P. Org. Biomol. Chem. 2021, 19, 2668–2675. doi:10.1039/d0ob02582h |
| 3. | Strecker, A. Justus Liebigs Ann. Chem. 1850, 75, 27–45. doi:10.1002/jlac.18500750103 |
| 20. | Soumya, P. K.; Vaishak, T. B.; Saranya, S.; Anilkumar, G. Appl. Organomet. Chem. 2021, 35, e6340. doi:10.1002/aoc.6340 |
| 42. | Akula, P. S.; Hong, B.-C.; Lee, G.-H. RSC Adv. 2018, 8, 19580–19584. doi:10.1039/c8ra03259a |
| 2. | Enders, D.; Shilvock, J. P. Chem. Soc. Rev. 2000, 29, 359–373. doi:10.1039/a908290e |
| 21. | Fairoosa, J.; Shamna, S.; Neetha, M.; Anilkumar, G. Appl. Organomet. Chem. 2021, 35, e6356. doi:10.1002/aoc.6356 |
| 43. | Echevarría, I.; Vaquero, M.; Quesada, R.; Espino, G. Inorg. Chem. Front. 2020, 7, 3092–3105. doi:10.1039/d0qi00580k |
| 18. | Neetha, M.; Afsina, C. M. A.; Aneeja, T.; Anilkumar, G. RSC Adv. 2020, 10, 33683–33699. doi:10.1039/d0ra05960a |
| 40. | Srivastava, V. P.; Yadav, A. K.; Yadav, L. D. S. Tetrahedron Lett. 2014, 55, 1788–1792. doi:10.1016/j.tetlet.2014.01.121 |
| 15. | Saranya, S.; Neetha, M.; Aneeja, T.; Anilkumar, G. Adv. Synth. Catal. 2020, 362, 4543–4551. doi:10.1002/adsc.202000827 |
| 19. | Li, Z.-l.; Sun, K.-k.; Cai, C. Org. Chem. Front. 2018, 5, 1848–1853. doi:10.1039/c8qo00322j |
| 41. | Dai, J.-J.; Zhang, W.-M.; Shu, Y.-J.; Sun, Y.-Y.; Xu, J.; Feng, Y.-S.; Xu, H.-J. Chem. Commun. 2016, 52, 6793–6796. doi:10.1039/c6cc01530a |
| 13. | Nielsen, M. A.; Nielsen, M. K.; Pittelkow, T. Org. Process Res. Dev. 2004, 8, 1059–1064. doi:10.1021/op0498823 |
| 14. | Koelsch, C. F.; Whitney, A. G. J. Org. Chem. 1941, 6, 795–803. doi:10.1021/jo01206a002 |
| 38. | Mishra, A.; Vats, T. K.; Deb, I. J. Org. Chem. 2016, 81, 6525–6534. doi:10.1021/acs.joc.6b01148 |
| 17. | Kanchana, U. S.; Mathew, T. V.; Anilkumar, G. J. Organomet. Chem. 2020, 920, 121337. doi:10.1016/j.jorganchem.2020.121337 |
| 39. | Kumar, P.; Varma, S.; Jain, S. L. J. Mater. Chem. A 2014, 2, 4514–4519. doi:10.1039/c3ta14783e |
| 24. | Davis, W. A.; Cava, M. P. J. Org. Chem. 1983, 48, 2774–2775. doi:10.1021/jo00164a030 |
| 25. | Wheland, R. C.; Martin, E. L. J. Org. Chem. 1975, 40, 3101–3109. doi:10.1021/jo00909a019 |
| 47. | Bhor, M. D.; Panda, A. G.; Nandurkar, N. S.; Bhanage, B. M. Tetrahedron Lett. 2008, 49, 6475–6479. doi:10.1016/j.tetlet.2008.09.002 |
| 48. | Wang, D.-J.; Chen, B.-Y.; Wang, Y.-Q.; Zhang, X.-W. Eur. J. Org. Chem. 2018, 1342–1346. doi:10.1002/ejoc.201701651 |
| 33. | Murahashi, S.-I.; Komiya, N.; Terai, H. Angew. Chem., Int. Ed. 2005, 44, 6931–6933. doi:10.1002/anie.200501496 |
| 34. | Murahashi, S.-I.; Nakae, T.; Terai, H.; Komiya, N. J. Am. Chem. Soc. 2008, 130, 11005–11012. doi:10.1021/ja8017362 |
| 31. | Wang, X.; Xiao, R.; Ai, J.; Cai, M. J. Chem. Res. 2017, 41, 576–580. doi:10.3184/174751917x15064232103065 |
| 32. | Murahashi, S.-I.; Komiya, N.; Terai, H.; Nakae, T. J. Am. Chem. Soc. 2003, 125, 15312–15313. doi:10.1021/ja0390303 |
| 29. | Reddy, K. H. V.; Satish, G.; Reddy, V. P.; Anil Kumar, B. S. P.; Nageswar, Y. V. D. RSC Adv. 2012, 2, 11084–11088. doi:10.1039/c2ra21630b |
| 30. | Kumar, S.; Kumar, P.; Jain, S. L. RSC Adv. 2013, 3, 24013–24016. doi:10.1039/c3ra44051f |
| 27. | Naota, T.; Takaya, H.; Murahashi, S.-I. Chem. Rev. 1998, 98, 2599–2660. doi:10.1021/cr9403695 |
| 49. | Wang, H.; Xu, D.; Guan, E.; Wang, L.; Zhang, J.; Wang, C.; Wang, S.; Xu, H.; Meng, X.; Yang, B.; Gates, B. C.; Xiao, F.-S. ACS Catal. 2020, 10, 6299–6308. doi:10.1021/acscatal.0c00485 |
| 28. | Verma, S.; Jain, S. L.; Sain, B. ChemCatChem 2011, 3, 1329–1332. doi:10.1002/cctc.201100111 |
© 2022 Aneeja et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.