Abstract
A series of tetraamino-bisthiourea chiral macrocycles containing two diarylthiourea and two chiral diamine units were synthesized by a fragment-coupling approach in high yields. Different chiral diamine units, including cyclohexanediamines and diphenylethanediamines were readily incorporated by both homo and hetero [1 + 1] macrocyclic condensation of bisamine and bisisothiocyanate fragments. With the easy synthesis, gram-scale of macrocycle products can be readily obtained. These chiral macrocycles were applied in catalyzing bioinspired decarboxylative Mannich reactions. Only 5 mol % of the optimal macrocycle catalyst efficiently catalyzed the decarboxylative addition of a broad scope of malonic acid half thioesters to isatin-derived ketimines with excellent yields and good enantioselectivity. The rigid macrocyclic framework and the cooperation between the thiourea and tertiary amine sites were found to be crucial for achieving efficient activation and stereocontrol. As shown in control experiments, catalysis with the acyclic analogues having the same structural motifs were non-selective.

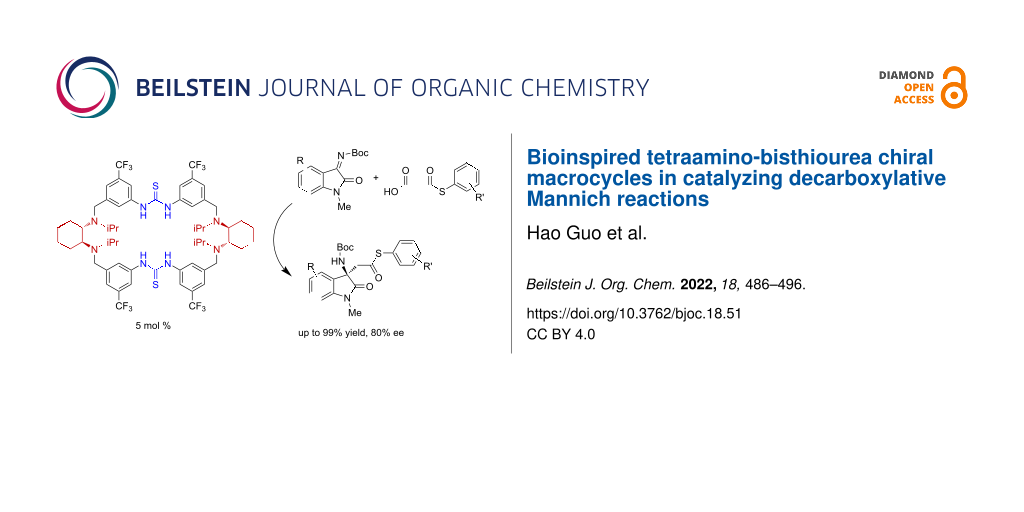
Graphical Abstract
Introduction
In the past decades, the development of supramolecular chemistry has enabled abundant host scaffolds and assembly tools for boosting catalytic processes, and stimulated the emergence of supramolecular catalysis [1-14]. Among which, macrocyclic compounds have attracted extensive attentions due to their enzyme-mimicking cavity and preorganized binding sites [4,6,15,16]. Various macrocyclic compounds including the privileged scaffolds like cyclodextrins [17-19], calixarenes [20-23], cucurbiturils [24,25], and cavitands [26,27] have been widely applied. While these conventional macrocycles can usually enable a confinement effect or serve as a supporting scaffold, they do not contain definite catalytic sites in their cyclic skeletons. When required, an additional catalytic functional group was commonly introduced through in-situ, noncovalent inclusion/encapsulation in the cavity or by covalent, post-functionalization of the macrocyclic scaffold. The encapsulated catalytic group could occupy the space for substrate entering, or has a risk to be squeezed out of the cavity under complex catalytic conditions. On the other hand, the covalently pendant catalytic group may reside far away from the center of the cavity.
On the other hand, we envisaged by use of tailored building units already containing definite catalytic sites to directly form a macrocycle scaffold could provide a different situation. In this way the catalytic functionalities are permanently installed within the macrocyclic skeleton by forming a persistent catalytic cavity. Following this idea, we have recently constructed a series of bisthiourea macrocycles [28-30]. Thiourea groups were introduced due to their superior anion binding property and potent electrophilic activation ability [31-36]. To incorporate extra functionality, tertiary amine groups can be also embedded as Lewis base sites for realizing electrophilic/nucleophilic cooperative catalysis [37-39]. For this purpose, one kind of tetraamino-bisthiourea chiral macrocycles were synthesized [30]. When applied in catalyzing the decarboxylative addition of phenyl β-ketoacids to cyclic imines bearing sulfamate heading group, an interesting substrate-induced assembly catalysis mode was uncovered [30]. To expand more applications, herein we report a systematic synthesis of tetraamino-bisthiourea chiral macrocycles and their performance in catalyzing the decarboxylative Mannich addition of malonic acid half thioesters (MAHTs) to isatin-derived ketimines. The macrocycle-enabled hydrogen-bonding activation network and the associated confined cavity could resemble the circumstance of the catalytic triad of Polyketide synthases (PKSs) [40-42] (Figure 1). On the other hand, the organocatalytic asymmetric decarboxylative addition reactions of MAHTs to imines provide an efficient means for accessing valuable chiral β-amino esters [43-52].
![[1860-5397-18-51-1]](/bjoc/content/figures/1860-5397-18-51-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Design of PKS-inspired multifunctional amino-thiourea macrocycle catalysts.
Figure 1: Design of PKS-inspired multifunctional amino-thiourea macrocycle catalysts.
Results and Discussion
Synthesis of macrocycles
The tetraamino-bisthiourea chiral macrocycles were synthesized by a stepwise strategy (Scheme 1). The easily available chiral diamines including 1,2-cyclohexanediamines and 1,2-diphenylethylenediamines were chosen as the linking components to afford Lewis base sites and also for introduction of chirality. Different alkyl substituents including methyl, n-propyl, isopropyl, and 3-pentyl were incorporated in order to tune the size and steric effect of the macrocyclic cavity and thus to enable diverse cavity environments. Among these macrocycles, M1, M5, M7, and M8 were previously synthesized [30] and the route can be similarly followed for the synthesis of the other macrocycles. To start the synthesis, enantiopure N,N’-disubstituted (S,S)-1,2-cyclohexanediamines 1a–d or (S,S)-1,2-diphenylethanediamines 1e,f were firstly reacted with two equiv 3-nitro-5-(trifluoromethyl)benzyl bromide (2) in the presence of a base to afford the dinitro compounds 3a–f in moderate to excellent yields (Scheme 1a). The diminished yield for product 3d was probably caused by the large steric hindrance of the 3-pentyl substituent. Reduction of the nitro groups by SnCl2 under acidic conditions gave the bisamine fragments 4a–f in 83–98% yields. The bisamine fragments were further converted to the bisisothiocyanates 5a–f by reaction with 1,1'-thiocarbonyldiimidazole in 66–89% yields.
![[1860-5397-18-51-i1]](/bjoc/content/inline/1860-5397-18-51-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of tetraamino-bisthiourea chiral macrocycles M1–M12. The synthesis of M1, M5, M7, and M8 was previously reported [30].
Scheme 1: Synthesis of tetraamino-bisthiourea chiral macrocycles M1–M12. The synthesis of M1, M5, M7, and M8 ...
Having the bisamine and bisisothiocyanate fragments in hand, macrocyclic condensations were then pursued. The homo-condensations between the homologous bisamine and bisisothiocyanate fragments were firstly tried (Scheme 1b). In the presence of an organic base, reactions between 4a–f and 5a–f went smoothly and afforded the desired macrocycle products M1–M6 in 35–72% yields. It is worth noting that common dilute conditions for macrocyclization reactions was not required here. Due to the very high efficiency, gram-scale preparation of the chiral macrocycles was readily achieved (see Supporting Information File 1). To enrich the diversity of the macrocyclic scaffolds, hetero-condensations between different bisamine and bisisothiocyanate fragments, including combination of different chiral configurations, were also investigated (Scheme 1c). Reactions between cyclohexanediamine-derived bisamine fragments 4a or the enantiomers ent-4a–c with diphenylethylenediamine-derived bisisothiocyanate fragments 5e,f afforded the desired hetero-combination macrocycles M7–M12 without additional difficulties. It should be noted that the incorporation of CF3 groups on the aryl moieties was to increase the acidity of thiourea so as to provide better hydrogen-bonding complexation and activation ability.
Catalytic reaction optimization
The synthesized macrocycles were then applied as catalysts in the decarboxylative addition of malonic acid half thioesters (MAHTs) to isatin-derived ketimines [48]. The reaction between ketimine 6a and MAHT 7a was initially performed in THF at room temperature with just 2 mol % loading of the chiral macrocycle catalysts (Table 1). All macrocycles were evaluated, and in all cases product 8a was obtained in moderate yields. Different diamine linking components and different substituents on the tertiary amine sites showed an important influence on the reaction stereoselectivity. The cyclohexanediamine-linking macrocycles M1–M4 afforded the product with overall higher enantiomeric excess (ee) (Table 1, entries 1–4). Among which the isopropyl-substituted macrocycle M3 gave the best selectivity, i.e., 42% ee. This suggested a suitable crowding cavity environment may be good for stereocontrol. The diphenylethylenediamine-linking macrocycles M5 and M6, however, gave very low ees (Table 1, entries 5 and 6). The hetero-macrocycles M7–M12 did not afford better selectivity as well (Table 1, entries 7–12). It is interesting to note that M9–M12 led to reversed selectivity, which may imply that the chiral cyclohexanediamine other than diphenylethylenediamine moiety governed the stereoselection process.
Table 1: Evaluation of different macrocycle catalystsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-51-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Cat. | Yield (%)b | ee (%)c |
| 1 | M1 | 32 | 9 |
| 2 | M2 | 43 | 11 |
| 3 | M3 | 41 | 42 |
| 4 | M4 | 49 | 29 |
| 5 | M5 | 48 | 4 |
| 6 | M6 | 51 | 0 |
| 7 | M7 | 45 | 13 |
| 8 | M8 | 39 | 9 |
| 9 | M9 | 31 | −4 |
| 10 | M10 | 42 | −12 |
| 11 | M11 | 31 | −12 |
| 12 | M12 | 41 | −4 |
aReaction conditions: 6a (0.2 mmol), 7a (0.3 mmol), 1 mL of THF; bisolated yields after column chromatography; cdetermined by HPLC analysis on a chiral stationary phase.
Using M3 as the optimal catalyst, the reaction solvent was then screened (Table 2). Ethyl ether was found to give a better conversion, but with decreased selectivity (Table 2, entry 2). The reaction in 1,4-dioxane afforded the product with a moderate yield and the best selectivity so far, 62% ee (Table 2, entry 3). To our delight, among the other ether solvents screened (Table 2, entries 4–7), cyclopentyl methyl ether (CPME) gave an excellent conversion (90% yield) and only a slightly diminished selectivity (58% ee). Reactions in other more polar solvents including toluene, ethyl acetate, halohydrocarbons, and acetonitrile gave overall very good conversions, but with very low selectivity except for the reaction in ethyl acetate which afforded the product in 43% ee (Table 2, entries 8–12).
Table 2: Evaluation of solventsa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-51-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Time (h) | Yield (%)b | ee (%)c |
| 1 | THF | 96 | 41 | 42 |
| 2 | Et2O | 96 | 73 | 34 |
| 3 | 1,4-dioxane | 96 | 52 | 62 |
| 4 | TBME | 96 | 79 | 33 |
| 5 | CPME | 96 | 90 | 58 |
| 6 | DME | 96 | 47 | 49 |
| 7 | EVE | 96 | 39 | 9 |
| 8 | toluene | 36 | 96 | 2 |
| 9 | EA | 84 | 81 | 43 |
| 10 | CH2Cl2 | 36 | 97 | 0 |
| 11 | CHCl3 | 36 | 93 | 1 |
| 12 | CH3CN | 84 | 71 | 12 |
aReaction conditions: 6a (0.2 mmol), 7a (0.3 mmol), 1 mL of solvent; bisolated yields after column chromatography; cdetermined by HPLC analysis on a chiral stationary phase. TBME: tert-butyl methyl ether; CPME: cyclopentyl methyl ether; DME: 1,2-dimethoxyethane; EVE: ethyl vinyl ether; EA: ethyl acetate.
Finally, the other reaction parameters, including catalyst loading, reaction temperature, and concentration were evaluated (Table 3). With CPME as the optimal solvent, increasing the loading of catalyst M3 from 2 mol % to 5 mol % led to an obviously more rapid conversion and furnished the product in 92% yield in 36 h (Table 3, entries 1 and 2). To our delight, the selectivity was also increased to 71% ee. However, further increasing the macrocycle loading to 10 mol % led to a diminished yield and nearly unchanged selectivity (Table 3, entry 3). The decreased yield for the addition product was due to the competitive decarboxylation of the sole MAHT substrate in the presence of a higher loading of the macrocycle containing tertiary amine basic sites. Decreasing the amount of MAHT substrate from 1.5 equiv to 1.0 equiv or increasing to 2.0 equiv did not give better outcome (Table 3, entries 4 and 5). Performing the reaction at 0 °C led to a very slow conversion, while at 40 °C the reaction became much faster but gave a diminished yield due to the competitive decarboxylation side reaction (Table 3, entries 6 and 7). In both cases, the enantioselectivity did not turn out to be better. A suitable reaction concentration (0.1–0.2 M) was found to be important. A very high or low reaction concentration led to decreased stereoselectivity probably due to the existence of catalyst aggregation or background reactions (Table 3, entries 8–10).
Table 3: Evaluation of catalyst loading, reaction temperature, and concentrationa.
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-51-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Cat. (mol %) | Temp. | Conc. [M]b | Time (h) | Yield (%)c | ee (%)d |
| 1 | M3 (2) | rt | 0.2 | 96 | 90 | 58 |
| 2 | M3 (5) | rt | 0.2 | 36 | 92 | 71 |
| 3 | M3 (10) | rt | 0.2 | 24 | 72 | 72 |
| 4e | M3 (5) | rt | 0.2 | 24 | 82 | 72 |
| 5f | M3 (5) | rt | 0.2 | 48 | 94 | 60 |
| 6 | M3 (5) | 0 °C | 0.2 | 120 | 65 | 63 |
| 7 | M3 (5) | 40 °C | 0.2 | 12 | 75 | 64 |
| 8 | M3 (5) | rt | 0.4 | 24 | 92 | 56 |
| 9 | M3 (5) | rt | 0.1 | 44 | 88 | 72 |
| 10 | M3 (5) | rt | 0.05 | 48 | 54 | 59 |
aReaction conditions: 6a (0.2 mmol) and 7a (0.3 mmol, 1.5 equiv) in CPME (cyclopentyl methyl ether) except otherwise noted; bconcentration of 6a; cisolated yields after column chromatography; ddetermined by HPLC analysis on a chiral stationary phase; e1.0 equiv 7a used; f2.0 equiv 7a used.
Substrate scope
Having established the optimal reaction conditions, the substrate scope was explored. Reactions of various isatin imines 6a–w with MAHT 7a were firstly investigated (Scheme 2). Different N-substituents on isatins caused a significant effect. For non-substituted (6b) or other substrates with larger substituents (6c–g), the corresponding products 8b–g were obtained in only moderate yields with decreased selectivity. Replacing the Boc-protecting group on the imine site by a Cbz group led to a largely decreased selectivity (8h). For a series of substrates with various substituents on the 5, 6, or 7-position, including 5-methyl (8i), 5-methoxy (8k), 6- or 7-fluoro (8l,m), 5,6-difluoro (8n), 5-, 6- or 7-chloro (8p–r), 5-, 6-, or 7-bromo (8t–v), and 7-trifluoromethyl (8w), all reactions completed within 36–48 h and afforded the products in good to excellent yields with 60–75% ee. In contrast, the reaction was disrupted by 4-substitution, and only trace conversion was observed for the 4-chloro or 4-bromo-substituted substrate (8o,s). This was probably caused by a steric effect as the 4-substituent is close to the imine reactive site and accordingly blocked it toward activation and nucleophilic attack. Interestingly, for the 5,7-dimethyl-substituted substrate, only trace conversion was observed as well (8j). This may be due to that the substrate was too bulky to fit within the macrocycle cavity.
![[1860-5397-18-51-i2]](/bjoc/content/inline/1860-5397-18-51-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of isatin imines. Reaction conditions: 6 (0.2 mmol), 7a (0.3 mmol), and 5 mol % M3 in 2 mL of CPME (cyclopentyl methyl ether).
Scheme 2: Substrate scope of isatin imines. Reaction conditions: 6 (0.2 mmol), 7a (0.3 mmol), and 5 mol % M3 ...
For MAHT substrates, various p- or m-substituents on the S-phenyl moiety caused negligible effects, and all the products were obtained in moderate to good yields with 69–80% ee (Scheme 3). For o-substitution, especially for the o-fluoro-substituent (8ag), however, only a moderate yield and low selectivity (12% ee) were obtained. For the S-naphthyl substrate, the reaction went smoothly as well and afforded the product in 65% yield with 68% ee (8al). The reactions for S-benzyl and S-ethyl substrates became sluggish and afforded the products in 34–48% yields in 96 h with very low selectivity (8am and 8an). For the S-tert-butyl substrate, only trace conversion was observed.
![[1860-5397-18-51-i3]](/bjoc/content/inline/1860-5397-18-51-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of MAHTs. Reaction conditions: 6a (0.2 mmol), 7 (0.3 mmol), and 5 mol % M3 in 2 mL of CPME (cyclopentyl methyl ether).
Scheme 3: Substrate scope of MAHTs. Reaction conditions: 6a (0.2 mmol), 7 (0.3 mmol), and 5 mol % M3 in 2 mL ...
Macrocyclic effect and catalytic mechanism
The above results showed that the tetraamino-bisthiourea chiral macrocycles can efficiently catalyze the decarboxylative addition reactions with good yields and enantioselectivity. To check the role of the macrocyclic framework, two acyclic compounds (9 and 3c) containing the similar structural motifs as the macrocycle catalyst were applied as catalysts in the reaction (Table 4). In compound 9, all the structural units of the macrocycle M3, including the two diarylthioureas and the four tertiary amine sites, were maximally maintained, except for one of the cyclohexanediamine units which was replaced by two dimethylamino groups to cut off the macrocyclic skeleton (for synthesis, see Supporting Information File 1). The compound 3c contains one cyclohexanediamine unit but no thiourea moieties. As shown in Table 4, macrocycle M3 catalyzed the reaction of 6a and 7a to afford 8a in 88% yield with 72% ee. Under the same conditions, compound 9 also efficiently catalyzed the reaction and promoted an excellent conversion, however, it furnished the product in nearly racemic form. This suggested that the macrocyclic framework is essential in enabling a defined chiral environment for efficient stereocontrol. On the other hand, compound 3c led to a much slower conversion and also nearly racemic product formation, indicating that the thiourea units must have engaged in activation and being cooperative with the tertiary amine sites.
Table 4: Evaluation of macrocyclic effecta.
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-51-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Cat. | Time (h) | Yield (%)b | ee (%)c |
| 1 | M3 (5 mol %) | 44 | 88 | 72 |
| 2 | 9 (5 mol %) | 44 | 92 | 3 |
| 3 | 3c (10 mol %) | 72 | 47 | −2 |
aReaction conditions: 6a (0.2 mmol), 7a (0.3 mmol), 2 mL of CPME (cyclopentyl methyl ether), room temperature; bisolated yields after column chromatography; cdetermined by HPLC analysis on a chiral stationary phase.
With the reaction outcomes and pronounced macrocyclic effect observed, a plausible catalytic mechanism is represented in Figure 2. The MAHT substrate is deprotonated by one of the tertiary amine sites, and the formed enolate intermediate can be stabilized by hydrogen bonding-mediated ion-pair interaction within the macrocyclic cavity. The imine substrate is activated by one or both of the two thiourea sites through hydrogen bonding to accept the enolate attack. The chiral environment provided by the (S,S)-cyclohexanediamine part governed the face-selective attack and led to the R-configurated product. In the last step, the decarboxylation leads to the enolate of the thioester, which is more basic than the MAHT-enolate and can thus be protonated by the ammonium fragment in the macrocycle. This leads to the neutral product, which can easily escape from the macrocyclic cavity, releasing the macrocycle catalyst to enter the next catalytic cycle. As suggested by the above control experiments, the rigid macrocyclic framework is crucial in organizing the multiple functional sites for cooperative binding and activation.
![[1860-5397-18-51-2]](/bjoc/content/figures/1860-5397-18-51-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, a series of multifunctional tetraamino-bisthiourea chiral macrocycles were efficiently synthesized. By using a modular fragment-coupling approach, different chiral diamine units, including the homo- and hetero-combination, can be easily incorporated. This provides a very rich structural diversity of the macrocycles and allows for fine tuning of the chiral cavity environments. With the short and high-yielding synthesis, gram-scale of macrocycle products can be readily obtained. This kind of macrocycles can efficiently catalyze the decarboxylative addition of malonic acid half thioesters to isatin-derived ketimines, affording a series of chiral β-amino ester products in excellent yields and good enantioselectivity. In contrast, reactions catalyzed by acyclic analogues containing very similar structural units were non-selective, suggesting the essential role of the rigid macrocyclic framework in realizing efficient stereocontrol. With the easy synthesis, rich structural diversity, cooperative binding and activation sites, we believe this type of biomimetic chiral macrocycles will find more applications as catalysts in other reactions.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 10.5 MB | Download |
References
-
van Leeuwen, P. W. N. M., Ed. Supramolecular Catalysis; Wiley-VCH: Weinheim, Germany, 2008. doi:10.1002/9783527621781
Return to citation in text: [1] -
Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340
Return to citation in text: [1] -
Meeuwissen, J.; Reek, J. N. H. Nat. Chem. 2010, 2, 615–621. doi:10.1038/nchem.744
Return to citation in text: [1] -
Dong, Z.; Luo, Q.; Liu, J. Chem. Soc. Rev. 2012, 41, 7890–7908. doi:10.1039/c2cs35207a
Return to citation in text: [1] [2] -
Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1660–1733. doi:10.1039/c3cs60027k
Return to citation in text: [1] -
Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1734–1787. doi:10.1039/c3cs60037h
Return to citation in text: [1] [2] -
Dydio, P.; Reek, J. N. H. Chem. Sci. 2014, 5, 2135–2145. doi:10.1039/c3sc53505c
Return to citation in text: [1] -
Leenders, S. H. A. M.; Gramage-Doria, R.; de Bruin, B.; Reek, J. N. H. Chem. Soc. Rev. 2015, 44, 433–448. doi:10.1039/c4cs00192c
Return to citation in text: [1] -
Brown, C. J.; Toste, F. D.; Bergman, R. G.; Raymond, K. N. Chem. Rev. 2015, 115, 3012–3035. doi:10.1021/cr4001226
Return to citation in text: [1] -
Blanco, V.; Leigh, D. A.; Marcos, V. Chem. Soc. Rev. 2015, 44, 5341–5370. doi:10.1039/c5cs00096c
Return to citation in text: [1] -
Vaquero, M.; Rovira, L.; Vidal-Ferran, A. Chem. Commun. 2016, 52, 11038–11051. doi:10.1039/c6cc04474c
Return to citation in text: [1] -
Davis, H. J.; Phipps, R. J. Chem. Sci. 2017, 8, 864–877. doi:10.1039/c6sc04157d
Return to citation in text: [1] -
Jiang, J.; Ouyang, G.; Zhang, L.; Liu, M. Chem. – Eur. J. 2017, 23, 9439–9450. doi:10.1002/chem.201700727
Return to citation in text: [1] -
Wang, K.; Jordan, J. H.; Hu, X.-Y.; Wang, L. Angew. Chem., Int. Ed. 2020, 59, 13712–13721. doi:10.1002/anie.202000045
Return to citation in text: [1] -
Wang, Q.-Q. Supramolecular Catalysis Using Organic Macrocycles. In Handbook of Macrocyclic Supramolecular Assembly; Liu, Y.; Chen, Y.; Zhang, H.-Y., Eds.; Springer: Singapore, 2019. doi:10.1007/978-981-13-1744-6_36-1
Return to citation in text: [1] -
Kauerhof, D.; Niemeyer, J. ChemPlusChem 2020, 85, 889–899. doi:10.1002/cplu.202000152
Return to citation in text: [1] -
Breslow, R.; Dong, S. D. Chem. Rev. 1998, 98, 1997–2012. doi:10.1021/cr970011j
Return to citation in text: [1] -
Takahashi, K. Chem. Rev. 1998, 98, 2013–2034. doi:10.1021/cr9700235
Return to citation in text: [1] -
Engeldinger, E.; Armspach, D.; Matt, D. Chem. Rev. 2003, 103, 4147–4174. doi:10.1021/cr030670y
Return to citation in text: [1] -
Homden, D. M.; Redshaw, C. Chem. Rev. 2008, 108, 5086–5130. doi:10.1021/cr8002196
Return to citation in text: [1] -
Le Poul, N.; Le Mest, Y.; Jabin, I.; Reinaud, O. Acc. Chem. Res. 2015, 48, 2097–2106. doi:10.1021/acs.accounts.5b00152
Return to citation in text: [1] -
Salvio, R.; Cacciapaglia, R.; Casnati, A. Calixarenes as Supramoleuclar Catalysts Endowed with Esterase and Phosphodiesterase Activity. In Calixarenes and Beyond; Neri, P.; Sessler, J. L.; Wang, M.-X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp 691–717. doi:10.1007/978-3-319-31867-7_26
Return to citation in text: [1] -
Yilmaz, M.; Sayin, S. Calixarenes in Organo and Biomimetic Catalysis. In Calixarenes and Beyond; Neri, P.; Sessler, J. L.; Wang, M.-X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp 719–742. doi:10.1007/978-3-319-31867-7_27
Return to citation in text: [1] -
Pemberton, B. C.; Raghunathan, R.; Volla, S.; Sivaguru, J. Chem. – Eur. J. 2012, 18, 12178–12190. doi:10.1002/chem.201202083
Return to citation in text: [1] -
Assaf, K. I.; Nau, W. M. Chem. Soc. Rev. 2015, 44, 394–418. doi:10.1039/c4cs00273c
Return to citation in text: [1] -
Hooley, R. J.; Rebek, J., Jr. Chem. Biol. 2009, 16, 255–264. doi:10.1016/j.chembiol.2008.09.015
Return to citation in text: [1] -
Yu, Y.; Rebek, J., Jr. Acc. Chem. Res. 2018, 51, 3031–3040. doi:10.1021/acs.accounts.8b00269
Return to citation in text: [1] -
Ning, R.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Chem. – Eur. J. 2018, 24, 4268–4272. doi:10.1002/chem.201800326
Return to citation in text: [1] -
Ning, R.; Zhou, H.; Nie, S.-X.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 10894–10898. doi:10.1002/anie.202003673
Return to citation in text: [1] -
Guo, H.; Zhang, L.-W.; Zhou, H.; Meng, W.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 2623–2627. doi:10.1002/anie.201910399
Return to citation in text: [1] [2] [3] [4] [5] -
Li, A.-F.; Wang, J.-H.; Wang, F.; Jiang, Y.-B. Chem. Soc. Rev. 2010, 39, 3729–3745. doi:10.1039/b926160p
Return to citation in text: [1] -
Amendola, V.; Fabbrizzi, L.; Mosca, L. Chem. Soc. Rev. 2010, 39, 3889–3915. doi:10.1039/b822552b
Return to citation in text: [1] -
Wittkopp, A.; Schreiner, P. R. Chem. – Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042
Return to citation in text: [1] -
Schreiner, P. R. Chem. Soc. Rev. 2003, 32, 289–296. doi:10.1039/b107298f
Return to citation in text: [1] -
Zhang, Z.; Schreiner, P. R. Chem. Soc. Rev. 2009, 38, 1187–1198. doi:10.1039/b801793j
Return to citation in text: [1] -
Park, Y.; Harper, K. C.; Kuhl, N.; Kwan, E. E.; Liu, R. Y.; Jacobsen, E. N. Science 2017, 355, 162–166. doi:10.1126/science.aal1875
Return to citation in text: [1] -
Takemoto, Y. Org. Biomol. Chem. 2005, 3, 4299–4306. doi:10.1039/b511216h
Return to citation in text: [1] -
Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/b719249e
Return to citation in text: [1] -
Fang, X.; Wang, C.-J. Chem. Commun. 2015, 51, 1185–1197. doi:10.1039/c4cc07909d
Return to citation in text: [1] -
Jez, J. M.; Austin, M. B.; Ferrer, J.-L.; Bowman, M. E.; Schröder, J.; Noel, J. P. Chem. Biol. 2000, 7, 919–930. doi:10.1016/s1074-5521(00)00041-7
Return to citation in text: [1] -
Austin, M. B.; Izumikawa, M.; Bowman, M. E.; Udwary, D. W.; Ferrer, J.-L.; Moore, B. S.; Noel, J. P. J. Biol. Chem. 2004, 279, 45162–45174. doi:10.1074/jbc.m406567200
Return to citation in text: [1] -
Zhang, Y.-M.; Hurlbert, J.; White, S. W.; Rock, C. O. J. Biol. Chem. 2006, 281, 17390–17399. doi:10.1074/jbc.m513199200
Return to citation in text: [1] -
Pan, Y.; Tan, C.-H. Synthesis 2011, 2044–2053. doi:10.1055/s-0030-1260607
Return to citation in text: [1] -
Wang, Z.-L. Adv. Synth. Catal. 2013, 355, 2745–2755. doi:10.1002/adsc.201300375
Return to citation in text: [1] -
Nakamura, S. Org. Biomol. Chem. 2014, 12, 394–405. doi:10.1039/c3ob42161a
Return to citation in text: [1] -
Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536
Return to citation in text: [1] -
Pan, Y.; Kee, C. W.; Jiang, Z.; Ma, T.; Zhao, Y.; Yang, Y.; Xue, H.; Tan, C.-H. Chem. – Eur. J. 2011, 17, 8363–8370. doi:10.1002/chem.201100687
Return to citation in text: [1] -
Hara, N.; Nakamura, S.; Sano, M.; Tamura, R.; Funahashi, Y.; Shibata, N. Chem. – Eur. J. 2012, 18, 9276–9280. doi:10.1002/chem.201200367
Return to citation in text: [1] [2] -
Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307
Return to citation in text: [1] -
Bahlinger, A.; Fritz, S. P.; Wennemers, H. Angew. Chem., Int. Ed. 2014, 53, 8779–8783. doi:10.1002/anie.201310532
Return to citation in text: [1] -
Nakamura, S.; Sano, M.; Toda, A.; Nakane, D.; Masuda, H. Chem. – Eur. J. 2015, 21, 3929–3932. doi:10.1002/chem.201406270
Return to citation in text: [1] -
Kaur, J.; Kumari, A.; Bhardwaj, V. K.; Chimni, S. S. Adv. Synth. Catal. 2017, 359, 1725–1734. doi:10.1002/adsc.201700011
Return to citation in text: [1]
| 1. | van Leeuwen, P. W. N. M., Ed. Supramolecular Catalysis; Wiley-VCH: Weinheim, Germany, 2008. doi:10.1002/9783527621781 |
| 2. | Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340 |
| 3. | Meeuwissen, J.; Reek, J. N. H. Nat. Chem. 2010, 2, 615–621. doi:10.1038/nchem.744 |
| 4. | Dong, Z.; Luo, Q.; Liu, J. Chem. Soc. Rev. 2012, 41, 7890–7908. doi:10.1039/c2cs35207a |
| 5. | Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1660–1733. doi:10.1039/c3cs60027k |
| 6. | Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1734–1787. doi:10.1039/c3cs60037h |
| 7. | Dydio, P.; Reek, J. N. H. Chem. Sci. 2014, 5, 2135–2145. doi:10.1039/c3sc53505c |
| 8. | Leenders, S. H. A. M.; Gramage-Doria, R.; de Bruin, B.; Reek, J. N. H. Chem. Soc. Rev. 2015, 44, 433–448. doi:10.1039/c4cs00192c |
| 9. | Brown, C. J.; Toste, F. D.; Bergman, R. G.; Raymond, K. N. Chem. Rev. 2015, 115, 3012–3035. doi:10.1021/cr4001226 |
| 10. | Blanco, V.; Leigh, D. A.; Marcos, V. Chem. Soc. Rev. 2015, 44, 5341–5370. doi:10.1039/c5cs00096c |
| 11. | Vaquero, M.; Rovira, L.; Vidal-Ferran, A. Chem. Commun. 2016, 52, 11038–11051. doi:10.1039/c6cc04474c |
| 12. | Davis, H. J.; Phipps, R. J. Chem. Sci. 2017, 8, 864–877. doi:10.1039/c6sc04157d |
| 13. | Jiang, J.; Ouyang, G.; Zhang, L.; Liu, M. Chem. – Eur. J. 2017, 23, 9439–9450. doi:10.1002/chem.201700727 |
| 14. | Wang, K.; Jordan, J. H.; Hu, X.-Y.; Wang, L. Angew. Chem., Int. Ed. 2020, 59, 13712–13721. doi:10.1002/anie.202000045 |
| 24. | Pemberton, B. C.; Raghunathan, R.; Volla, S.; Sivaguru, J. Chem. – Eur. J. 2012, 18, 12178–12190. doi:10.1002/chem.201202083 |
| 25. | Assaf, K. I.; Nau, W. M. Chem. Soc. Rev. 2015, 44, 394–418. doi:10.1039/c4cs00273c |
| 30. | Guo, H.; Zhang, L.-W.; Zhou, H.; Meng, W.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 2623–2627. doi:10.1002/anie.201910399 |
| 20. | Homden, D. M.; Redshaw, C. Chem. Rev. 2008, 108, 5086–5130. doi:10.1021/cr8002196 |
| 21. | Le Poul, N.; Le Mest, Y.; Jabin, I.; Reinaud, O. Acc. Chem. Res. 2015, 48, 2097–2106. doi:10.1021/acs.accounts.5b00152 |
| 22. | Salvio, R.; Cacciapaglia, R.; Casnati, A. Calixarenes as Supramoleuclar Catalysts Endowed with Esterase and Phosphodiesterase Activity. In Calixarenes and Beyond; Neri, P.; Sessler, J. L.; Wang, M.-X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp 691–717. doi:10.1007/978-3-319-31867-7_26 |
| 23. | Yilmaz, M.; Sayin, S. Calixarenes in Organo and Biomimetic Catalysis. In Calixarenes and Beyond; Neri, P.; Sessler, J. L.; Wang, M.-X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp 719–742. doi:10.1007/978-3-319-31867-7_27 |
| 48. | Hara, N.; Nakamura, S.; Sano, M.; Tamura, R.; Funahashi, Y.; Shibata, N. Chem. – Eur. J. 2012, 18, 9276–9280. doi:10.1002/chem.201200367 |
| 17. | Breslow, R.; Dong, S. D. Chem. Rev. 1998, 98, 1997–2012. doi:10.1021/cr970011j |
| 18. | Takahashi, K. Chem. Rev. 1998, 98, 2013–2034. doi:10.1021/cr9700235 |
| 19. | Engeldinger, E.; Armspach, D.; Matt, D. Chem. Rev. 2003, 103, 4147–4174. doi:10.1021/cr030670y |
| 43. | Pan, Y.; Tan, C.-H. Synthesis 2011, 2044–2053. doi:10.1055/s-0030-1260607 |
| 44. | Wang, Z.-L. Adv. Synth. Catal. 2013, 355, 2745–2755. doi:10.1002/adsc.201300375 |
| 45. | Nakamura, S. Org. Biomol. Chem. 2014, 12, 394–405. doi:10.1039/c3ob42161a |
| 46. | Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536 |
| 47. | Pan, Y.; Kee, C. W.; Jiang, Z.; Ma, T.; Zhao, Y.; Yang, Y.; Xue, H.; Tan, C.-H. Chem. – Eur. J. 2011, 17, 8363–8370. doi:10.1002/chem.201100687 |
| 48. | Hara, N.; Nakamura, S.; Sano, M.; Tamura, R.; Funahashi, Y.; Shibata, N. Chem. – Eur. J. 2012, 18, 9276–9280. doi:10.1002/chem.201200367 |
| 49. | Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307 |
| 50. | Bahlinger, A.; Fritz, S. P.; Wennemers, H. Angew. Chem., Int. Ed. 2014, 53, 8779–8783. doi:10.1002/anie.201310532 |
| 51. | Nakamura, S.; Sano, M.; Toda, A.; Nakane, D.; Masuda, H. Chem. – Eur. J. 2015, 21, 3929–3932. doi:10.1002/chem.201406270 |
| 52. | Kaur, J.; Kumari, A.; Bhardwaj, V. K.; Chimni, S. S. Adv. Synth. Catal. 2017, 359, 1725–1734. doi:10.1002/adsc.201700011 |
| 4. | Dong, Z.; Luo, Q.; Liu, J. Chem. Soc. Rev. 2012, 41, 7890–7908. doi:10.1039/c2cs35207a |
| 6. | Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1734–1787. doi:10.1039/c3cs60037h |
| 15. | Wang, Q.-Q. Supramolecular Catalysis Using Organic Macrocycles. In Handbook of Macrocyclic Supramolecular Assembly; Liu, Y.; Chen, Y.; Zhang, H.-Y., Eds.; Springer: Singapore, 2019. doi:10.1007/978-981-13-1744-6_36-1 |
| 16. | Kauerhof, D.; Niemeyer, J. ChemPlusChem 2020, 85, 889–899. doi:10.1002/cplu.202000152 |
| 30. | Guo, H.; Zhang, L.-W.; Zhou, H.; Meng, W.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 2623–2627. doi:10.1002/anie.201910399 |
| 37. | Takemoto, Y. Org. Biomol. Chem. 2005, 3, 4299–4306. doi:10.1039/b511216h |
| 38. | Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/b719249e |
| 39. | Fang, X.; Wang, C.-J. Chem. Commun. 2015, 51, 1185–1197. doi:10.1039/c4cc07909d |
| 30. | Guo, H.; Zhang, L.-W.; Zhou, H.; Meng, W.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 2623–2627. doi:10.1002/anie.201910399 |
| 31. | Li, A.-F.; Wang, J.-H.; Wang, F.; Jiang, Y.-B. Chem. Soc. Rev. 2010, 39, 3729–3745. doi:10.1039/b926160p |
| 32. | Amendola, V.; Fabbrizzi, L.; Mosca, L. Chem. Soc. Rev. 2010, 39, 3889–3915. doi:10.1039/b822552b |
| 33. | Wittkopp, A.; Schreiner, P. R. Chem. – Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042 |
| 34. | Schreiner, P. R. Chem. Soc. Rev. 2003, 32, 289–296. doi:10.1039/b107298f |
| 35. | Zhang, Z.; Schreiner, P. R. Chem. Soc. Rev. 2009, 38, 1187–1198. doi:10.1039/b801793j |
| 36. | Park, Y.; Harper, K. C.; Kuhl, N.; Kwan, E. E.; Liu, R. Y.; Jacobsen, E. N. Science 2017, 355, 162–166. doi:10.1126/science.aal1875 |
| 40. | Jez, J. M.; Austin, M. B.; Ferrer, J.-L.; Bowman, M. E.; Schröder, J.; Noel, J. P. Chem. Biol. 2000, 7, 919–930. doi:10.1016/s1074-5521(00)00041-7 |
| 41. | Austin, M. B.; Izumikawa, M.; Bowman, M. E.; Udwary, D. W.; Ferrer, J.-L.; Moore, B. S.; Noel, J. P. J. Biol. Chem. 2004, 279, 45162–45174. doi:10.1074/jbc.m406567200 |
| 42. | Zhang, Y.-M.; Hurlbert, J.; White, S. W.; Rock, C. O. J. Biol. Chem. 2006, 281, 17390–17399. doi:10.1074/jbc.m513199200 |
| 28. | Ning, R.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Chem. – Eur. J. 2018, 24, 4268–4272. doi:10.1002/chem.201800326 |
| 29. | Ning, R.; Zhou, H.; Nie, S.-X.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 10894–10898. doi:10.1002/anie.202003673 |
| 30. | Guo, H.; Zhang, L.-W.; Zhou, H.; Meng, W.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 2623–2627. doi:10.1002/anie.201910399 |
| 26. | Hooley, R. J.; Rebek, J., Jr. Chem. Biol. 2009, 16, 255–264. doi:10.1016/j.chembiol.2008.09.015 |
| 27. | Yu, Y.; Rebek, J., Jr. Acc. Chem. Res. 2018, 51, 3031–3040. doi:10.1021/acs.accounts.8b00269 |
| 30. | Guo, H.; Zhang, L.-W.; Zhou, H.; Meng, W.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2020, 59, 2623–2627. doi:10.1002/anie.201910399 |
© 2022 Guo et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.