Abstract
Dibenzo[g,p]chrysene (DBC), which consists of a twisted naphthalene core with four fused benzene rings, is a promising framework for organic electronic materials. Therefore, the research for structure–property relationships is important for the design of DBC-based materials. Here, the electrochemical and spectroscopic properties of DBC derivatives were investigated, and the effects of substituents and torsion of the naphthalene moiety were examined based on density functional theory (DFT) calculations. All the substituted DBC derivatives showed higher oxidation potentials than that for DBC-H, even for compounds that contained an electron-donating group such as DBC-Me and DBC-SMe. DFT calculations clearly indicate that these higher oxidation potentials are due to the ineffective conjugation of the MeO group, which is oriented perpendicular to the benzene ring because of the steric repulsion of substituents on both sides. More specifically, the inductive effect of the MeO group is dominant rather than the mesomeric effect when the substituent is located at both sides of the MeO group. Concerning the torsion of the naphthalene moiety, the twisting results in a slight increase in the HOMO and a slight lowering of the LUMO. The twisting effect is much smaller than the conjugation effect of the MeO group. Absorption spectra of all the substituted DBC derivatives also showed a red-shift as compared to that for DBC-H. Concerning the luminescence, a strong photoluminescence was observed for DBC-H and DBC-Si.
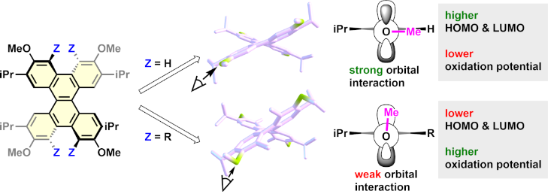
Graphical Abstract
Introduction
Polycyclic aromatic hydrocarbons (PAHs) have attracted interest as potential electronic and optoelectronic materials [1-12]. Non-planar PAHs have been extensively investigated from the viewpoint of their synthetic challenge and/or for the development of functional organic materials [13-22]. Among such PAHs, twisted acenes are an interesting class of compounds due to their characteristic structures and conjugation systems [23-25]. Dibenzo[g,p]chrysene (DBC), which consists of a twisted naphthalene core with four fused benzene rings (Figure 1a) [26], is a promising framework for serving as organic semiconductors, dyes, liquid crystals, and light-emitting materials. A number of substituted DBCs have been reported in this context [27-46]. To develop charge-transport materials, Rathore et al. reported on the stability of radical cations of DBCs with MeO groups located at X and/or Y (MeO-DBC-1, MeO-DBC-2, and MeO-DBC-3, Figure 1b) [43]. The first oxidation potential (Eox1) of MeO-DBC-1 was reported to be 0.40 V (based on Fc/Fc+), which is 0.48 V lower than that of DBC. In contrast, when a MeO group is introduced at the X position (MeO-DBC-2), the Eox1 is lower by only 0.15 V than that of DBC. It has also been reported that the oxidation potential of MeO-DBC-3, in which the MeO groups are attached at both X and Y, is 0.06 V higher than that for MeO-DBC-1. These remarkable substituent effects are an interesting and important finding for molecular design, but the effects of X and Z substituents and the twisting of the naphthalene moiety have not been reported.
![[1860-5397-18-96-1]](/bjoc/content/figures/1860-5397-18-96-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) DBC. b) Dependence of Eox1 on the position of the MeO groups [43]. c) Previous work [52]. d) This work.
Figure 1: a) DBC. b) Dependence of Eox1 on the position of the MeO groups [43]. c) Previous work [52]. d) This work.
We previously studied the synthesis of solution-processable DBC derivatives with various substituents attached [47-51]. We also recently reported on a synthetic strategy for preparing DBC derivatives using DBC-H with four isopropyl groups at X as a key template for the derivatization (Figure 1c). Based on this strategy, various substituents were introduced at Z to produce DBC-Br, DBC-Me, DBC-SMe, DBC-S(O)2Me, and DBC-Si (Figure 1c) [52]. The structures of all these derivatives were determined by X-ray crystallographic analysis, in which torsion angles were varied in a range from 31.8º (DBC-Si) to 57.4º (DBC-S(O)2Me) [52]. These DBC derivatives have four methoxy moieties at the Y position, which aroused our interest concerning the stability of those oxidation states.
Herein, we report on the electrochemical and spectroscopic properties of the DBC derivatives, where the effects of substituents and torsion were examined with the aid of DFT calculations. Consequently, the findings revealed that the substitution at the Z position induces a change in the conformation of the MeO groups, making the conjugation of the MeO groups ineffective, thus resulting in the lowering of both HOMO and LUMO energy levels. Concerning the twisting, the effect to the HOMO and LUMO energy levels was found to be small. We anticipate that the impact of diverse substituents and torsion angles on the chemical properties would be beneficial in terms of creating DBC-based materials.
Results and Discussion
Electrochemical properties
Cyclic voltammograms (CVs) and square-wave voltammograms (SWVs) were measured for DBC-H, DBC-Me, DBC-SMe, DBC-Br, DBC-S(O)2Me, and DBC-Si (Figure 2) [53]. Table 1 summarizes the first and second oxidation potentials based on Fc/Fc+ (Eox1 and Eox2) determined from the SWVs, together with the torsion angles determined from the X-ray crystal structures [52], the HOMO and LUMO levels determined from DFT calculations [52,54] and estimated based on Eox1. The voltammogram of DBC-H exhibited a reversible, two-step, two-electron redox process, with Eox1 and Eox2 values of 0.34 V and 0.72 V, respectively (Figure 2a). The value of Eox1 is 0.06 V lower than that of MeO-DBC-1 which does not contain isopropyl groups. This is in contrast to MeO-DBC-3, in which four MeO groups are introduced in place of the isopropyl groups, which has a 0.06 V higher oxidation potential than that of MeO-DBC-1. This indicates that alkyl substituents in the X position are effective in stabilizing the radical cation, thus making it more susceptible to oxidation. Unlike DBC-H, an irreversible voltammogram was observed in case of DBC-Me (Figure 2b). The first oxidation potential obtained from the SWV was 0.51 V, which is 0.17 V higher than that of DBC-H. This higher oxidation potential is somewhat surprising, which is discussed in the next paragraph based on DFT calculations. The CV of DBC-SMe showed a reversible two-electron redox process, with Eox1 and Eox2 values of 0.41 V and 0.88 V, respectively (Figure 2c) [53]. It is interesting to note that DBC-SMe exhibited a higher oxidation potential than DBC-H despite the electron-donating nature due to mesomeric effects based on lone pairs of sulfur atoms. In the CV of DBC-Br, a one-electron redox was observed as a reversible process, but a second redox process was not observed (Figure 2d). On the other hand, both the first and second oxidation processes were observed in the SWV of DBC-Br (Eox1 and Eox2 are 0.79 V and 1.15 V, respectively). DBC-S(O)2Me with the electron-withdrawing substituents resulted in a reversible oxidation wave, but only a one-electron redox process could be observed due to the limitations of the solvent (Figure 2e). The potential of 0.98 V is the highest among the compounds measured in this study. To investigate the reduction behaviour, DBC-S(O)2Me was measured in the low potential region. A peak, which appeared to be the one-electron reduction peak, was observed at −2.25 V (see Figure S1 in Supporting Information File 1). Finally, the CV of the silole-fused DBC-Si was investigated and the results indicated a reversible two-electron redox process (Eox1 and Eox2 are 0.43 V and 0.82 V, respectively, Figure 2f). These values for Eox1 and Eox2 for DBC-Si are slightly higher than those of DBC-H. The obtained electrochemical data were nearly consistent with the trend of the values for HOMO obtained based on DFT calculations.
![[1860-5397-18-96-2]](/bjoc/content/figures/1860-5397-18-96-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: CVs and SWVs of DBC derivatives in CH2Cl2 (≈1.0 × 10−3 M, see Supporting Information File 1 for details) including 5.0 × 10−2 M NBu4BF4 as a supporting electrolyte under Ar at 298 K (working electrode: Pt, scan rate: 100 mV/s and 40 mV/s for CV and SWV measurements, respectively).
Figure 2: CVs and SWVs of DBC derivatives in CH2Cl2 (≈1.0 × 10−3 M, see Supporting Information File 1 for details) including 5.0 × 10−2 M ...
Table 1: Electrochemical data, torsion angles determined from the X-ray crystal structures, and HOMO and LUMO levels for DBC derivativesa.
| compounds | Eox1 [V]b | Eox2 [V]b | torsion angle [°]c | HOMO [eV]d (the estimated values based on experimental data in parentheses)e | LUMO [eV]d |
| DBC-H | 0.34 | 0.72 | 36.9 | −4.64 (−5.4) | −0.87 |
| DBC-Me | 0.51 | 0.96 | 55.4 | −4.81 (−5.6) | −1.22 |
| DBC-SMe | 0.41 | 0.88 | 57.4 | −5.00 (−5.5) | −1.42 |
| DBC-Br | 0.79 | 1.15 | 56.1 | −5.24 (−5.9) | −1.71 |
| DBC-S(O)2Me | 0.98 | – | 57.4 | −5.56 (−6.1) | −2.00 |
| DBC-Si | 0.43 | 0.82 | 31.8 | −4.80 (−5.5) | −1.09 |
aConcentration: Around 1.0 × 10−3 M in CH2Cl2 (for detailed values, see Supporting Information File 1) containing 5.0 × 10−2 M NBu4BF4 as a supporting electrolyte. SWVs were recorded at a platinum electrode at 298 K under Ar. bBased on Fc/Fc+. cThe values obtained from X-ray crystallographic analyses [52]. dThe values obtained from DFT calculations at B3LYP6-31G(d,p) [52,54]. eThe energy values of HOMO were estimated based on the following equation EHOMO = −(Eox1 vs Fc+/Fc + 5.1) [55].
Theoretical calculations
DFT calculations were performed to clarify the reasons for the oxidation potentials [52,54,56]. To investigate the effects of the torsion of the naphthalene moiety and the conformation of the MeO group on the oxidation potential of these materials, hypothetical compounds DBC-H(56°)-1 and DBC-H(56°)-2 were created, respectively. In DBC-H(56°)-1, the atoms are fixed except for the Me group of DBC-Me (torsion angle = 56.5°), and the Me group is changed to H. DBC-H(56°)-2 is the same structure as DBC-H(56°)-1 except for the MeO group conformation. Optimizations of DBC-H(56°)-1 and DBC-H(56°)-2 based on DFT calculations were performed by fixing the atoms, as described above [56]. The conformations of the MeO group in DBC-H(56°)-1 and DBC-H(56°)-2 are nearly perpendicular (98.3°) to and parallel (179.8°) to the benzene ring, respectively (Figure 3 and Table 2). The results were compared to those for DBC-H and DBC-Me (Figure 3). To examine the torsional effect, DBC-H (torsion angle = 39.0°) and DBC-H(56°)-2 (torsion angle = 56.5°) were compared. The HOMO level of the highly twisted DBC-H(56°)-2 was 0.09 eV higher than that of the less twisted DBC-H. Conversely, the LUMO level of the highly twisted DBC-H(56°)-2 was 0.05 eV lower than the less twisted DBC-H. As a result, the HOMO–LUMO gap of DBC-H(56°)-2 becomes smaller than that of DBC-H. This is consistent with the trend reported for twisted acenes [57]. The conformational effect of the MeO group was investigated by comparison of DBC-H(56°)-1 (perpendicular to the benzene ring, 98.3°) with DBC-H(56°)-2 (parallel to the benzene ring, 179.8°). Consequently, both the HOMO and LUMO levels of DBC-H(56°)-2 are higher than those of DBC-H(56°)-1 by −0.40 eV and −0.37 eV, respectively. This is likely attributed by the effect of conjugation for the orbital of an oxygen atom as shown in the schematic drawing in Figure 3. When the conformation of the MeO group is almost parallel to the benzene ring, the strong orbital interaction between the orbitals on the oxygen and adjacent carbon atoms is possible in HOMO (the orbital drawings are also shown in Figure S2 in Supporting Information File 1). In this case, the mesomeric effect of an oxygen atom is dominant. On the other hand, when the conformation of the MeO group is almost perpendicular to the benzene ring, the interaction between orbitals on the oxygen and adjacent carbon atoms becomes weak in the case of HOMO. In this case, the inductive effect of an oxygen atom can be dominant. Thus, the substituents at the Z position allow the MeO group to be oriented perpendicular to the benzene ring, which results in the lowering of both the HOMO and LUMO (Figure 3). In DBC-Me, the lowering of the HOMO based on the inductive effect offsets the increase in HOMO due to the electron-donating nature of the Me group. This can account for the observed higher Eox1 for DBC-Me than that for DBC-H.
![[1860-5397-18-96-3]](/bjoc/content/figures/1860-5397-18-96-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: DFT-optimized structures, orbital drawings of HOMO, schematic drawings of orbital interaction, and energy diagrams for DBC-H, DBC-H(56°)-1, DBC-H(56°)-2, and DBC-Me.
Figure 3: DFT-optimized structures, orbital drawings of HOMO, schematic drawings of orbital interaction, and ...
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-96-i1.svg?max-width=637&scale=1.0)
Other derivatives were also examined. The dihedral angles are summarized in Table 2. The MeS group is an electron-donating group and may increase the HOMO, but the HOMO level of DBC-SMe is lower than that of DBC-H, as shown in the electrochemical study and by DFT calculations (Table 1). This is considered to be due to the contribution of the inductive effect of the MeO group by ineffective conjugation. In DBC-Br and DBC-S(O)2Me, both the HOMO and LUMO are lower, which can be attributed to the combined effects of their electron-withdrawing by Br and S(O)2Me groups and ineffective conjugation of the MeO group. In the case of DBC-Si, where the dihedral angle of the MeO group is 156.6°, both the HOMO and LUMO are lower than those for DBC-H. Although it is not perpendicular, the lower energy levels for HOMO and LUMO can be accounted for by the ineffective conjugation of the MeO group.
Spectroscopic properties
Absorption and photoluminescence spectra and the simulations of absorption based on TD-DFT calculations [58] are shown in Figure 4 (see Figure S3 in Supporting Information File 1 for excited spectra). The spectral data are summarized in Table 3. The TD-DFT calculations reproduce the absorption spectra quite well. The longest absorption peak is attributed to the transition from HOMO to LUMO and HOMO-1 to LUMO+1 (see Tables S1–S6 in Supporting Information File 1). The trend for the order of optical band gap is roughly consistent with that of the HOMO–LUMO gap obtained from DFT calculation [52].
![[1860-5397-18-96-4]](/bjoc/content/figures/1860-5397-18-96-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Absorption (solid line) and photoluminescence (dotted red line) spectra (upper graphs) in CH2Cl2 and simulations of absorption based on TD-DFT calculations [lower graphs, TD-B3LYP-D3/6-31G(d,p)//B3LYP/6-31G(d,p)] for DBC derivatives.
Figure 4: Absorption (solid line) and photoluminescence (dotted red line) spectra (upper graphs) in CH2Cl2 an...
Table 3: Absorption and photoluminescence spectral data of DBC derivatives in CH2Cl2.
| compounds |
absorption λmax [nm]
molar absorption coefficient ε [M−1·cm−1] in parentheses |
optical band gapa
[eV] |
photoluminescence λmax
[nm] |
quantum yield
[%]b |
| DBC-H |
363
(16200) |
2.95 | 416 | 28 |
| DBC-Me |
381
(20300) |
2.91 | 427 | 21 |
| DBC-SMe |
384
(16500) |
2.88 | 433 | 3 |
| DBC-Br |
386
(16100) |
2.86 | –c | –c |
| DBC-S(O)2Me |
380
(12900) |
2.82 | 455 | 6 |
| DBC-Si |
368
(9500) |
2.97 | 413 | 11 |
aEstimated from the absorption edge. bMeasured based on the absolute quantum yield method using an integrating sphere. cToo weak photoluminescence to measure.
In the photoluminescence spectra, the luminescence of DBC-Br was very weak. On the other hand, DBC-H, DBC-Me, and DBC-Si showed relatively strong photoluminescences with quantum yields of 28%, 21%, and 11%, respectively (Table 3). The photoluminescence wavelengths were shifted toward longer wavelengths in the order of DBC-Si, DBC-H, DBC-Me, DBC-SMe, and DBC-S(O)2Me. Of these, the Stokes shift for DBC-S(O)2Me was the largest, which is due to the electron-withdrawing nature of the S(O)2Me group.
Conclusion
The electrochemical and spectroscopic properties of DBC derivatives were investigated, and the effects of substituents and torsion of the naphthalene moiety were discussed based on DFT calculations. It was also found that introducing a substituent at Z position resulted in a higher oxidation potential than that for DBC-H, even for compounds that contained electron-donating groups, such as DBC-Me and DBC-SMe. DFT calculations clearly indicate that this is due to the ineffective conjugation of the MeO group which is oriented perpendicular to the aromatic ring because of the steric repulsion of substituents on both sides. More specifically, the inductive effect of the MeO group is dominant rather than the mesomeric effect when the substituent is present at the Z position. Concerning the torsion of the naphthalene moiety, the twisting caused a slight increase in the HOMO and a slight lowering of the LUMO. The twisting effect is much smaller than the conjugation effect of the MeO group. Absorption spectra of all the substituted DBC derivatives also showed a red-shift as compared to that for DBC-H. Concerning photoluminescence, a strong photoluminescence was observed for DBC-H and DBC-Si. The findings reported in this study will be useful for the molecular design of such materials, and could lead to electronic material applications in the future.
Supporting Information
| Supporting Information File 1: Figures S1–S3, Tables S1–S6, general, experimental procedure, and cartesian coordinates of optimized structures obtained based on the theoretical calculation. | ||
| Format: PDF | Size: 859.6 KB | Download |
References
-
Clar, E. Polycyclic hydrocarbons; Academic Press: London, UK, 1964; Vol. I.II.
Return to citation in text: [1] -
Harvey, R. G. Polycyclic Aromatic Hydrocarbons; Wiley-VCH: New York, NY, USA, 1997.
Return to citation in text: [1] -
Fetzer, J. C. Large Polycyclic Aromatic Hydrocarbons, 1st ed.; Chemistry and Analysis, Vol. 158; John Wiley & Sons: Hoboken, NJ, USA, 2000.
Return to citation in text: [1] -
Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z
Return to citation in text: [1] -
Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c
Return to citation in text: [1] -
Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045
Return to citation in text: [1] -
Pisula, W.; Feng, X.; Müllen, K. Chem. Mater. 2011, 23, 554–567. doi:10.1021/cm102252w
Return to citation in text: [1] -
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Sun, Z.; Ye, Q.; Chi, C.; Wu, J. Chem. Soc. Rev. 2012, 41, 7857–7889. doi:10.1039/c2cs35211g
Return to citation in text: [1] -
Zhang, D.; Duan, L. J. Phys. Chem. Lett. 2019, 10, 2528–2537. doi:10.1021/acs.jpclett.9b00526
Return to citation in text: [1] -
Tian, D.; Chen, Y. Adv. Opt. Mater. 2021, 9, 2002264. doi:10.1002/adom.202002264
Return to citation in text: [1] -
Li, Q.; Zhang, Y.; Xie, Z.; Zhen, Y.; Hu, W.; Dong, H. J. Mater. Chem. C 2022, 10, 2411–2430. doi:10.1039/d1tc04866j
Return to citation in text: [1] -
Yao, T.; Yu, H.; Vermeij, R. J.; Bodwell, G. J. Pure Appl. Chem. 2008, 80, 533–546. doi:10.1351/pac200880030533
Return to citation in text: [1] -
Petrukhina, M. A.; Scott, L. T., Eds. Fragments of Fullerenes and Carbon Nanotubes; John Wiley & Sons: Hoboken, NJ, USA, 2011. doi:10.1002/9781118011263
Return to citation in text: [1] -
Wu, Y.-T.; Siegel, J. S. Chem. Rev. 2006, 106, 4843–4867. doi:10.1021/cr050554q
Return to citation in text: [1] -
Tsefrikas, V. M.; Scott, L. T. Chem. Rev. 2006, 106, 4868–4884. doi:10.1021/cr050553y
Return to citation in text: [1] -
Gingras, M. Chem. Soc. Rev. 2013, 42, 968–1006. doi:10.1039/c2cs35154d
Return to citation in text: [1] -
Amaya, T.; Hirao, T. Chem. Rec. 2015, 15, 310–321. doi:10.1002/tcr.201402078
Return to citation in text: [1] -
Ball, M.; Zhong, Y.; Wu, Y.; Schenck, C.; Ng, F.; Steigerwald, M.; Xiao, S.; Nuckolls, C. Acc. Chem. Res. 2015, 48, 267–276. doi:10.1021/ar500355d
Return to citation in text: [1] -
Segawa, Y.; Ito, H.; Itami, K. Nat. Rev. Mater. 2016, 1, 15002. doi:10.1038/natrevmats.2015.2
Return to citation in text: [1] -
Chen, C.-F.; Shen, Y. Helicene Chemistry; Springer: Berlin, Heidelberg, 2017. doi:10.1007/978-3-662-53168-6
Return to citation in text: [1] -
Saito, M.; Shinokubo, H.; Sakurai, H. Mater. Chem. Front. 2018, 2, 635–661. doi:10.1039/c7qm00593h
Return to citation in text: [1] -
Pascal, R. A., Jr. Chem. Rev. 2006, 106, 4809–4819. doi:10.1021/cr050550l
Return to citation in text: [1] -
Rickhaus, M.; Mayor, M.; Juríček, M. Chem. Soc. Rev. 2016, 45, 1542–1556. doi:10.1039/c5cs00620a
Return to citation in text: [1] -
Ma, S.; Gu, J.; Lin, C.; Luo, Z.; Zhu, Y.; Wang, J. J. Am. Chem. Soc. 2020, 142, 16887–16893. doi:10.1021/jacs.0c08555
and references cited therein.
Return to citation in text: [1] -
Herbstein, F. H. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1979, 35, 1661–1670. doi:10.1107/s0567740879007354
Return to citation in text: [1] -
Tokito, S.; Noda, K.; Fujikawa, H.; Taga, Y.; Kimura, M.; Shimada, K.; Sawaki, Y. Appl. Phys. Lett. 2000, 77, 160–162. doi:10.1063/1.126910
Return to citation in text: [1] -
Yamaguchi, S.; Swager, T. M. J. Am. Chem. Soc. 2001, 123, 12087–12088. doi:10.1021/ja016692o
Return to citation in text: [1] -
Kumar, S.; Varshney, S. K. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 2002, 378, 59–64. doi:10.1080/713738586
Return to citation in text: [1] -
Li, C.-W.; Wang, C.-I.; Liao, H.-Y.; Chaudhuri, R.; Liu, R.-S. J. Org. Chem. 2007, 72, 9203–9207. doi:10.1021/jo701504m
Return to citation in text: [1] -
Chaudhuri, R.; Hsu, M.-Y.; Li, C.-W.; Wang, C.-I.; Chen, C.-J.; Lai, C. K.; Chen, L.-Y.; Liu, S.-H.; Wu, C.-C.; Liu, R.-S. Org. Lett. 2008, 10, 3053–3056. doi:10.1021/ol801029x
Return to citation in text: [1] -
Shimizu, M.; Nagao, I.; Tomioka, Y.; Hiyama, T. Angew. Chem., Int. Ed. 2008, 47, 8096–8099. doi:10.1002/anie.200803213
Return to citation in text: [1] -
Navale, T. S.; Zhai, L.; Lindeman, S. V.; Rathore, R. Chem. Commun. 2009, 2857–2859. doi:10.1039/b903133b
Return to citation in text: [1] -
Mori, T.; Fujita, K.; Kimura, M. J. Photopolym. Sci. Technol. 2010, 23, 317–322. doi:10.2494/photopolymer.23.317
Return to citation in text: [1] -
Tsuji, H.; Ueda, Y.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2010, 132, 11854–11855. doi:10.1021/ja1059119
Return to citation in text: [1] -
Navale, T. S.; Thakur, K.; Rathore, R. Org. Lett. 2011, 13, 1634–1637. doi:10.1021/ol200069c
Return to citation in text: [1] -
Mochida, K.; Kawasumi, K.; Segawa, Y.; Itami, K. J. Am. Chem. Soc. 2011, 133, 10716–10719. doi:10.1021/ja202975w
Return to citation in text: [1] -
Ueda, Y.; Tsuji, H.; Tanaka, H.; Nakamura, E. Chem. – Asian J. 2014, 9, 1623–1628. doi:10.1002/asia.201402102
Return to citation in text: [1] -
Hashimoto, S.; Ikuta, T.; Shiren, K.; Nakatsuka, S.; Ni, J.; Nakamura, M.; Hatakeyama, T. Chem. Mater. 2014, 26, 6265–6271. doi:10.1021/cm503102d
Return to citation in text: [1] -
Suzuki, N.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 4984–4987. doi:10.1021/acs.orglett.5b02426
Return to citation in text: [1] -
Liu, X.-Y.; Tang, X.; Zhao, Y.; Zhao, D.; Fan, J.; Liao, L.-S. Dyes Pigm. 2017, 146, 234–239. doi:10.1016/j.dyepig.2017.06.036
Return to citation in text: [1] -
Song, S.; Huang, G.; Kojima, T.; Nakae, T.; Uno, H.; Sakaguchi, H. Chem. Lett. 2017, 46, 1525–1527. doi:10.1246/cl.170614
Return to citation in text: [1] -
Ivanov, M. V.; Talipov, M. R.; Navale, T. S.; Rathore, R. J. Phys. Chem. C 2018, 122, 2539–2545. doi:10.1021/acs.jpcc.7b11232
Return to citation in text: [1] [2] [3] -
Wang, S.; Yang, P.; Chang, K.; Lv, W.; Mi, B.; Song, J.; Zhao, X.; Gao, Z. Org. Electron. 2019, 74, 269–275. doi:10.1016/j.orgel.2019.07.022
Return to citation in text: [1] -
Kogashi, K.; Matsuno, T.; Sato, S.; Isobe, H. Angew. Chem., Int. Ed. 2019, 58, 7385–7389. doi:10.1002/anie.201902893
Return to citation in text: [1] -
Suzuki, Y.; Tohnai, N.; Saeki, A.; Hisaki, I. Chem. Commun. 2020, 56, 13369–13372. doi:10.1039/d0cc06081j
Return to citation in text: [1] -
Yoshida, N.; Kamiguchi, S.; Sakao, K.; Akasaka, R.; Fujii, Y.; Maruyama, T.; Iwasawa, T. Tetrahedron Lett. 2020, 61, 152033. doi:10.1016/j.tetlet.2020.152033
Return to citation in text: [1] -
Yoshida, N.; Kamiguchi, S.; Fujii, Y.; Sakao, K.; Maruyama, T.; Tokai, S.; Matsumoto, Y.; Taguchi, Y.; Akasaka, R.; Iwasawa, T. Tetrahedron Lett. 2020, 61, 152406. doi:10.1016/j.tetlet.2020.152406
Return to citation in text: [1] -
Fujii, Y.; Maruyama, T.; Akasaka, R.; Sakao, K.; Tokai, S.; Taguchi, Y.; Matsumoto, Y.; Kamiguchi, S.; Yoshida, N.; Iwasawa, T. Tetrahedron Lett. 2021, 65, 152758. doi:10.1016/j.tetlet.2020.152758
Return to citation in text: [1] -
Fujii, Y.; Taguchi, Y.; Tokai, S.; Matsumoto, Y.; Yoshida, N.; Iwasawa, T. Tetrahedron 2021, 95, 132353. doi:10.1016/j.tet.2021.132353
Return to citation in text: [1] -
Yoshida, N.; Akasaka, R.; Awakura, Y.; Amaya, T.; Iwasawa, T. Eur. J. Org. Chem. 2021, 5343–5347. doi:10.1002/ejoc.202100869
Return to citation in text: [1] -
Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
The voltammograms including the low potential region for DBC-SMe and DBC-S(O)2Me are shown in Figure S1 (Supporting Information File 1). Other DBC derivatives did not show any peaks in the low potential region.
Return to citation in text: [1] [2] -
The HOMO and LUMO energy levels for DBC-H, DBC-Me, DBC-SMe, DBC-Br, DBC-S(O)2Me, and DBC-Si based on DFT calculations were reported in a previous paper together with the optimized structure and HOMO and LUMO orbital drawings (Supporting Information File 1 in reference [52]).
Return to citation in text: [1] [2] [3] -
Cardona, C. M.; Li, W.; Kaifer, A. E.; Stockdale, D.; Bazan, G. C. Adv. Mater. (Weinheim, Ger.) 2011, 23, 2367–2371. doi:10.1002/adma.201004554
Return to citation in text: [1] -
DFT calculations for DBC-H(56°)-1 and DBC-H(56°)-2 were performed in this study.
Return to citation in text: [1] [2] -
Bedi, A.; Gidron, O. Acc. Chem. Res. 2019, 52, 2482–2490. doi:10.1021/acs.accounts.9b00271
Return to citation in text: [1] -
TD-DFT calculations for DBC-H, DBC-Me, DBC-SMe, DBC-Br, DBC-S(O)2Me, and DBC-Si were performed in this study. Tables S1–S6 in Supporting Information File 1 show the wavelengths and oscillator strengths for the DBC derivatives.
Return to citation in text: [1]
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 54. | The HOMO and LUMO energy levels for DBC-H, DBC-Me, DBC-SMe, DBC-Br, DBC-S(O)2Me, and DBC-Si based on DFT calculations were reported in a previous paper together with the optimized structure and HOMO and LUMO orbital drawings (Supporting Information File 1 in reference [52]). |
| 56. | DFT calculations for DBC-H(56°)-1 and DBC-H(56°)-2 were performed in this study. |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 54. | The HOMO and LUMO energy levels for DBC-H, DBC-Me, DBC-SMe, DBC-Br, DBC-S(O)2Me, and DBC-Si based on DFT calculations were reported in a previous paper together with the optimized structure and HOMO and LUMO orbital drawings (Supporting Information File 1 in reference [52]). |
| 55. | Cardona, C. M.; Li, W.; Kaifer, A. E.; Stockdale, D.; Bazan, G. C. Adv. Mater. (Weinheim, Ger.) 2011, 23, 2367–2371. doi:10.1002/adma.201004554 |
| 1. | Clar, E. Polycyclic hydrocarbons; Academic Press: London, UK, 1964; Vol. I.II. |
| 2. | Harvey, R. G. Polycyclic Aromatic Hydrocarbons; Wiley-VCH: New York, NY, USA, 1997. |
| 3. | Fetzer, J. C. Large Polycyclic Aromatic Hydrocarbons, 1st ed.; Chemistry and Analysis, Vol. 158; John Wiley & Sons: Hoboken, NJ, USA, 2000. |
| 4. | Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z |
| 5. | Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c |
| 6. | Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045 |
| 7. | Pisula, W.; Feng, X.; Müllen, K. Chem. Mater. 2011, 23, 554–567. doi:10.1021/cm102252w |
| 8. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 9. | Sun, Z.; Ye, Q.; Chi, C.; Wu, J. Chem. Soc. Rev. 2012, 41, 7857–7889. doi:10.1039/c2cs35211g |
| 10. | Zhang, D.; Duan, L. J. Phys. Chem. Lett. 2019, 10, 2528–2537. doi:10.1021/acs.jpclett.9b00526 |
| 11. | Tian, D.; Chen, Y. Adv. Opt. Mater. 2021, 9, 2002264. doi:10.1002/adom.202002264 |
| 12. | Li, Q.; Zhang, Y.; Xie, Z.; Zhen, Y.; Hu, W.; Dong, H. J. Mater. Chem. C 2022, 10, 2411–2430. doi:10.1039/d1tc04866j |
| 27. | Tokito, S.; Noda, K.; Fujikawa, H.; Taga, Y.; Kimura, M.; Shimada, K.; Sawaki, Y. Appl. Phys. Lett. 2000, 77, 160–162. doi:10.1063/1.126910 |
| 28. | Yamaguchi, S.; Swager, T. M. J. Am. Chem. Soc. 2001, 123, 12087–12088. doi:10.1021/ja016692o |
| 29. | Kumar, S.; Varshney, S. K. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 2002, 378, 59–64. doi:10.1080/713738586 |
| 30. | Li, C.-W.; Wang, C.-I.; Liao, H.-Y.; Chaudhuri, R.; Liu, R.-S. J. Org. Chem. 2007, 72, 9203–9207. doi:10.1021/jo701504m |
| 31. | Chaudhuri, R.; Hsu, M.-Y.; Li, C.-W.; Wang, C.-I.; Chen, C.-J.; Lai, C. K.; Chen, L.-Y.; Liu, S.-H.; Wu, C.-C.; Liu, R.-S. Org. Lett. 2008, 10, 3053–3056. doi:10.1021/ol801029x |
| 32. | Shimizu, M.; Nagao, I.; Tomioka, Y.; Hiyama, T. Angew. Chem., Int. Ed. 2008, 47, 8096–8099. doi:10.1002/anie.200803213 |
| 33. | Navale, T. S.; Zhai, L.; Lindeman, S. V.; Rathore, R. Chem. Commun. 2009, 2857–2859. doi:10.1039/b903133b |
| 34. | Mori, T.; Fujita, K.; Kimura, M. J. Photopolym. Sci. Technol. 2010, 23, 317–322. doi:10.2494/photopolymer.23.317 |
| 35. | Tsuji, H.; Ueda, Y.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2010, 132, 11854–11855. doi:10.1021/ja1059119 |
| 36. | Navale, T. S.; Thakur, K.; Rathore, R. Org. Lett. 2011, 13, 1634–1637. doi:10.1021/ol200069c |
| 37. | Mochida, K.; Kawasumi, K.; Segawa, Y.; Itami, K. J. Am. Chem. Soc. 2011, 133, 10716–10719. doi:10.1021/ja202975w |
| 38. | Ueda, Y.; Tsuji, H.; Tanaka, H.; Nakamura, E. Chem. – Asian J. 2014, 9, 1623–1628. doi:10.1002/asia.201402102 |
| 39. | Hashimoto, S.; Ikuta, T.; Shiren, K.; Nakatsuka, S.; Ni, J.; Nakamura, M.; Hatakeyama, T. Chem. Mater. 2014, 26, 6265–6271. doi:10.1021/cm503102d |
| 40. | Suzuki, N.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 4984–4987. doi:10.1021/acs.orglett.5b02426 |
| 41. | Liu, X.-Y.; Tang, X.; Zhao, Y.; Zhao, D.; Fan, J.; Liao, L.-S. Dyes Pigm. 2017, 146, 234–239. doi:10.1016/j.dyepig.2017.06.036 |
| 42. | Song, S.; Huang, G.; Kojima, T.; Nakae, T.; Uno, H.; Sakaguchi, H. Chem. Lett. 2017, 46, 1525–1527. doi:10.1246/cl.170614 |
| 43. | Ivanov, M. V.; Talipov, M. R.; Navale, T. S.; Rathore, R. J. Phys. Chem. C 2018, 122, 2539–2545. doi:10.1021/acs.jpcc.7b11232 |
| 44. | Wang, S.; Yang, P.; Chang, K.; Lv, W.; Mi, B.; Song, J.; Zhao, X.; Gao, Z. Org. Electron. 2019, 74, 269–275. doi:10.1016/j.orgel.2019.07.022 |
| 45. | Kogashi, K.; Matsuno, T.; Sato, S.; Isobe, H. Angew. Chem., Int. Ed. 2019, 58, 7385–7389. doi:10.1002/anie.201902893 |
| 46. | Suzuki, Y.; Tohnai, N.; Saeki, A.; Hisaki, I. Chem. Commun. 2020, 56, 13369–13372. doi:10.1039/d0cc06081j |
| 53. | The voltammograms including the low potential region for DBC-SMe and DBC-S(O)2Me are shown in Figure S1 (Supporting Information File 1). Other DBC derivatives did not show any peaks in the low potential region. |
| 26. | Herbstein, F. H. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1979, 35, 1661–1670. doi:10.1107/s0567740879007354 |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 23. | Pascal, R. A., Jr. Chem. Rev. 2006, 106, 4809–4819. doi:10.1021/cr050550l |
| 24. | Rickhaus, M.; Mayor, M.; Juríček, M. Chem. Soc. Rev. 2016, 45, 1542–1556. doi:10.1039/c5cs00620a |
| 25. |
Ma, S.; Gu, J.; Lin, C.; Luo, Z.; Zhu, Y.; Wang, J. J. Am. Chem. Soc. 2020, 142, 16887–16893. doi:10.1021/jacs.0c08555
and references cited therein. |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 13. | Yao, T.; Yu, H.; Vermeij, R. J.; Bodwell, G. J. Pure Appl. Chem. 2008, 80, 533–546. doi:10.1351/pac200880030533 |
| 14. | Petrukhina, M. A.; Scott, L. T., Eds. Fragments of Fullerenes and Carbon Nanotubes; John Wiley & Sons: Hoboken, NJ, USA, 2011. doi:10.1002/9781118011263 |
| 15. | Wu, Y.-T.; Siegel, J. S. Chem. Rev. 2006, 106, 4843–4867. doi:10.1021/cr050554q |
| 16. | Tsefrikas, V. M.; Scott, L. T. Chem. Rev. 2006, 106, 4868–4884. doi:10.1021/cr050553y |
| 17. | Gingras, M. Chem. Soc. Rev. 2013, 42, 968–1006. doi:10.1039/c2cs35154d |
| 18. | Amaya, T.; Hirao, T. Chem. Rec. 2015, 15, 310–321. doi:10.1002/tcr.201402078 |
| 19. | Ball, M.; Zhong, Y.; Wu, Y.; Schenck, C.; Ng, F.; Steigerwald, M.; Xiao, S.; Nuckolls, C. Acc. Chem. Res. 2015, 48, 267–276. doi:10.1021/ar500355d |
| 20. | Segawa, Y.; Ito, H.; Itami, K. Nat. Rev. Mater. 2016, 1, 15002. doi:10.1038/natrevmats.2015.2 |
| 21. | Chen, C.-F.; Shen, Y. Helicene Chemistry; Springer: Berlin, Heidelberg, 2017. doi:10.1007/978-3-662-53168-6 |
| 22. | Saito, M.; Shinokubo, H.; Sakurai, H. Mater. Chem. Front. 2018, 2, 635–661. doi:10.1039/c7qm00593h |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 54. | The HOMO and LUMO energy levels for DBC-H, DBC-Me, DBC-SMe, DBC-Br, DBC-S(O)2Me, and DBC-Si based on DFT calculations were reported in a previous paper together with the optimized structure and HOMO and LUMO orbital drawings (Supporting Information File 1 in reference [52]). |
| 47. | Yoshida, N.; Kamiguchi, S.; Sakao, K.; Akasaka, R.; Fujii, Y.; Maruyama, T.; Iwasawa, T. Tetrahedron Lett. 2020, 61, 152033. doi:10.1016/j.tetlet.2020.152033 |
| 48. | Yoshida, N.; Kamiguchi, S.; Fujii, Y.; Sakao, K.; Maruyama, T.; Tokai, S.; Matsumoto, Y.; Taguchi, Y.; Akasaka, R.; Iwasawa, T. Tetrahedron Lett. 2020, 61, 152406. doi:10.1016/j.tetlet.2020.152406 |
| 49. | Fujii, Y.; Maruyama, T.; Akasaka, R.; Sakao, K.; Tokai, S.; Taguchi, Y.; Matsumoto, Y.; Kamiguchi, S.; Yoshida, N.; Iwasawa, T. Tetrahedron Lett. 2021, 65, 152758. doi:10.1016/j.tetlet.2020.152758 |
| 50. | Fujii, Y.; Taguchi, Y.; Tokai, S.; Matsumoto, Y.; Yoshida, N.; Iwasawa, T. Tetrahedron 2021, 95, 132353. doi:10.1016/j.tet.2021.132353 |
| 51. | Yoshida, N.; Akasaka, R.; Awakura, Y.; Amaya, T.; Iwasawa, T. Eur. J. Org. Chem. 2021, 5343–5347. doi:10.1002/ejoc.202100869 |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 58. | TD-DFT calculations for DBC-H, DBC-Me, DBC-SMe, DBC-Br, DBC-S(O)2Me, and DBC-Si were performed in this study. Tables S1–S6 in Supporting Information File 1 show the wavelengths and oscillator strengths for the DBC derivatives. |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 53. | The voltammograms including the low potential region for DBC-SMe and DBC-S(O)2Me are shown in Figure S1 (Supporting Information File 1). Other DBC derivatives did not show any peaks in the low potential region. |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 43. | Ivanov, M. V.; Talipov, M. R.; Navale, T. S.; Rathore, R. J. Phys. Chem. C 2018, 122, 2539–2545. doi:10.1021/acs.jpcc.7b11232 |
| 56. | DFT calculations for DBC-H(56°)-1 and DBC-H(56°)-2 were performed in this study. |
| 43. | Ivanov, M. V.; Talipov, M. R.; Navale, T. S.; Rathore, R. J. Phys. Chem. C 2018, 122, 2539–2545. doi:10.1021/acs.jpcc.7b11232 |
| 52. | Kamiguchi, S.; Akasaka, R.; Yoshida, N.; Imai, T.; Yamaoka, Y.; Amaya, T.; Iwasawa, T. Tetrahedron Lett. 2022, 92, 153664. doi:10.1016/j.tetlet.2022.153664 |
| 57. | Bedi, A.; Gidron, O. Acc. Chem. Res. 2019, 52, 2482–2490. doi:10.1021/acs.accounts.9b00271 |
© 2022 Imai et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.