Abstract
Histone deacetylases (HDACs) play an essential role in the transcriptional regulation of cells through the deacetylation of nuclear histone and non-histone proteins and are promising therapeutic targets for the treatment of various diseases. Here, the synthesis of new compounds in which a hydroxamic acid residue is attached to differently substituted pyrimidine rings via a methylene group bridge of varying length as potential HDAC inhibitors is described. The target compounds were obtained by alkylation of 2-(alkylthio)pyrimidin-4(3H)-ones with ethyl 2-bromoethanoate, ethyl 4-bromobutanoate, or methyl 6-bromohexanoate followed by aminolysis of the obtained esters with hydroxylamine. Oxidation of the 2-methylthio group to the methylsulfonyl group and following treatment with amines resulted in the formation of the corresponding 2-amino-substituted derivatives, the ester group of which reacted with hydroxylamine to give the corresponding hydroxamic acids. The synthesized hydroxamic acids were tested as inhibitors of the HDAC4 and HDAC8 isoforms. Among the synthesized pyrimidine-based hydroxamic acids N-hydroxy-6-[6-methyl-2-(methylthio)-5-propylpyrimidin-4-yloxy]hexanamide was found to be the most potent inhibitor of both the HDAC4 and HDAC8 isoforms, with an IC50 of 16.6 µM and 1.2 µM, respectively.

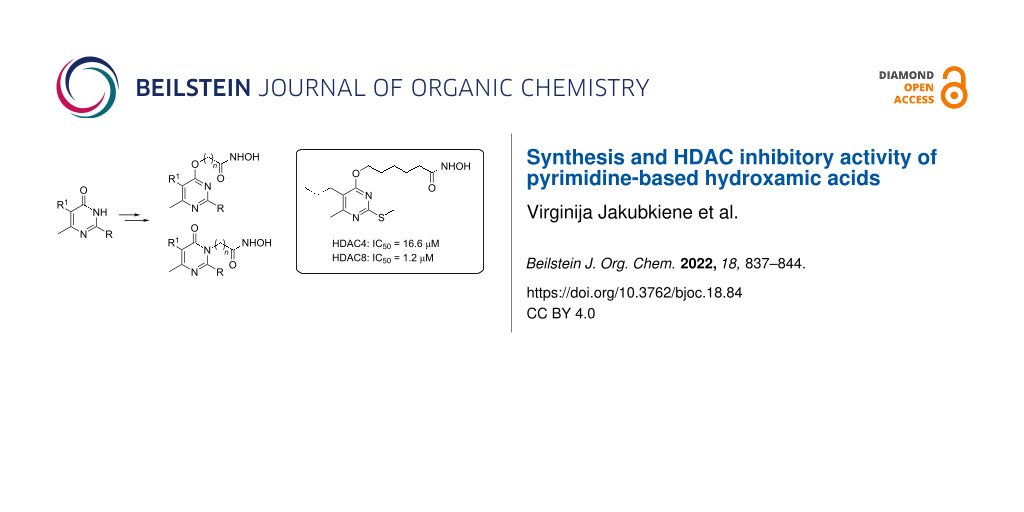
Graphical Abstract
Introduction
Histone deacetylases (HDACs) are a family of intracellular proteins responsible for removing acetyl groups in histones. This function is essential in the transcription of DNA, as histones with acetyl groups do not interact as strongly with DNA, which opens the chromatin for transcription. HDACs also interact with other cellular proteins to regulate vital functions such as cell differentiation or apoptosis. They have also been shown to play a significant role in pathologies such as cancer, neurodegenerative diseases, and metabolic disorders [1,2]. HDACs are structurally divided into 18 isoforms, which are grouped into 4 classes. The 11 isoforms belonging to classes I, II, and IV are dependent on the Zn2+ ion in their catalytic site, while the remaining 7 isoforms of class III, known as sirtuins, are dependent on the NAD+ coenzyme [3,4]. According to current knowledge, HDAC inhibitors usually have several structural subunits: a zinc chelating group, a hydrophobic linker, and a hydrophobic (usually aromatic) cap [1,2,5]. One of the most commonly used zinc chelating groups in HDACs inhibitors is a hydroxamic acid moiety (–CONHOH) [6-15]. The ability of hydroxamic acids to form chelates with various metal cations, including the Zn2+ ion found in the catalytic center of most HDAC proteins, gives them good biological activity in inhibiting these proteins. To date, three HDAC inhibitor drugs containing this functional group in their structure have been approved [6,7] (Figure 1).
![[1860-5397-18-84-1]](/bjoc/content/figures/1860-5397-18-84-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: FDA-approved HDAC inhibitors with a hydroxamic acid moiety.
Figure 1: FDA-approved HDAC inhibitors with a hydroxamic acid moiety.
However, the main disadvantage of many HDAC inhibitors is their low selectivity: most of them interact with all HDAC isoforms of groups I, II, and IV. Thus, efforts are currently underway to synthesize inhibitors selective for certain HDAC isoforms [2]. On the other hand, pyrimidines represent an important group of heterocyclic compounds exhibiting a broad spectrum of biological activity [16-20]. Pyrimidine-based hydroxamic acids with HDAC inhibitory activity have also been described [21,22].
Herein, we report on the synthesis of novel pyrimidine derivatives in which the hydroxamic acid fragment is linked to a variably substituted pyrimidine moiety via a methylene group bridge of varying length and the evaluation of their HDAC inhibitory activity.
Results and Discussion
Chemistry
Commonly, hydroxamic acids (N-hydroxyamides) are prepared by coupling activated carboxylic acids with O-protected hydroxylamine [23-25] or by treatment of esters with hydroxylamine [26-30]. In this work, the pyrimidine-based hydroxamic acids were synthesized by aminolysis of the corresponding esters. The required esters 3 and 4 were obtained by alkylation of pyrimidinones 1 and 2 with ethyl 2-bromoethanoate in triethylamine in the presence of tetrabutylammonium bromide at 50–60 °C in 73% and 70% yields, respectively (Scheme 1).
![[1860-5397-18-84-i1]](/bjoc/content/inline/1860-5397-18-84-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of compounds 3–18. Reagents and conditions: (a) ethyl 2-bromoethanoate, TBAB, TEA, 50–60 °C, 0.5 h; (b) oxone, DMF, 40 °C, 0.5 h; (c) corresponding amine, DMSO, 50–70 °C, 0.5 h; (d) H2O, DMSO, 100 °C, 0.5 h; (e) H2O, reflux, 1 h; (f) aqueous NaOH, dioxane, rt, 12 h, then, conc. HCl to pH 2; (g) NH2OH.HCl, KOH, H2O (H2O/MeOH for 7), 0–5 °C, 1–4 h (rt, 96 h for 7), then, conc. HCl to pH 5–6.
Scheme 1: Synthesis of compounds 3–18. Reagents and conditions: (a) ethyl 2-bromoethanoate, TBAB, TEA, 50–60 ...
In order to functionalize the 2nd position of the pyrimidine ring, the 2-methylthio group of compound 3 was oxidized to the better leaving 2-methylsulfonyl group. In our previous work [31] we investigated the oxidation of some 2-methylthiopyrimidines with m-CPBA and oxone and found that oxone gave better results, so we have chosen it for this reaction. Thus, heating compound 3 in dimethylformamide at 40 °C for 0.5 h with oxone gave compound 5 in 80% yield. In the NMR spectra of compound 5, the peaks of the methylsulfonyl group are downfield shifted by 0.8 ppm and 24.8 ppm in the 1H and 13C NMR spectra, respectively, in comparison with the signals of the methylthio group of compound 3 (see Supporting Information File 1, Figures S1, S2 and S5, S6). Heating compound 5 with primary and secondary amines in dimethyl sulfoxide at 50–70 °C for 0.5 h gave the corresponding (2-substituted pyrimidin-4-yloxy)acetates 6–9. In order to replace the 2-methylsulfonyl group with a hydroxy group, we tested several techniques. It was found that heating compound 5 with water in dimethyl sulfoxide at 100 °C or boiling for 1 h in water led to hydrolysis of both 2-methylsulfonyl and 4-(ethoxycarbonyl)methoxy groups with the formation of 6-methyluracil (10) in 40% and 56% yields, respectively. Compound 11 was successfully synthesized by stirring compound 5 with aqueous sodium hydroxide solution in dioxane at room temperature for 12 h. Hydroxamic acids 12–18 were synthesized by the interaction of esters 3, 4, and 6–9, with hydroxylamine in water at 0–5 °C (in case of ester 7, in a mixture of water and methanol 1:1 at room temperature).
To investigate the effect of an alkyl substituent at the fifth position of the pyrimidine ring and the length of the methylene bridge linking the pyrimidine ring and the hydroxamic acid residue on the HDAC activity, hydroxamic acids 25–31 were synthesized (Scheme 2).
![[1860-5397-18-84-i2]](/bjoc/content/inline/1860-5397-18-84-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of compounds 20–31. Reagents and conditions: (a) ethyl 2-bromoethanoate (for 22) (or ethyl 4-bromobutanoate (for 20, 23), or methyl 6-bromohexanoate (for 21, 24)), TBAB, TEA, 50–60 °C, 0.5–4 h; (b) NH2OH.HCl, KOH, MeOH, 0–5 °C, 1 h, then ester 20–24, 0–5 °C,1 h, then conc. HCl to pH 5–6.
Scheme 2: Synthesis of compounds 20–31. Reagents and conditions: (a) ethyl 2-bromoethanoate (for 22) (or ethy...
In contrast to the synthesis of esters 3 and 4, alkylation of pyrimidinones 1 and 19 with the corresponding bromoesters in triethylamine in the presence of tetrabutylammonium bromide at 50–60 °C afforded mixtures of the O-alkylation 20a–24a and N(3)-alkylation 20b–24b products (Table 1, entries 1–5), which are easily separated by column chromatography. The overall yields are reasonable with the exception of the synthesis 24a and 24b. To improve the yield of the alkylation reaction of compound 19 with methyl 6-bromohexanoate and select the conditions favoring the formation of the O-isomer, a weak base, potassium carbonate, and a bipolar aprotic solvent, dimethylformamide, were used. The reaction was carried out at room temperature for 120 hours and a significant increase in the overall yield (from 52 to 71%) and selectivity (from 1.4:1 to 2.5:1) in favor of the O-isomer 24a were observed (compare entries 5 and 6 in Table 1). The best alkylation results were obtained when the alkylation reaction was carried out at 50–60 °C. These reaction conditions proved to be the most successful with an overall reaction yield as high as 82% and a ratio of O- to N-isomers 24a to 24b as high as 3.3:1 (Table 1, entry 7).
Table 1: Alkylation of pyrimidin-4(3H)-ones 1 and 19 with bromoesters (1.1 equiv).
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-84-i3.svg?max-width=637&scale=1.0)
|
||||||||
| entry | substr. | R2 | R3 | n | solvent (TBAB or base) | reaction temp., °C (time, h) | alkylation product (yield, %) | |
| O-isomer | N-isomer | |||||||
| 1 | 1 | H | Et | 3 | TEA (TBAB) | 50–60 (4) | 20a (45) | 20b (31) |
| 2 | 1 | H | Me | 5 | TEA (TBAB) | 50–60 (4) | 21a (50) | 21b (28) |
| 3 | 19 | n-Pr | Et | 1 | TEA (TBAB) | 50–60 (4) | 22a (62) | 22b (12) |
| 4 | 19 | n-Pr | Et | 3 | TEA (TBAB) | 50–60 (4) | 23a (37) | 23b (32) |
| 5 | 19 | n-Pr | Me | 5 | TEA (TBAB) | 50–60 (4) | 24a (30) | 24b (22) |
| 6 | 19 | n-Pr | Me | 5 | DMF (K2CO3) | rt (120) | 24a (51) | 24b (20) |
| 7 | 19 | n-Pr | Me | 5 | DMF (K2CO3) | 50–60 (4) | 24a (63) | 24b (19) |
As expected, the polar aprotic solvent dimethylformamide promoted the reaction of the electrophile with the more negatively charged oxygen atom by well solvating the cation. Thus, this method allows the synthesis of the O-isomer 24a in an acceptable 63% yield, though the alkylation reaction is not very selective. Hydroxamic acids 25–31 were obtained in 52–92% yields in the usual way by treating the corresponding esters with hydroxylamine in methanol at 0–5 °C (Scheme 2).
It is known that there are two possible tautomeric forms of each hydroxamic acid: the keto and enol tautomer. Furthemore, each tautomer can adopt an E or Z conformation (Figure 2) [32-36].
![[1860-5397-18-84-2]](/bjoc/content/figures/1860-5397-18-84-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The conformational and tautomeric forms of hydroxamic acids according to [36].
Figure 2: The conformational and tautomeric forms of hydroxamic acids according to [36].
The results of NMR spectra and theoretical calculations showed that hydroxamic acids tend to adopt the more stable keto-E and keto-Z conformations [36]. In the 1H NMR spectra of the synthesized hydroxamic acids 12–18 and 25–31 two sets of proton signals from the NH and OH groups of the hydroxamic acid residue are observed. Following data [36], we assigned the more intense peaks to the keto-E form and the less intense ones to the keto-Z form. It is interesting to note that two proton signals of the OCH2 groups are also observed in the 1H NMR spectra of compounds 12, 13, 16–18, 27, and 30. The fact that the ratio of the intensities of these signals is the same as the ratio of the intensities of the signals of the NH and OH groups of the hydroxamic acid moiety suggests that two OCH2 group signals are also due to the two isomeric forms. The fragment of the 1H NMR spectrum of compound 12 is presented in Figure 3 (full NMR spectra of the compounds are given in Supporting Information File 1).
![[1860-5397-18-84-3]](/bjoc/content/figures/1860-5397-18-84-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Fragment of the 1H NMR spectrum in DMSO-d6 of compound 12.
Figure 3: Fragment of the 1H NMR spectrum in DMSO-d6 of compound 12.
In vitro HDAC4 and HDAC8 inhibitory activities of the synthesized compounds
Compounds 12–18 and 25–31 were tested as inhibitors of HDAC4 and HDAC8 isoforms according to the procedures described previously [37]. The activity data of the tested compounds are presented in Table 2. A comparison of the IC50 values for compounds 12–18, which differ only in the substituent at the second position of the pyrimidine ring, reveals that the presence of a methylthio group at this position gives compound 12 a slight advantage in the inhibition of the HDAC4 isoform, while the presence of a more bulky substituent at the second position of the pyrimidine ring favors the inhibition of HDAC8 isoforms (Table 2, compounds 13–15 and 17). However, most of the compounds tested, with the exception of 29, were inactive or showed weak inhibitory effect on the HDAC4 isoform.
Table 2: Inhibitory activities (IC50) of tested compounds against HDAC4 and HDAC8.
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-84-i4.svg?max-width=637&scale=1.0)
|
||||||
| compound | R | R1 | R2 | n | IC50a, μM | |
| HDAC4 | HDAC8 | |||||
| 12 | SMe | 38 | 28 | |||
| 13 | SEt | ≥100 | 13 | |||
| 14 | NHCH2CH=CH2 | ≥100 | 15 | |||
| 15 | NHCH2C6H5 | ≥100 | 14 | |||
| 16 | NMe2 | ≥100 | 35 | |||
| 17 | pyrrolidin-1-yl | ≥100 | 13 | |||
| 18 | OH | ≥100 | 60 | |||
| 25 | H | 3 | >50 | 5.65 | ||
| 26 | H | 5 | >50 | 2.05 | ||
| 27 | n-Pr | 1 | 38 | 1.4 | ||
| 28 | n-Pr | 3 | >35 | 2.4 | ||
| 29 | n-Pr | 5 | 16.6 | 1.2 | ||
| 30 | 1 | >35 | >35 | |||
| 31 | 3 | >35 | >35 | |||
| vorinostat | 27b | 5.3b | ||||
aIC50 values were determined by measuring the inhibition of enzymatic activity using BOC-LYS-(TFA)-AMC as a substrate, 100 mM TRIS, 300 mM KCl, pH 8.0 buffer, at 30 °C. bTaken from ref [38].
Comparison of the potency of the HDAC8 isoform inhibitory activity of compounds 12, 25, and 26 with that of compounds 27–29 suggests that the propyl substituent at the position 5 of the pyrimidine ring is favorable for this effect. Extension of the methylene bridge connecting the pyrimidine ring to the hydroxamic acid residue to the same series of compounds also increases the ability of the compounds to inhibit the HDAC8 isoform, except in the case of compound 28. It should be noted that both, HDAC4 and HDAC8 isoforms were most inhibited by compound 29 (IC50 16.6 and 1.2 μM, respectively). The structure of this most active compound is distinguished by the fact that the compound not only has a propyl substituent at position 5 of the pyrimidine ring, but also has the longest pentamethylene group bridge connecting the pyrimidine ring to the hydroxamic acid residue.
Conclusion
In summary, starting with corresponding 2-(alkylthio)pyrimidin-4(3H)-ones we have developed an efficient synthesis of hitherto unknown pyrimidine-based hydroxamic acids, wherein the hydroxamic acid residue is attached to the pyrimidine ring by a methylene linker of varying length. The 1H NMR spectra show that the synthesized hydroxamic acids in solution exist as an equilibrium mixture of two isomeric forms. The inhibitory activity of the synthesized hydroxamic acids on HDAC4 and HDAC8 isoforms shows that most of them are prone to inhibit HDAC8 isoform rather than HDAC4. The most potent inhibitor of both the HDAC4 and HDAC8 isoforms was found to be N-hydroxy-6-[6-methyl-2-(methylthio)-5-propylpyrimidin-4-yloxy]hexanamide (29) with an IC50 of 16.6 and 1.2 µM, respectively. We believe that the obtained data on HDAC inhibitory activity of the synthesized pyrimidine-based hydroxamic acids will be useful in the future design of potent HDAC inhibitors.
Experimental
Melting points were determined in open capillaries with a digital melting point IA9100series apparatus (ThermoFisher Scientific). All reactions and purity of the synthesized compounds were monitored by TLC using silica gel 60 F254 aluminum plates (Merck). Visualization was accomplished by UV light. Column chromatography was performed using silica gel 60 (0.040–0.063 mm) (Merck). NMR spectra were recorded on a Bruker Ascend 400 spectrometer (400 MHz and 100 MHz for 1H and 13C, respectively). 1H NMR and 13C NMR were referenced to residual solvent peaks. High-resolution mass spectrometry (HRMS) analyses were carried out on a Dual-ESI Q-TOF 6520 (Agilent Technologies) mass spectrometer.
The starting compounds 1 [39], 2 [39] and 19 [40] were prepared following the reported methods. Synthetic details, characterization and analytical data as well as 1H and 13C NMR spectra of all synthesized compounds are presented in the Supporting Information File 1.
General procedure for the synthesis of esters 3, 4, and 20–24
To a mixture of pyrimidinone 1, 2 or 19 (1 mmol) and TBAB (0.032 g, 0.1 mmol) in TEA (0.4 mL) at 50–60 °C the corresponding bromoester (1.1 mmol) was added dropwise under stirring. The reaction mixture was stirred at this temperature for 0.5–4 h (for details see Supporting Information File 1), then cooled to room temperature, and poured into ice water (30 mL). In case of esters 3 and 4, the resultant precipitate was collected by filtration, washed with cold water, dried, and recrystallized from hexane. In case of esters 20–24, the resultant mixture was extracted with chloroform (3 × 10 mL). The organic solutions were combined, washed with brine, dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by column chromatography.
General procedure for the synthesis of esters 6–9
To a suspension of compound 5 (0.274 g, 1 mmol) in dry DMSO (0.3 mL) the corresponding amine (2 mmol; in the case of 8 – the solution of dimethylamine in DMSO) was added. The reaction mixture was stirred at 50–70 °C for 0.5 h, then cooled to room temperature, and poured into ice water (10 mL). The resultant precipitate was collected by filtration, washed with water, dried, and recrystallized from hexane.
6-Methyluracil (10)
Method A. To a suspension of compound 5 (0.274 g, 1 mmol) in DMSO (0.3 mL) H2O (0.036 g, 2 mmol) was added. The reaction mixture was stirred at 100 °C for 0.5 h, then cooled to room temperature, and poured into ice water (5 mL). The resultant precipitate was collected by filtration, washed with ice water, and dried to yield white powder.
Method B. A mixture of compound 5 (0.274 g, 1 mmol) and H2O (5 mL) was heated at reflux for 1 h, then cooled to 0 °C. The resultant precipitate was collected by filtration, washed with ice water, and dried to yield 0.07 g (56%) of compound 10.
Ethyl (6-methyl-2-oxo-1,2-dihydropyrimidin-4-yloxy]acetate (11)
To a solution of compound 5 (0.274 g, 1 mmol) in dioxane (25 mL) a 1 M aqueous solution of NaOH (2 mL) was added. The reaction mixture was stirred at room temperature for 12 h, then water (15 mL) was added to the mixture, and stirring was continued for 5 min. The reaction mixture was acidified with conc. HCl to pH 2 and extracted with DCM. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by column chromatography to give 0.155 g (73%) of compound 11.
General procedure for the synthesis of hydroxamic acids 12–18
To a mixture of esters 3, 4, 6–9, or 11 (1 mmol) and NH2OH·HCl (0.204 g, 3 mmol) in H2O (3 mL) (in the case of ester 7 in a mixture H2O/MeOH 1:1, 6 mL) at 0–5 °C a solution of KOH (0.392 g, 7 mmol) in H2O (1 mL) was added dropwise under stirring. The reaction mixture was stirred at this temperature for 1–4 h (in the case of ester 7 it was stirred at room temperature for 96 h), then acidified with conc. HCl to pH 5–6. The resultant precipitate was collected by filtration, washed with ice water, and dried to yield compounds 12–18.
General procedure for the synthesis of hydroxamic acids 25–31
A mixture of NH2OH·HCl (0.556 g, 8 mmol), KOH (0.672 g, 12 mmol), and methanol (5 mL) was stirred at room temperature for 0.5 h, then cooled to 0–5 °C and filtered. To the filtrate the corresponding ester 20–24 (1 mmol) was added. The reaction mixture was stirred at 0–5 °C for 1 h, then the solvent was removed under reduced pressure, the residue was dissolved in water, cooled to 0–5 °C and acidified with conc. HCl to pH 5–6. The resultant precipitate was collected by filtration, washed with ice water and dried (compound 30 was recrystallized from a mixture of ethanol/water 1:1). In case of compound 29, after acidification, the reaction mixture was extracted with chloroform (3 × 10 mL), the organic solutions were combined, washed with brine, dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by column chromatography.
Supporting Information
| Supporting Information File 1: Compounds characterization and analytical data; HDAC enzyme activity assay; references; NMR spectra. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Roche, J.; Bertrand, P. Eur. J. Med. Chem. 2016, 121, 451–483. doi:10.1016/j.ejmech.2016.05.047
Return to citation in text: [1] [2] -
Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039
Return to citation in text: [1] [2] [3] -
Dokmanovic, M.; Clarke, C.; Marks, P. A. Mol. Cancer Res. 2007, 5, 981–989. doi:10.1158/1541-7786.mcr-07-0324
Return to citation in text: [1] -
Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Eur. J. Med. Chem. 2020, 208, 112831. doi:10.1016/j.ejmech.2020.112831
Return to citation in text: [1] -
De Vreese, R.; D'hooghe, M. Eur. J. Med. Chem. 2017, 135, 174–195. doi:10.1016/j.ejmech.2017.04.013
Return to citation in text: [1] -
He, X.; Hui, Z.; Xu, L.; Bai, R.; Gao, Y.; Wang, Z.; Xie, T.; Ye, X.-Y. Eur. J. Med. Chem. 2022, 227, 113946. doi:10.1016/j.ejmech.2021.113946
Return to citation in text: [1] [2] -
Qiu, X.; Zhu, L.; Wang, H.; Tan, Y.; Yang, Z.; Yang, L.; Wan, L. Bioorg. Med. Chem. 2021, 52, 116510. doi:10.1016/j.bmc.2021.116510
Return to citation in text: [1] [2] -
Liu, W.; Liang, Y.; Si, X. Eur. J. Med. Chem. 2020, 205, 112679. doi:10.1016/j.ejmech.2020.112679
Return to citation in text: [1] -
Liu, Q.; Zhang, B.; Wang, Y.; Wang, X.; Gou, S. Eur. J. Med. Chem. 2022, 229, 114058. doi:10.1016/j.ejmech.2021.114058
Return to citation in text: [1] -
Anh, D. T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E. J.; Jun, H. W.; Kang, J. S.; Kwon, J.-H.; Dung, D. T. M.; Anh, V. T.; Hue, V. T. M.; Han, S.-B.; Nam, N.-H. Bioorg. Chem. 2020, 101, 103988. doi:10.1016/j.bioorg.2020.103988
Return to citation in text: [1] -
Tavares, M. T.; Costa de Almeida, L.; Kronenberger, T.; Monteiro Ferreira, G.; Fujii de Divitiis, T.; Franco Zannini Junqueira Toledo, M.; Mariko Aymoto Hassimotto, N.; Agostinho Machado-Neto, J.; Veras Costa-Lotufo, L.; Parise-Filho, R. Bioorg. Med. Chem. 2021, 35, 116085. doi:10.1016/j.bmc.2021.116085
Return to citation in text: [1] -
He, F.; Ran, Y.; Li, X.; Wang, D.; Zhang, Q.; Lv, J.; Yu, C.; Qu, Y.; Zhang, X.; Xu, A.; Wei, C.; Chou, C. J.; Wu, J. Bioorg. Chem. 2020, 103, 104109. doi:10.1016/j.bioorg.2020.104109
Return to citation in text: [1] -
Trivedi, P.; Adhikari, N.; Amin, S. A.; Bobde, Y.; Ganesh, R.; Jha, T.; Ghosh, B. Eur. J. Pharm. Sci. 2019, 138, 105046. doi:10.1016/j.ejps.2019.105046
Return to citation in text: [1] -
Dong, Y.; Hu, H.; Sun, Y.; Qin, M.; Gong, P.; Hou, Y.; Zhao, Y. Bioorg. Med. Chem. Lett. 2020, 30, 127610. doi:10.1016/j.bmcl.2020.127610
Return to citation in text: [1] -
Pan, Z.; Li, X.; Wang, Y.; Jiang, Q.; Jiang, L.; Zhang, M.; Zhang, N.; Wu, F.; Liu, B.; He, G. J. Med. Chem. 2020, 63, 3678–3700. doi:10.1021/acs.jmedchem.9b02178
Return to citation in text: [1] -
N, J. B.; Goudgaon, N. M. J. Mol. Struct. 2021, 1246, 131168. doi:10.1016/j.molstruc.2021.131168
Return to citation in text: [1] -
Nadar, S.; Khan, T. Chem. Biol. Drug Des. 2022, in press. doi:10.1111/cbdd.14001
Return to citation in text: [1] -
Filho, E. V.; Pinheiro, E. M. C.; Pinheiro, S.; Greco, S. J. Tetrahedron 2021, 92, 132256. doi:10.1016/j.tet.2021.132256
Return to citation in text: [1] -
Ayati, A.; Moghimi, S.; Toolabi, M.; Foroumadi, A. Eur. J. Med. Chem. 2021, 221, 113523. doi:10.1016/j.ejmech.2021.113523
Return to citation in text: [1] -
Bunev, A. S.; Khochenkov, D. A.; Khochenkova, Y. A.; Machkova, Y. S.; Varakina, E. V.; Gasanov, R. E.; Troshina, M. A.; Avdyakova, O. S. Synth. Commun. 2021, 51, 2521–2527. doi:10.1080/00397911.2021.1939383
Return to citation in text: [1] -
Pan, T.; Dan, Y.; Guo, D.; Jiang, J.; Ran, D.; Zhang, L.; Tian, B.; Yuan, J.; Yu, Y.; Gan, Z. Eur. J. Med. Chem. 2021, 224, 113672. doi:10.1016/j.ejmech.2021.113672
Return to citation in text: [1] -
Huang, Y.; Dong, G.; Li, H.; Liu, N.; Zhang, W.; Sheng, C. J. Med. Chem. 2018, 61, 6056–6074. doi:10.1021/acs.jmedchem.8b00393
Return to citation in text: [1] -
Chu-Farseeva, Y.-y.; Mustafa, N.; Poulsen, A.; Tan, E. C.; Yen, J. J. Y.; Chng, W. J.; Dymock, B. W. Eur. J. Med. Chem. 2018, 158, 593–619. doi:10.1016/j.ejmech.2018.09.024
Return to citation in text: [1] -
Mehndiratta, S.; Lin, M.-H.; Wu, Y.-W.; Chen, C.-H.; Wu, T.-Y.; Chuang, K.-H.; Chao, M.-W.; Chen, Y.-Y.; Pan, S.-L.; Chen, M.-C.; Liou, J.-P. Eur. J. Med. Chem. 2020, 185, 111725. doi:10.1016/j.ejmech.2019.111725
Return to citation in text: [1] -
Rees, S. W. P.; Leung, E.; Reynisson, J.; Barker, D.; Pilkington, L. I. Bioorg. Chem. 2021, 114, 105152. doi:10.1016/j.bioorg.2021.105152
Return to citation in text: [1] -
Liu, Y.; Ruan, Z.; Wang, Y.; Huang, S.-H.; Hong, R. Tetrahedron 2019, 75, 1767–1773. doi:10.1016/j.tet.2018.12.026
Return to citation in text: [1] -
Cheng, G.; Wang, Z.; Yang, J.; Bao, Y.; Xu, Q.; Zhao, L.; Liu, D. Bioorg. Chem. 2019, 84, 410–417. doi:10.1016/j.bioorg.2018.12.011
Return to citation in text: [1] -
Raudszus, R.; Nowotny, R.; Gertzen, C. G. W.; Schöler, A.; Krizsan, A.; Gockel, I.; Kalwa, H.; Gohlke, H.; Thieme, R.; Hansen, F. K. Bioorg. Med. Chem. 2019, 27, 115039. doi:10.1016/j.bmc.2019.07.055
Return to citation in text: [1] -
Yuan, Z.; Chen, S.; Gao, C.; Dai, Q.; Zhang, C.; Sun, Q.; Lin, J.-S.; Guo, C.; Chen, Y.; Jiang, Y. Bioorg. Chem. 2019, 87, 200–208. doi:10.1016/j.bioorg.2019.03.027
Return to citation in text: [1] -
Chen, L.; Zhang, Y.; Wang, C.; Tang, Z.; Meng, Q.; Sun, H.; Qi, Y.; Ma, X.; Li, L.; Li, Y.; Xu, Y. Bioorg. Chem. 2021, 114, 105045. doi:10.1016/j.bioorg.2021.105045
Return to citation in text: [1] -
Jakubkiene, V.; Vaiciunaite, E.; Kriukaite, K.; Didzgalvis, J.; Tumkevicius, S. Synth. Commun. 2018, 48, 1974–1985. doi:10.1080/00397911.2018.1474229
Return to citation in text: [1] -
Brown, D. A.; Glass, W. K.; Mageswaran, R.; Girmay, B. Magn. Reson. Chem. 1988, 26, 970–973. doi:10.1002/mrc.1260261107
Return to citation in text: [1] -
Sałdyka, M.; Mielke, Z. Vib. Spectrosc. 2007, 45, 46–54. doi:10.1016/j.vibspec.2007.06.003
Return to citation in text: [1] -
Cheshmedzhieva, D.; Toshev, N.; Gerova, M.; Petrov, O.; Dudev, T. J. Mol. Model. 2018, 24, 114. doi:10.1007/s00894-018-3651-6
Return to citation in text: [1] -
Arora, R.; Issar, U.; Kakkar, R. Comput. Theor. Chem. 2017, 1105, 18–26. doi:10.1016/j.comptc.2017.02.014
Return to citation in text: [1] -
Adiguzel, E.; Yilmaz, F.; Emirik, M.; Ozil, M. J. Mol. Struct. 2017, 1127, 403–412. doi:10.1016/j.molstruc.2016.07.081
Return to citation in text: [1] [2] [3] [4] -
Meyer-Almes, F.-J. Chem. Biol. Drug Des. 2019, 93, 1214–1250. doi:10.1111/cbdd.13449
Return to citation in text: [1] -
Kleinschek, A.; Meyners, C.; Digiorgio, E.; Brancolini, C.; Meyer‐Almes, F.-J. ChemMedChem 2016, 11, 2598–2606. doi:10.1002/cmdc.201600528
Return to citation in text: [1] -
Kotaiah, S.; Ramadevi, B.; Naidu, A.; Dubey, P. K. Asian J. Chem. 2013, 25, 9869–9871. doi:10.14233/ajchem.2013.15532
Return to citation in text: [1] [2] -
Chi, Y. F.; Chang, K.-J. J. Am. Chem. Soc. 1938, 60, 1721–1723. doi:10.1021/ja01275a005
Return to citation in text: [1]
| 39. | Kotaiah, S.; Ramadevi, B.; Naidu, A.; Dubey, P. K. Asian J. Chem. 2013, 25, 9869–9871. doi:10.14233/ajchem.2013.15532 |
| 38. | Kleinschek, A.; Meyners, C.; Digiorgio, E.; Brancolini, C.; Meyer‐Almes, F.-J. ChemMedChem 2016, 11, 2598–2606. doi:10.1002/cmdc.201600528 |
| 39. | Kotaiah, S.; Ramadevi, B.; Naidu, A.; Dubey, P. K. Asian J. Chem. 2013, 25, 9869–9871. doi:10.14233/ajchem.2013.15532 |
| 1. | Roche, J.; Bertrand, P. Eur. J. Med. Chem. 2016, 121, 451–483. doi:10.1016/j.ejmech.2016.05.047 |
| 2. | Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039 |
| 6. | He, X.; Hui, Z.; Xu, L.; Bai, R.; Gao, Y.; Wang, Z.; Xie, T.; Ye, X.-Y. Eur. J. Med. Chem. 2022, 227, 113946. doi:10.1016/j.ejmech.2021.113946 |
| 7. | Qiu, X.; Zhu, L.; Wang, H.; Tan, Y.; Yang, Z.; Yang, L.; Wan, L. Bioorg. Med. Chem. 2021, 52, 116510. doi:10.1016/j.bmc.2021.116510 |
| 36. | Adiguzel, E.; Yilmaz, F.; Emirik, M.; Ozil, M. J. Mol. Struct. 2017, 1127, 403–412. doi:10.1016/j.molstruc.2016.07.081 |
| 6. | He, X.; Hui, Z.; Xu, L.; Bai, R.; Gao, Y.; Wang, Z.; Xie, T.; Ye, X.-Y. Eur. J. Med. Chem. 2022, 227, 113946. doi:10.1016/j.ejmech.2021.113946 |
| 7. | Qiu, X.; Zhu, L.; Wang, H.; Tan, Y.; Yang, Z.; Yang, L.; Wan, L. Bioorg. Med. Chem. 2021, 52, 116510. doi:10.1016/j.bmc.2021.116510 |
| 8. | Liu, W.; Liang, Y.; Si, X. Eur. J. Med. Chem. 2020, 205, 112679. doi:10.1016/j.ejmech.2020.112679 |
| 9. | Liu, Q.; Zhang, B.; Wang, Y.; Wang, X.; Gou, S. Eur. J. Med. Chem. 2022, 229, 114058. doi:10.1016/j.ejmech.2021.114058 |
| 10. | Anh, D. T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E. J.; Jun, H. W.; Kang, J. S.; Kwon, J.-H.; Dung, D. T. M.; Anh, V. T.; Hue, V. T. M.; Han, S.-B.; Nam, N.-H. Bioorg. Chem. 2020, 101, 103988. doi:10.1016/j.bioorg.2020.103988 |
| 11. | Tavares, M. T.; Costa de Almeida, L.; Kronenberger, T.; Monteiro Ferreira, G.; Fujii de Divitiis, T.; Franco Zannini Junqueira Toledo, M.; Mariko Aymoto Hassimotto, N.; Agostinho Machado-Neto, J.; Veras Costa-Lotufo, L.; Parise-Filho, R. Bioorg. Med. Chem. 2021, 35, 116085. doi:10.1016/j.bmc.2021.116085 |
| 12. | He, F.; Ran, Y.; Li, X.; Wang, D.; Zhang, Q.; Lv, J.; Yu, C.; Qu, Y.; Zhang, X.; Xu, A.; Wei, C.; Chou, C. J.; Wu, J. Bioorg. Chem. 2020, 103, 104109. doi:10.1016/j.bioorg.2020.104109 |
| 13. | Trivedi, P.; Adhikari, N.; Amin, S. A.; Bobde, Y.; Ganesh, R.; Jha, T.; Ghosh, B. Eur. J. Pharm. Sci. 2019, 138, 105046. doi:10.1016/j.ejps.2019.105046 |
| 14. | Dong, Y.; Hu, H.; Sun, Y.; Qin, M.; Gong, P.; Hou, Y.; Zhao, Y. Bioorg. Med. Chem. Lett. 2020, 30, 127610. doi:10.1016/j.bmcl.2020.127610 |
| 15. | Pan, Z.; Li, X.; Wang, Y.; Jiang, Q.; Jiang, L.; Zhang, M.; Zhang, N.; Wu, F.; Liu, B.; He, G. J. Med. Chem. 2020, 63, 3678–3700. doi:10.1021/acs.jmedchem.9b02178 |
| 37. | Meyer-Almes, F.-J. Chem. Biol. Drug Des. 2019, 93, 1214–1250. doi:10.1111/cbdd.13449 |
| 1. | Roche, J.; Bertrand, P. Eur. J. Med. Chem. 2016, 121, 451–483. doi:10.1016/j.ejmech.2016.05.047 |
| 2. | Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039 |
| 5. | De Vreese, R.; D'hooghe, M. Eur. J. Med. Chem. 2017, 135, 174–195. doi:10.1016/j.ejmech.2017.04.013 |
| 36. | Adiguzel, E.; Yilmaz, F.; Emirik, M.; Ozil, M. J. Mol. Struct. 2017, 1127, 403–412. doi:10.1016/j.molstruc.2016.07.081 |
| 3. | Dokmanovic, M.; Clarke, C.; Marks, P. A. Mol. Cancer Res. 2007, 5, 981–989. doi:10.1158/1541-7786.mcr-07-0324 |
| 4. | Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Eur. J. Med. Chem. 2020, 208, 112831. doi:10.1016/j.ejmech.2020.112831 |
| 36. | Adiguzel, E.; Yilmaz, F.; Emirik, M.; Ozil, M. J. Mol. Struct. 2017, 1127, 403–412. doi:10.1016/j.molstruc.2016.07.081 |
| 23. | Chu-Farseeva, Y.-y.; Mustafa, N.; Poulsen, A.; Tan, E. C.; Yen, J. J. Y.; Chng, W. J.; Dymock, B. W. Eur. J. Med. Chem. 2018, 158, 593–619. doi:10.1016/j.ejmech.2018.09.024 |
| 24. | Mehndiratta, S.; Lin, M.-H.; Wu, Y.-W.; Chen, C.-H.; Wu, T.-Y.; Chuang, K.-H.; Chao, M.-W.; Chen, Y.-Y.; Pan, S.-L.; Chen, M.-C.; Liou, J.-P. Eur. J. Med. Chem. 2020, 185, 111725. doi:10.1016/j.ejmech.2019.111725 |
| 25. | Rees, S. W. P.; Leung, E.; Reynisson, J.; Barker, D.; Pilkington, L. I. Bioorg. Chem. 2021, 114, 105152. doi:10.1016/j.bioorg.2021.105152 |
| 31. | Jakubkiene, V.; Vaiciunaite, E.; Kriukaite, K.; Didzgalvis, J.; Tumkevicius, S. Synth. Commun. 2018, 48, 1974–1985. doi:10.1080/00397911.2018.1474229 |
| 21. | Pan, T.; Dan, Y.; Guo, D.; Jiang, J.; Ran, D.; Zhang, L.; Tian, B.; Yuan, J.; Yu, Y.; Gan, Z. Eur. J. Med. Chem. 2021, 224, 113672. doi:10.1016/j.ejmech.2021.113672 |
| 22. | Huang, Y.; Dong, G.; Li, H.; Liu, N.; Zhang, W.; Sheng, C. J. Med. Chem. 2018, 61, 6056–6074. doi:10.1021/acs.jmedchem.8b00393 |
| 32. | Brown, D. A.; Glass, W. K.; Mageswaran, R.; Girmay, B. Magn. Reson. Chem. 1988, 26, 970–973. doi:10.1002/mrc.1260261107 |
| 33. | Sałdyka, M.; Mielke, Z. Vib. Spectrosc. 2007, 45, 46–54. doi:10.1016/j.vibspec.2007.06.003 |
| 34. | Cheshmedzhieva, D.; Toshev, N.; Gerova, M.; Petrov, O.; Dudev, T. J. Mol. Model. 2018, 24, 114. doi:10.1007/s00894-018-3651-6 |
| 35. | Arora, R.; Issar, U.; Kakkar, R. Comput. Theor. Chem. 2017, 1105, 18–26. doi:10.1016/j.comptc.2017.02.014 |
| 36. | Adiguzel, E.; Yilmaz, F.; Emirik, M.; Ozil, M. J. Mol. Struct. 2017, 1127, 403–412. doi:10.1016/j.molstruc.2016.07.081 |
| 16. | N, J. B.; Goudgaon, N. M. J. Mol. Struct. 2021, 1246, 131168. doi:10.1016/j.molstruc.2021.131168 |
| 17. | Nadar, S.; Khan, T. Chem. Biol. Drug Des. 2022, in press. doi:10.1111/cbdd.14001 |
| 18. | Filho, E. V.; Pinheiro, E. M. C.; Pinheiro, S.; Greco, S. J. Tetrahedron 2021, 92, 132256. doi:10.1016/j.tet.2021.132256 |
| 19. | Ayati, A.; Moghimi, S.; Toolabi, M.; Foroumadi, A. Eur. J. Med. Chem. 2021, 221, 113523. doi:10.1016/j.ejmech.2021.113523 |
| 20. | Bunev, A. S.; Khochenkov, D. A.; Khochenkova, Y. A.; Machkova, Y. S.; Varakina, E. V.; Gasanov, R. E.; Troshina, M. A.; Avdyakova, O. S. Synth. Commun. 2021, 51, 2521–2527. doi:10.1080/00397911.2021.1939383 |
| 40. | Chi, Y. F.; Chang, K.-J. J. Am. Chem. Soc. 1938, 60, 1721–1723. doi:10.1021/ja01275a005 |
| 2. | Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039 |
| 26. | Liu, Y.; Ruan, Z.; Wang, Y.; Huang, S.-H.; Hong, R. Tetrahedron 2019, 75, 1767–1773. doi:10.1016/j.tet.2018.12.026 |
| 27. | Cheng, G.; Wang, Z.; Yang, J.; Bao, Y.; Xu, Q.; Zhao, L.; Liu, D. Bioorg. Chem. 2019, 84, 410–417. doi:10.1016/j.bioorg.2018.12.011 |
| 28. | Raudszus, R.; Nowotny, R.; Gertzen, C. G. W.; Schöler, A.; Krizsan, A.; Gockel, I.; Kalwa, H.; Gohlke, H.; Thieme, R.; Hansen, F. K. Bioorg. Med. Chem. 2019, 27, 115039. doi:10.1016/j.bmc.2019.07.055 |
| 29. | Yuan, Z.; Chen, S.; Gao, C.; Dai, Q.; Zhang, C.; Sun, Q.; Lin, J.-S.; Guo, C.; Chen, Y.; Jiang, Y. Bioorg. Chem. 2019, 87, 200–208. doi:10.1016/j.bioorg.2019.03.027 |
| 30. | Chen, L.; Zhang, Y.; Wang, C.; Tang, Z.; Meng, Q.; Sun, H.; Qi, Y.; Ma, X.; Li, L.; Li, Y.; Xu, Y. Bioorg. Chem. 2021, 114, 105045. doi:10.1016/j.bioorg.2021.105045 |
© 2022 Jakubkiene et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.