Abstract
1,3-Dimethyl-2,3-dihydrobenzo[d]imidazoles, 1H, and 1,1',3,3'-tetramethyl-2,2',3,3'-tetrahydro-2,2'-bibenzo[d]imidazoles, 12, are of interest as n-dopants for organic electron-transport materials. Salts of 2-(4-(dimethylamino)phenyl)-4,7-dimethoxy-, 2-cyclohexyl-4,7-dimethoxy-, and 2-(5-(dimethylamino)thiophen-2-yl)benzo[d]imidazolium (1g–i+, respectively) have been synthesized and reduced with NaBH4 to 1gH, 1hH, and 1iH, and with Na:Hg to 1g2 and 1h2. Their electrochemistry and reactivity were compared to those derived from 2-(4-(dimethylamino)phenyl)- (1b+) and 2-cyclohexylbenzo[d]imidazolium (1e+) salts. E(1+/1•) values for 2-aryl species are less reducing than for 2-alkyl analogues, i.e., the radicals are stabilized more by aryl groups than the cations, while 4,7-dimethoxy substitution leads to more reducing E(1+/1•) values, as well as cathodic shifts in E(12•+/12) and E(1H•+/1H) values. Both the use of 3,4-dimethoxy and 2-aryl substituents accelerates the reaction of the 1H species with PC61BM. Because 2-aryl groups stabilize radicals, 1b2 and 1g2 exhibit weaker bonds than 1e2 and 1h2 and thus react with 6,13-bis(triisopropylsilylethynyl)pentacene (VII) via a “cleavage-first” pathway, while 1e2 and 1h2 react only via “electron-transfer-first”. 1h2 exhibits the most cathodic E(12•+/12) value of the dimers considered here and, therefore, reacts more rapidly than any of the other dimers with VII via “electron-transfer-first”. Crystal structures show rather long central C–C bonds for 1b2 (1.5899(11) and 1.6194(8) Å) and 1h2 (1.6299(13) Å).
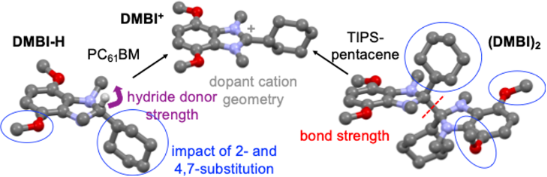
Graphical Abstract
Introduction
Electrical doping of organic semiconductors can play an important role in tuning the properties of organic semiconductors for a variety of applications [1-5]. The most straightforward n-dopants for doping electron-transporting materials are simple one-electron reductants; however, to be effective for a wide range of semiconductors, they must exhibit low ionization energies and thus air sensitivity. One approach to circumvent this issue is to identify systems where the electron-transfer process is coupled to other chemical reactions, increasing the kinetic stability of the dopant to air, and thus increasing its ease of storage and handling.
Arguably, the most widely investigated air-inert n-dopants are 1,3-dimethyl-2,3-dihydrobenzo[d]imidazoles (DMBI-H, 1H, Figure 1); these species have been known for decades (e.g., 1aH, one of the simplest such derivatives, was first reported in 1954 [6]), but were only introduced in n-dopants in 2010, when Bao and co-workers reported the use of N-DMBI-H (1bH, Figure 1) to n-dope fullerenes [7]. Although widely used, due to their facile synthesis, structural tunability, and good air stability in the solid state, 1H derivatives are relatively limited in dopant strength and their reactivity with organic semiconductors (SC) does not depend solely on the SC reduction potential, since the first step, at least in many cases, is a hydride transfer rather than an electron transfer [8,9]. Moreover, as well forming the desired semiconductor radical anion SC•−, and the stable DMBI+ (1+) species, a hydrogen atom must be lost from the dopant, in some cases resulting in the incorporation of hydrogen-reduced side products into the semiconductor film [9], although in other cases it may be lost as H2 [8,10,11].
![[1860-5397-19-121-1]](/bjoc/content/figures/1860-5397-19-121-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: DMBI+, DMBI-H, and (DMBI)2 derivatives discussed in this work (new compounds in red).
Figure 1: DMBI+, DMBI-H, and (DMBI)2 derivatives discussed in this work (new compounds in red).
The first report of a (DMBI)2 dimer (12, Figure 1) was of 1a2 in 1984 [12]. More recently, dimers 1b2–1f2 (Figure 1) have been used as n-dopants [13-20]. They behave similarly to the closed-shell dimers formed by certain 19-electron transition-metal sandwich compounds [21-23], exhibiting moderate air stability and acting as quite strong dopants, reacting with semiconductors more rapidly and predictably than hydride donors such as the corresponding 1H species [8], cleanly only to give SC•– and the corresponding monomeric cations. However, 12 dopants offer the possibility of more planar dopant ions than the organometallic dimers, which can be advantageous [19].
Although the impact of different 2-aryl Y groups on the reactivity of 1H species have been examined [9,24], there has been no direct comparison of the solution reactivity (or doping behavior) of 1H or 12 reductants with Y = aryl substituents to that of their Y = alkyl counterparts, while there has also been limited effort on examining the effects of substituents on the benzimidazole 6-membered ring in either class of reductant [16,24]. Furthermore, there has been little work on Y = 2-thienyl 1H derivatives. Here, we report two new dimers (1g2 and 1h2) and three new hydride donors (1gH, 1hH, 1iH). We also report crystal structures of several of these compounds and of several salts of the corresponding 1+ cations, and compare the electrochemistry and reactivity of these species.
Results and Discussion
Synthesis
Although an unsymmetrical 12-like molecule, 2-diethoxyphosphoryl-1,1',3,3'-tetramethyl-2,2',3,3'-tetrahydro-2,2'-bibenzo[d]imidazole, has been obtained from addition of HPO3Et2 across the central C=C bond of bis(1,3-dimethylbenzoimidazolinidin-2-ylidene) [25], 12 dimers have generally been obtained by reductive electrochemical or chemical dimerization of 1+ cations [12,13,16,19,26]. 1H derivatives can be obtained in a number of ways, including direct condensation of N,N'-dimethylphenylene-1,2-diamine derivatives with the appropriate aldehydes, YCHO [24,27], or borohydride reduction of 1+ salts [24]. The cations conversely can be obtained from 1H derivatives, for example through hydride abstraction by Ph3C+ [13]. Alternatively, they can also be obtained by condensation of N,N'-dimethylphenylene-1,2-diamine derivatives with acid chlorides, YCOCl, or through the methylation of 2-substituted benzoimidazoles [24], which in turn can be obtained from condensation between phenylene-1,2-diamines and carboxylic acids YCO2H [28], oxidative condensation between YCHO and phenylene-1,2-diamines [29], or reductive condensation between YCHO and 2-nitroanilines [24].
In this work we condensed the appropriate YCHO aldehyde (II) and 1,2-diaminobenzene (I) derivatives in the presence of sodium metabisulfite (Na2S2O5) [29] to obtain the corresponding substituted benzimidazoles (III) in essentially quantitative yield (Scheme 1). In the absence of Na2S2O5, but under otherwise similar conditions, we obtained in some cases the imines in which one of the amino groups condenses with the aldehyde but where the subsequent second condensation and oxidation does not take place, i.e., structures of type IV (Scheme 1), which are known to be converted to benzimidazoles by various oxidants and/or catalysts [30-32]. The benzimidazoles were then doubly methylated with iodomethane (or methyl tosylate) to afford the benzimidazolium iodides (or tosylates), 1+I− (or 1+OTs−), which were metathesized to the corresponding hexafluorophosphates, 1+PF6−. Either I− or PF6− salt can then be converted to the corresponding 1H derivative using NaBH4 in MeOH. The PF6− salts are somewhat more soluble than the iodides in THF, so were reductively dimerized to 12 in THF using Na:Hg, although reduction of 1i+PF6− failed to afford 1i2. As we have noted before for other 12 species, amides (V, Scheme 1) are encountered as both byproducts of dimer synthesis and dimer decomposition products [14]. V derivatives have also been obtained as pyrolysis products of a variety of Y = aryl 1H derivatives [33], while Vb has also been found to be both a solution decomposition product of 1bH [27,34] and a beneficial additive for a 1bH-doped polymer [27], and has been crystallographically characterized [34]. In the case of molecules with aryl Y-substituents – 1b2 and 1g2 – the room-temperature 1H and 13C NMR spectra (see Supporting Information File 1, Figures S2, S26 and S27, and reference [26]) display more resonances than expected based on the highest symmetry possible for the molecule indicating that the sample represents neither solely a high-symmetry conformer, nor a mixture of rapidly exchanging lower symmetry conformers. In the case of 1b2 all the proton resonances are rather broad, and variable-temperature experiments (see Supporting Information File 1, Figure S2) showed further broadening and then coalescence of some of these peaks on increasing the temperature, consistent with the room-temperature spectrum being affected by restricted rotation; interestingly the crystal structure of 1b2 contains molecules with two very different conformations (see below).
![[1860-5397-19-121-i1]](/bjoc/content/inline/1860-5397-19-121-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of DMBI-H and (DMBI)2 derivatives and structures of side products.
Scheme 1: Synthesis of DMBI-H and (DMBI)2 derivatives and structures of side products.
The 12 dimers are somewhat more sensitive to air than the corresponding 1H hydrides, but are all sufficiently stable as solids that they can briefly be handled in air, for example, for weighing. The solids do decompose slowly in air, although we have not quantified this; in inert atmosphere, however, they are completely stable (at least 4 months for solid 1b2). Both 1H and 12 derivatives decompose more rapidly on exposure to air in solution. In CDCl3 decomposition is rapid, consistent with the reactivity of many reductants with that solvent. In C6D6 these compounds are more stable, allowing, for example, rapid acquisition of a 1H NMR spectrum; however, handling under nitrogen is advisable as these species completely decompose to V (and perhaps 1+ species) on timescales of hours to days (see Supporting Information File 1, Figures S3–S5).
Crystal structures
We have determined the structures of two 12 dimers, four 1H derivatives (including 1bH, the structure of which has previously been reported, but with somewhat lower precision than in the present work [34]), and three salts of 1+ cations using single-crystal X-ray diffraction. Here, we briefly discuss some of the more interesting structural findings; a more detailed comparison of structural parameters is given in the Supporting Information File 1, Table S2. In particular, we are aware of only two previously reported crystal structures of DMBI dimers [14], although several related structures of organic dimers, including those of benzothiazoline, benzoxazoline, acridanyl, morpholinonyl dimers (22–52, respectively, Figure 2) have been reported in different chemical contexts [35-38]. The crystal structure of (N-DMBI)2, 1b2 (Figure 3), contains two crystallographically inequivalent molecules that are geometrically rather different from each other. One of the molecules has crystallographic inversion (Ci) symmetry, and approximate molecular C2h symmetry, and so has a perfectly staggered conformation around the central C–C bond and thus a Y–C–C–Y torsion angle of precisely 180°; the structure closely resembles those of the two inequivalent molecules in the structure of the previously reported Y = ferrocenyl derivative, 1c2 [14], or the molecule in the structure 22 [35], all three of which also have Ci symmetry. The other conformer present, although also staggered, has no crystallographic, or even approximate molecular, symmetry and is characterized by a Y–C–C–Y torsion angle of 60.3°. The conformation found in the structure of the Y = cyclohexyl, R’ = OMe derivative, 1h2 (Figure 3), is somewhat similar to that previously reported for its non-methoxylated analogue 1e2 [14]; the 1h2 molecule does not have the crystallographic C2 symmetry of the latter, but does have approximate molecular C2 symmetry, while the Y–C–C–Y torsion angles for 1h2 and 1e2 are 149.4° and 140.3°, respectively, and thus both intermediate between the perfectly staggered (180° torsion) and neighboring eclipsed conformation (120°). The imidazole rings in the previously reported and present dimer structures are mostly somewhat folded towards a puckered envelope conformation, generally with the Y group in a pseudo-axial position and the 1,3-methyl groups and the central C–C bond in pseudo-equatorial positions, although for one of the monomers in the unsymmetrical conformer in the structure of (N-DMBI)2, 1b2, the Y and central bond are pseudo-equatorial and pseudo-axial, respectively. However, this folding is generally much less pronounced than in 1H derivatives (see below, Figure 4, and Table S2 in Supporting Information File 1) presumably since in the dimers both 2-substituents (Y and the other monomer unit) are fairly bulky, whereas in the hydrides there is a large difference in bulk between the hydridic H-atom and theY-group and thus a strong preference for Y to occupy a pseudo-equatorial position.
![[1860-5397-19-121-2]](/bjoc/content/figures/1860-5397-19-121-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystallographically characterized molecules related to DMBI dimers.
Figure 2: Crystallographically characterized molecules related to DMBI dimers.
![[1860-5397-19-121-3]](/bjoc/content/figures/1860-5397-19-121-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structures from the single crystal structures of 1b2 (two crystallographically inequivalent molecules, left and center) and 1h2 (right), shown with 50% thermal ellipsoids and excluding disorder in 1h2 and hydrogen atoms for clarity.
Figure 3: Molecular structures from the single crystal structures of 1b2 (two crystallographically inequivale...
As with other 12 species [14] and related organic [35,37,38] and organometallic dimers [22,39-46], the central C–C bond of the present dimers are rather long compared to typical C–C bonds, although not remarkably so given that these are hexasubstituted ethane derivatives. Values of 1.5899(11) and 1.6194(8) Å are found for the symmetrical and unsymmetrical conformers of 1b2, respectively, while a value of 1.6299(13) Å is found for 1h2; these may be compared to hexasubstituted central C–C bond length values of 1.595(5) and 1.601(5) Å for the two inequivalent molecules of the Y = Fc, R = R' = H derivative 1c2 [14], 1.640(4) Å for the Y = cyclohexyl, R = R' = H derivative 1e2 [14], 1.573 Å for 22 [35], and 1.591 Å for 52 [38], while (PhEt2C)2, a simple hexasubstituted ethane, exhibits a central C–C bond length of 1.635 Å [47]. The tetrasubstituted central C–C bond of 42 is also rather long (1.58 Å) [37]. Bridged benzoxazoline dimers, 32, have, on the other hand, relatively short C–C central bonds, perhaps due to the influence of the propanediyl tether; the hexasubstituted bond of 3c2 is only 1.549(6) Å in length, while the tetrasubstituted bonds of 3a2 and 3b2 are even shorter [36].
The crystallographically determined central C–C bond lengths for 1b2 are shorter than that previously reported for the Y = cyclohexyl, R = R' = H derivative 1e2 (1.640(4) Å) [14], despite DFT calculations indicating that the former dimer is considerably more weakly bonded [8,14] and kinetic evidence for the “cleavage-first” mechanism occurring in doping reactions using 1b2 but not 1e2 (see below). We have previously noted a similar lack of correlation between bond length and bond dissociation energy in comparing the structures of 1c2 (Y = Fc; R = R' = H) and 1e2 (Y = cyclohexyl; R = R' = H) [14], and in comparing those of different organometallic dimers [22,46]. As noted in our previous work [14,22,46], the bond length depends on orbital overlap and steric strain in the dimer, whereas dissociation energetics also depend on the stability of the monomeric odd-electron species, which vary considerably; in the case of 1• radicals an important factor is the ability of the Y substituent to delocalize spin density.
The 1H structures (Figure 4) are similar to those of other DMBI-H structures in the literature [34,48-50] (and are compared in more detail in Supporting Information File 1, Table S2); in all cases the imidazole ring is folded in a “puckered envelope” conformation with the 2-Y and 1,3-dimethyl substituents in pseudo-equatorial positions and the reactive hydridic 2-H-atom pesudo-axial. The cation structures (Figure 5) give some insight into the variety of dopant-ion shapes and sizes that can be afforded by these types of dopants. The angle between the imidazolium ring and the aromatic ring of the 1g+I− is 41.5°, close to the range of values previously reported for 1b+ salts (42.5–52.5°) [19,34] and for salts of Y = Ph, R = R' = H cations with different counterions (42.0–54.9°) [51-53]. As expected, owing to reduced steric interactions associated with the five-membered rather than six-membered aromatic ring, the structure of 1i+PF6− contains a somewhat more planar cation (31.9°). Finally, we note that the new structures reported here mean that the 1b and 1h systems join the 1c (Y = Fc; R = R' = H) system [50] as families for which 1+, 1H, and 12 members are all crystallographically characterized.
![[1860-5397-19-121-4]](/bjoc/content/figures/1860-5397-19-121-4.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 4: Molecular structures from the single crystal structures of 1bH (upper left), 1gH (upper right), 1hH (lower left), and 1iH (lower right), shown with 50% thermal ellipsoids and excluding hydrogen atoms for clarity, except for the hydridic 2-hydrogen atoms (located and refined for 1bH, geometrically placed for the others).
Figure 4: Molecular structures from the single crystal structures of 1bH (upper left), 1gH (upper right), 1hH...
![[1860-5397-19-121-5]](/bjoc/content/figures/1860-5397-19-121-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Structures of the cations from the single crystal structures of 1g+I− (left), 1h+PF6− (center), and 1i+PF6− (right), shown with 50% thermal ellipsoids and excluding hydrogen atoms and counter anions.
Figure 5: Structures of the cations from the single crystal structures of 1g+I− (left), 1h+PF6− (center), and ...
Electrochemistry
The 1+, 1H, and 12 species were investigated using cyclic voltammetry in THF/0.1 M Bu4NPF6 at a scan rate of 50 mV s−1. The voltammograms (shown for one series of compounds in Figure 6) were qualitatively similar to those reported and shown elsewhere for other compounds of the same classes [9,13,19,24], and the redox potentials are summarized in Table 1. The cations exhibit features assigned to E(1+/1•) that are non-reversible owing to the rapid dimerization of 1•. These values are important in determining the overall thermodynamic reducing power of the dimers according to:
![[1860-5397-19-121-i2]](/bjoc/content/inline/1860-5397-19-121-i2.svg?max-width=590&scale=1.18182)
where ΔGdiss(12) is the free-energy change for dissociation of 12 to 1• (dissociation energetics are not estimated in the present work, but have been estimated using DFT calculations for 1b–e2 in previous works [8,14] and, in favorable cases, can be experimentally estimated using electron spin resonance [14] or using dissociation and dimerization barriers from reaction kinetics and variable scan-rate electrochemistry, respectively [54]) and where F is the Faraday constant. Similarly, at least for cases where the reactive hydrides of 1H derivatives are ultimately lost as H2, the strength of 1H dopants is given by:
![[1860-5397-19-121-i3]](/bjoc/content/inline/1860-5397-19-121-i3.svg?max-width=590&scale=1.18182)
where ΔGdiss(1H) is the free-energy change for dissociation of 1H to 1• and H• (again, not discussed in this work), and ΔGdiss(H2) the free-energy change for dissociation of dihydrogen. The values of E(1+/1•) are also relevant to the kinetics of steps in doping reactions that involve 1•, in particular for doping reactions in which the initial step is dimer dissociation and the second step is an electron transfer from 1• to SC (or SC•−). The E(1+/1•) potentials for the Y = 4-dimethylaminophenyl 1b+/1b• and 1g+/1g• systems are both somewhat less reducing than those for their Y = cyclohexyl counterparts, 1e+/1e• and 1h+/1h•, respectively. These differences are also similar to those previously seen in the comparison of Y = metallocenyl systems 1c+/1c• and 1d+/1d• with 1e+/1e• (and in the DFT-calculated ionization energies of 1c–e•) [14,50] and are perhaps surprising since 4-(dimethylamino)phenyl and metallocenyl groups are π-donors, unlike cyclohexyl, and thus might be expected to be better able to stabilize an adjacent cation. However, aryl and metallocenyl substituents also stabilize adjacent radicals more effectively than alkyl groups and this effect is presumably dominant in the present case. The importance of radical stabilization may in part be because the positive charges in Y = H or alkyl 1+ ions is already substantially stabilized by the aromaticity of the benzimidazolium ions, whereas the spin densities of the corresponding 1• radicals are highly localized; indeed DFT calculations for the Y = alkyl 1e• derivative indicate spin density almost entirely on the 2-position of the five-membered ring, while for Y = aryl and metallocenyl examples 1b•, 1c•, and 1d• there is substantial spin delocalization onto the Y-substituents [14,55]. Different extents of deviation from planarity in cations and radicals, as well as inductive effects, may also play a role.
![[1860-5397-19-121-6]](/bjoc/content/figures/1860-5397-19-121-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Cyclic voltammograms (50 mV s−1, THF, 0.1 M Bu4NPF6) of 1g+PF6–, 1gH, and 1g2, in each case containing ferrocene as an internal reference. Black arrows indicate the starting point and scan initial direction for each voltammograms. Note that the oxidation peak of 1g2 is seen in the voltammogram of 1g+PF6– following scanning of the reduction peak, while the reduction peak of the cation is seen in the voltammograms of both 1gH and 1g2 following scanning of the irreversible oxidation peaks.
Figure 6: Cyclic voltammograms (50 mV s−1, THF, 0.1 M Bu4NPF6) of 1g+PF6–, 1gH, and 1g2, in each case contain...
Table 1: Electrochemical potentials (V) for DMBI derivativesa.
| Ered(1+/1•) | Eox(1H•+/1H) | Eox(12•+/12) | |
| 1b (Y = C6H4-4-NMe2; R = R' = H) | −2.38b | −0.13c | −0.75b |
| 1c (Y = Fc; R = R' = H) | −2.24d | −0.06e | −0.89d |
| 1d (Y = Rc; R = R' = H) | −2.29d | −0.07e | −0.59d |
| 1e (Y = cy-C6H11; R = R' = H) | −2.45d | −0.06 | −0.64d |
| 1g (Y = C6H4-4-NMe2; R = H; R' = OMe) | −2.42 | −0.22 | −0.87 |
| 1h (Y = cy-C6H11; R = H; R' = OMe) | −2.56 | −0.11 | −0.92 |
| 1i (Y = 2-C4H3S-5-NMe2; R = R' = H) | –2.05 | −0.22 | – |
avs FeCp2+/0 in THF, 0.1 M Bu4NPF6; bdata from reference [19]; cdata from reference [8]; ddata from reference [14]; edata from reference [50].
The 1i+/1i• (Y = 5-(dimethylamino)-2-thienyl; R = R' = H) potential is less reducing than that of 1b+/1b• (Y = 4-dimethylaminophenyl; R = R' = H). 5-(Dimethylamino)-2-thienyl is more strongly π-donating than 4-dimethylaminophenyl, at least according to NMR and DFT data for molecules in which the (hetero)aryl group is more or less coplanar with a π-acceptor [56], although some tabulated Swain–Lupton substituent constants do suggest phenyl can be a stronger π-donor than thienyl towards another aryl ring [57]. Presumably inductive effects destabilizing 1i+, different extents of planarization, and improved radical stabilization by the 5-(dimethylamino)-2-thienyl susbtituent play a role. As expected, R' = OMe groups on the six-membered benzimidazolium ring do have a net cation-stabilizing effect, resulting in 1g• and 1h• being more reducing monomers than their non-methoxylated analogues 1b• and 1e•, respectively.
Cyclic voltammograms of both 1H and 12 both reveal irreversible oxidations (with the corresponding 1+ reductions seen in subsequent reductive cycles, see Figure 6 for examples). These 1H•+/1H and 12•+/12 potentials are relevant to the air stability of the hydrides and dimers, respectively, as well as to other processes in which 1H or 12 acts as an electron donor, such as the initation step proposed for the radical-chain dehalogenation of α-dihaloketones by a 1H derivative [58] and dimer n-doping reactions that proceed via the “ET-first” mechanism (see below). In all cases the dimers are more easily oxidized, consistent with their greater air sensitivity. The impact of the Y-substituents on both 1H•+/1H and 12•+/12 potentials is not straightforward; one would expect π-conjugated substituents to make little contribution to the HOMO of either 1H or 12 (as shown in calculated molecular orbitals for several examples [14,50,55,59,60]) and so the dependence of these potentials on Y is likely to be due to a combination of inductive effects and perhaps steric effects on the molecular conformation. As expected, methoxy R' substituents lead to 1H•+/1H and 12•+/12 potentials that are more reducing than those for analogous species without these groups. 1h2 (Y = cyclohexyl, R = H, R' = MeO) is the most easily oxidized DMBI dimer that we have examined to date; however, it is a little less easily oxidized than [RuCp*(1,3,5-Me3C6H3)]2 (−1.09 V) [61] and, like [RuCp*(1,3,5-Me3C6H3)]2, can still be handled in air.
Reactivity
To compare the reactivity of the new compounds towards relevant organic semiconductors (SC), we have examined the reactions of the 1H derivatives with the solubilized fullerene PC61BM (VI, Figure 7) and that of the 12 derivatives with 6,13-bis(triisopropylsilylethynyl)pentacene (TIPS-pentacene, VII, Figure 7), since we have previously found that these dopant class/SC combinations often react on a timescale suitable for monitoring using UV–vis–NIR spectroscopy (1H derivatives do not react significantly with VII in solution at room temperature, while the reactions of 12 derivatives and VI are very rapid) [9,14,50,61]. Figure 8a compares the evolution of the absorbance at 1030 nm, corresponding to a VI•– absorption maximum, when doping excess VI with 1H derivatives in chlorobenzene at 293 K in the absence of light, air, and water. In each case the reaction is apparently first order in dopant, consistent with the rate law:
![[1860-5397-19-121-i4]](/bjoc/content/inline/1860-5397-19-121-i4.svg?max-width=590&scale=1.18182)
previously demonstrated for 1bH and VI [9]. The rate constants, k, obtained assuming this rate law are shown in Table 2 (the value for 3b being similar to that previously determined [9]). One can anticipate, extending the Hammond postulate, that increased driving forces should correlate with reduced barriers and increased rate constants. Values of k do not correlate with the 1H•+/1H potentials, but, at least when comparing aryl and alkyl Y substituents and when comparing R' = H and R' = OMe examples, do correlate with the expected stability of the resultant 1+ cations, which is also expected to correlate with the hydride donor strength of 1H. This is consistent with previous findings that the first and rate-determining step of several 1H/SC reactions, including 1H/VI reactions, is not an electron transfer, but a hydride transfer [8,9]. There is conflicting evidence in the literature regarding the π-donor characteristics of phenyl and thienyl groups [56,57], while thienyl is more inductively electron-withdrawing [57], as noted in the electrochemical section; however, the observed rate constants for 1bH and 1iH suggest that 5-dimethylamino-2-thienyl affords less net charge stabilization than 4-dimethylaminophenyl.
![[1860-5397-19-121-7]](/bjoc/content/figures/1860-5397-19-121-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Acceptors used to examine reactivity of DMBI-H and (DMBI)2 derivatives.
Figure 7: Acceptors used to examine reactivity of DMBI-H and (DMBI)2 derivatives.
![[1860-5397-19-121-8]](/bjoc/content/figures/1860-5397-19-121-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: a) Temporal evolution of the absorbance at 1030 nm, corresponding to an absorption maximum of VI•–, when PC61BM (VI, 2.7 mM) is reacted with different 1H derivatives (0.4 mM) in chlorobenzene at room temperature. b) Temporal evolution of the absorbance at 750 nm, corresponding to one of the absorption maxima of VII•–, when TIPS-pentacene (VII, 0.026 mM) is reacted with different 12 derivatives (0.37 mM) in chlorobenzene at room temperature.
Figure 8: a) Temporal evolution of the absorbance at 1030 nm, corresponding to an absorption maximum of VI•–,...
Two reaction pathways have been established for the oxidation of organometallic and organic dimers. A “cleavage-first” mechanism, whereby the dimer is in equilibrium with a small concentration of the corresponding odd-electron monomer, which can then rapidly react with an acceptor such as an organic semiconductor (SC) through an exergonic electron transfer (ET), has been observed for the reactions of several relatively weakly bonded dimeric dopants (the Y = metallocenyl DMBI dimers 1c2 and 1d2 as well as various organometallic dimers) with VII [14,46,61], as well as in the oxidation of bis(3,5,5-trimethyl-2-morpholinon-3-yl), 52 (Figure 2), by isatin derivatives [62]. In the alternative “ET-first” mechanism the first step is an endergonic dimer-to-SC ET; subsequent rapid cleavage of the odd-electron dimer cation affords the stable monomer cation and an odd-electron monomer, the latter then undergoing an exergonic ET to another SC molecule. The “ET-first” mechanism occurs in parallel with the “cleavage-first” mechanism for many of the VII doping reactions mentioned above and is the only mechanism seen for dimeric dopants that are more strongly bound (1e2, as well as various organometallic dimers including [RuCp*(1,3,5-Me3C6H3)]2) [14,46,61], as well as being observed in different contexts in, for example, the oxidation of 42 by various quinone derivatives [63]. For both mechanisms, the first steps are typically rate determining and thus, in general, the rate law is:
![[1860-5397-19-121-i5]](/bjoc/content/inline/1860-5397-19-121-i5.svg?max-width=590&scale=1.18182)
where k1 and k2 are rate constants for the first steps of the “cleavage first” and “ET-first” pathways respectively, k1 being negligible in the case of strongly bound dimers.
Figure 8b compares the evolution of one of the distinctive VII •– absorptions when doping VII with excess 12 derivatives in chlorobenzene at 293 K in the absence of light, air, and water. In the case of the Y = 4-dimethylaminophenyl dimers 1b2 and 1g2, the VII•– absorption grows in and then falls approximately linearly at a comparable rate. This type of plot is a signature of dimer/VII combinations for which the “cleavage-first” pathway is important and has previously been seen for the reactions of VII with 1c2, 1d2, (RhCp*Cp)2, and one of the isomers of [RuCp*{1,4-(Me2N)2C6H4}]2, all of which are calculated to be relatively weakly bonded [14,46,61]. Specifically, this behavior is consistent with a “cleavage-first” mechanism in which the initial cleavage is rate determining and for which the resultant one-electron monomers are capable of reducing both VII to VII•– (−1.55 V) and VII•– to VII2– (−1.93 V); since the cleavage is rate determining, VII will be converted to VII•– and then, when excess dimer is used, to VII2– with a comparable rate constant. Indeed spectra obtained at long-reaction times (see Supporting Information File 1, Figure S7) are similar to those previously attributed to VII2– [14,46,61], such as the reaction product of VII and Na:K. On the other hand, when only the “ET-first” mechanism is operative, the conversion of VII•– to VII2– will be much slower, if it is even observable, than the initial formation of VII•– from VII due to the considerably greater endergonicity expected for this step. This is seen for the solution reaction of 1h2, where, as in the case of non-methoxylated analogue 1e2, only the formation of VII•– is seen and the growth in its absorbance can be fitted as first order in VII. Returning to the case of 1b2 and 1g2, we note that the rise in VII•– absorption is neither zero-order nor first-order in VII, consistent with both mechanisms contributing, as previously demonstrated by more extensive investigations in the case of 1c2, 1d2, and (RhCp*Cp)2 [14,61]. Thus, the Y = alkyl derivative (1h2, “ET-first” only) appears to be more strongly bonded than its Y = aryl counterparts (1b2, 1g2, both mechanisms), consistent with previous DFT calculations for 1b2 and 1e2 (ΔUdiss = 163 and 210 kJ mol–1, respectively) and with the expected impact of the different Y substituents on monomer radical stability. In addition, the reaction of 1h2 and VII to form VII•– under the conditions used in the present study is complete much sooner than reactions using 1b2 or 1g2, consistent with the ET-first reaction of 3h2 being more rapid than that for either 1b2 or 1g2. Furthermore, the presumed “cleavage-first” reductions of VII•– to VII2– proceed only slightly faster for 1g2 than for its non-methoxylated analogue 1b2, suggesting the OMe groups only slightly weaken the bond in the latter and that the difference in the rates of formation of VII•– with these two dimers is largely due to differences in the rate of the 12-to-VII ET reaction. Furthermore, the ordering of ET-first rates (1h2 > 1g2 > 1b2 > 1e2, that for 1e2 being estimated by extrapolating previously reported parameters to the present conditions of temperature and concentration) reflecting the trend expected based on the E(12•+/12) values of Table 1.
It is worth noting that, although we see evidence for the “cleavage-first” mechanism in the reactions of 1b2 and 1g2 with VII at these specific concentrations, the “ET-first” mechanism will dominate these reactions (as well as those of the same dopants with more readily reduced SCs) under typical doping conditions, where SC and sub-stoichiometric dimer are mixed in solution prior to spin-coating at much higher concentrations. However, as we have previously noted, there are potential advantages and disadvantages for dimers for which the cleavage-first pathway is viable and those for which it is not. For the former class, doping in solution will proceed as long as E(SC/SC•–) is less reducing than E(1+/0.512), whereas in the latter this limit can only be reached as long as the 12-to-SC ET step is kinetically feasible under the reaction conditions. Moreover, for a given monomer redox potential, E(1+/1•), a weakly bound dimer will be thermodynamically stronger (Equation 1) although, in some cases the effects of structural change on E(1+/1•) and ΔGdiss(12) partially cancel one another, as in the comparison of 1b2 vs 1e2 or 1g2 vs 1h2 (i.e., for Y = 4-dimethylaminophenyl, dimers are more weakly bound and monomers less reducing that for Y = cyclohexyl). Conversely, the combination of a strongly bound dimer and an acceptor with E(SC/SC•−) with the reach of E(1+/0.512), but sufficiently cathodic that ET is very slow, could permit activation of doping by an external stimulus, such as photoexcitation, when desired, for example subsequent to processing.
Conclusion
In conclusion we have reported a number of new DMBI-H and (DMBI)2 reductants and compared their structures, electrochemistry, and reactivity with those of previously reported analogues. The structures show similar features to other related compounds, notably the dimers show long central C–C bonds. The E(1+/1•) potentials depend strongly on the 2-substituents (Y), become increasing reducing (more negative) in the order Y = 5-(dimethylamino)thiophen-2-yl < 4-(dimethylamino)phenyl < cyclohexyl, indicating the effects of radical stabilization are more important than those of cation stabilization, while the E(1H•+/1H) and E(12•+/12) potentials are less strongly and clearly affected by the 2-substituents. On the other hand, methoxy R’ substituents lead to more reducing values of E(1+/1•), E(1H•+/1H), and E(12•+/12) than for R’ = H analogues. The reaction rates of 1H with PC61BM (VI) increase in the order Y = cyclohexyl < 5-(dimethylamino)thiophen-2-yl < 4-(dimethylamino)phenyl and R’ = H < MeO, broadly consistent with the anticipated influence of these substituents on the DMBI+ stability, as expected for a hydride-transfer reaction. The rates of reactions of the dimers with TIPS-pentacene (VII) follow a more complex pattern: examples with Y = cyclohexyl react solely via an “electron-transfer-first” mechanism, consistent with a relatively strongly bonded dimer, whereas Y = 4-(dimethylamino)phenyl derivatives also react by a “cleavage-first” mechanism, consistent with a weaker central bond, which in turn is consistent with stabilization of the monomeric radicals by the 2-aryl substituents. The Y = cyclohexyl, R’ = OMe dimer reacts most rapidly with TIPS-pentacene via the “ET-first” mechanism, consistent with this dimer also exhibiting the most cathodic value of E(12•+/12). Overall, this study gives insight into how substituents have different effects on the reactivity of DMBI-H derivatives and of (DMBI)2 species, and may help provide guidance for dopant selection and for future dopant design.
Supporting Information
| Supporting Information File 1: Synthetic and other experimental procedures, details of crystal-structure determinations, variable-temperature NMR data, stability data, optical spectra for reactivity studies, and NMR spectra of new compounds. | ||
| Format: PDF | Size: 2.4 MB | Download |
Funding
The work was primarily supported by the National Science Foundation through award DMR-1807797/2216857. Diffraction studies at New Mexico Highlands University were supported by the National Science Foundation’s PREM program through DMR-2122108. SJ was supported by Fulbright-Nehru Postdoctoral Fellowship from the United States-India Educational Foundation and Institute of International Education for (grant no. 2266/FNPDR/2017). This work was also authored in part by the National Renewable Energy Laboratory (NREL), operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308; specifically, some of the data analysis was carried out as part of a Laboratory Directed Research and Development (LDRD) Program at NREL. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.
References
-
Walzer, K.; Maennig, B.; Pfeiffer, M.; Leo, K. Chem. Rev. 2007, 107, 1233–1271. doi:10.1021/cr050156n
Return to citation in text: [1] -
Russ, B.; Glaudell, A.; Urban, J. J.; Chabinyc, M. L.; Segalman, R. A. Nat. Rev. Mater. 2016, 1, 16050. doi:10.1038/natrevmats.2016.50
Return to citation in text: [1] -
Lüssem, B.; Keum, C.-M.; Kasemann, D.; Naab, B.; Bao, Z.; Leo, K. Chem. Rev. 2016, 116, 13714–13751. doi:10.1021/acs.chemrev.6b00329
Return to citation in text: [1] -
Wang, Z.-K.; Liao, L.-S. Adv. Opt. Mater. 2018, 6, 1800276. doi:10.1002/adom.201800276
Return to citation in text: [1] -
Barlow, S.; Marder, S. R.; Lin, X.; Zhang, F.; Kahn, A. Electrial Doping of Organic Semiconductors with Molecular Oxidants and Reductants. In Conjugated Polymers, 4th ed.; Skotheim, T. A.; Reynolds, J.; Thompson, B. C., Eds.; CRC Press: Boca Raton, FL, USA, 2019. doi:10.1201/9780429190520-2
Return to citation in text: [1] -
Wahl, H. Bull. Soc. Chim. Fr. 1954, 251–253.
Return to citation in text: [1] -
Wei, P.; Oh, J. H.; Dong, G.; Bao, Z. J. Am. Chem. Soc. 2010, 132, 8852–8853. doi:10.1021/ja103173m
Return to citation in text: [1] -
Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Guo, H.; Yang, C.-Y.; Zhang, X.; Motta, A.; Feng, K.; Xia, Y.; Shi, Y.; Wu, Z.; Yang, K.; Chen, J.; Liao, Q.; Tang, Y.; Sun, H.; Woo, H. Y.; Fabiano, S.; Facchetti, A.; Guo, X. Nature 2021, 599, 67–73. doi:10.1038/s41586-021-03942-0
Return to citation in text: [1] -
Pallini, F.; Mattiello, S.; Manfredi, N.; Mecca, S.; Fedorov, A.; Sassi, M.; Al Kurdi, K.; Ding, Y.-F.; Pan, C.-K.; Pei, J.; Barlow, S.; Marder, S. R.; Nguyen, T.-Q.; Beverina, L. J. Mater. Chem. A 2023, 11, 8192–8201. doi:10.1039/d3ta00231d
Return to citation in text: [1] -
Ludvík, J.; Pragst, F.; Volke, J. J. Electroanal. Chem. Interfacial Electrochem. 1984, 180, 141–156. doi:10.1016/0368-1874(84)83576-5
Return to citation in text: [1] [2] -
Naab, B. D.; Zhang, S.; Vandewal, K.; Salleo, A.; Barlow, S.; Marder, S. R.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2014, 26, 4268–4272. doi:10.1002/adma.201400668
Return to citation in text: [1] [2] [3] [4] -
Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] -
Naab, B. D.; Gu, X.; Kurosawa, T.; To, J. W. F.; Salleo, A.; Bao, Z. Adv. Electron. Mater. 2016, 2, 1600004. doi:10.1002/aelm.201600004
Return to citation in text: [1] -
Yuan, D.; Huang, D.; Zhang, C.; Zou, Y.; Di, C.-a.; Zhu, X.; Zhu, D. ACS Appl. Mater. Interfaces 2017, 9, 28795–28801. doi:10.1021/acsami.7b07282
Return to citation in text: [1] [2] [3] -
Schwarze, M.; Gaul, C.; Scholz, R.; Bussolotti, F.; Hofacker, A.; Schellhammer, K. S.; Nell, B.; Naab, B. D.; Bao, Z.; Spoltore, D.; Vandewal, K.; Widmer, J.; Kera, S.; Ueno, N.; Ortmann, F.; Leo, K. Nat. Mater. 2019, 18, 242–248. doi:10.1038/s41563-018-0277-0
Return to citation in text: [1] -
Al Kurdi, K.; Gregory, S. A.; Jhulki, S.; Conte, M.; Barlow, S.; Yee, S. K.; Marder, S. R. Mater. Adv. 2020, 1, 1829–1834. doi:10.1039/d0ma00406e
Return to citation in text: [1] -
Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lungwitz, D.; Joy, S.; Mansour, A. E.; Opitz, A.; Karunasena, C.; Li, H.; Panjwani, N. A.; Moudgil, K.; Tang, K.; Behrends, J.; Barlow, S.; Marder, S. R.; Brédas, J.-L.; Graham, K.; Koch, N.; Kahn, A. J. Phys. Chem. Lett. 2023, 14, 5633–5640. doi:10.1021/acs.jpclett.3c01022
Return to citation in text: [1] -
Guo, S.; Kim, S. B.; Mohapatra, S. K.; Qi, Y.; Sajoto, T.; Kahn, A.; Marder, S. R.; Barlow, S. Adv. Mater. (Weinheim, Ger.) 2012, 24, 699–703. doi:10.1002/adma.201103238
Return to citation in text: [1] -
Mohapatra, S. K.; Fonari, A.; Risko, C.; Yesudas, K.; Moudgil, K.; Delcamp, J. H.; Timofeeva, T. V.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2014, 20, 15385–15394. doi:10.1002/chem.201404007
Return to citation in text: [1] [2] [3] [4] -
Mohapatra, S. K.; Marder, S. R.; Barlow, S. Acc. Chem. Res. 2022, 55, 319–332. doi:10.1021/acs.accounts.1c00612
Return to citation in text: [1] -
Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Küçükbay, H.; Çetinkaya, E.; Çetinkaya, B.; Lappert, M. F. Synth. Commun. 1997, 27, 4059–4066. doi:10.1080/00397919708005451
Return to citation in text: [1] -
Pham, P. H.; Barlow, S.; Marder, S. R.; Luca, O. R. Chem Catal. 2023, 3, 100675. doi:10.1016/j.checat.2023.100675
Return to citation in text: [1] [2] -
Pallini, F.; Mattiello, S.; Cassinelli, M.; Rossi, P.; Mecca, S.; Tan, W. L.; Sassi, M.; Lanzani, G.; McNeill, C. R.; Caironi, M.; Beverina, L. ACS Appl. Energy Mater. 2022, 5, 2421–2429. doi:10.1021/acsaem.1c03893
Return to citation in text: [1] [2] [3] -
Lim, C.-H.; Ilic, S.; Alherz, A.; Worrell, B. T.; Bacon, S. S.; Hynes, J. T.; Glusac, K. D.; Musgrave, C. B. J. Am. Chem. Soc. 2019, 141, 272–280. doi:10.1021/jacs.8b09653
Return to citation in text: [1] -
Ghosh, R.; Kushwaha, A.; Das, D. J. Phys. Chem. B 2017, 121, 8786–8794. doi:10.1021/acs.jpcb.7b05947
Return to citation in text: [1] [2] -
Crippa, G. B.; Maffei, S. Gazz. Chim. Ital. 1941, 71, 194–200.
Return to citation in text: [1] -
Balachandran, K. S.; George, M. V. Indian J. Chem. 1973, 11, 1267–1271.
Return to citation in text: [1] -
Speier, G.; Párkányi, L. J. Org. Chem. 1986, 51, 218–221. doi:10.1021/jo00352a016
Return to citation in text: [1] -
Reddy, A. P. R.; Veeranagaiah, V.; Ratnam, C. V. Indian J. Chem. 1985, B24, 367–371.
Return to citation in text: [1] -
Bardagot, O.; Aumaître, C.; Monmagnon, A.; Pécaut, J.; Bayle, P.-A.; Demadrille, R. Appl. Phys. Lett. 2021, 118, 203904. doi:10.1063/5.0047637
Return to citation in text: [1] [2] [3] [4] [5] -
Miler-Srenger, E. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1973, 29, 1119–1124. doi:10.1107/s0567740873003973
Return to citation in text: [1] [2] [3] [4] -
Ramirez-Montes, P. I.; Ochoa, M. E.; Rodríguez, V.; Santillan, R.; García-Ortega, H.; Rodríguez, P.; Farfán, N. Tetrahedron Lett. 2012, 53, 5887–5890. doi:10.1016/j.tetlet.2012.08.094
Return to citation in text: [1] [2] -
Preuss, J.; Zanker, V.; Gieren, A. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1977, 33, 2317–2319. doi:10.1107/s0567740877008346
Return to citation in text: [1] [2] [3] -
Haltiwanger, R. C.; Koch, T. H.; Olesen, J. A.; Kim, C. S.; Kim, N. K. J. Am. Chem. Soc. 1977, 99, 6327–6331. doi:10.1021/ja00461a026
Return to citation in text: [1] [2] [3] -
Andrianov, V. G.; Struchkov, Y. T.; Petrakova, V. A.; Vol'kenau, N. A. Koord. Khim. 1986, 12, 978–980.
Return to citation in text: [1] -
Gaudet, M. V.; Hanson, A. W.; White, P. S.; Zaworotko, M. J. Organometallics 1989, 8, 286–293. doi:10.1021/om00104a004
Return to citation in text: [1] -
Lee, S.; Lovelace, S. R.; Arford, D. J.; Geib, S. J.; Weber, S. G.; Cooper, N. J. J. Am. Chem. Soc. 1996, 118, 4190–4191. doi:10.1021/ja952217c
Return to citation in text: [1] -
Hsu, S. C. N.; Yeh, W.-Y.; Lee, G.-H.; Peng, S.-M. J. Am. Chem. Soc. 1998, 120, 13250–13251. doi:10.1021/ja982773h
Return to citation in text: [1] -
Hitchcock, P. B.; Lappert, M. F.; Protchenko, A. V. J. Am. Chem. Soc. 2001, 123, 189–190. doi:10.1021/ja005580e
Return to citation in text: [1] -
Shao, L.; Geib, S. J.; Badger, P. D.; Cooper, N. J. J. Am. Chem. Soc. 2002, 124, 14812–14813. doi:10.1021/ja027081e
Return to citation in text: [1] -
Tamm, M.; Bannenberg, T.; Fröhlich, R.; Grimme, S.; Gerenkamp, M. Dalton Trans. 2004, 482–491. doi:10.1039/b314347c
Return to citation in text: [1] -
Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Kratt, G.; Beckhaus, H.-D.; Lindner, H. J.; Rüchardt, C. Chem. Ber. 1983, 116, 3235–3263. doi:10.1002/cber.19831160921
Return to citation in text: [1] -
Beauchamp, A. L.; Montgrain, F.; Wuest, J. D. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1987, 43, 1557–1560. doi:10.1107/s0108270187091108
Return to citation in text: [1] -
Mas-Marzá, E.; Poyatos, M.; Sanaú, M.; Peris, E. Inorg. Chem. 2004, 43, 2213–2219. doi:10.1021/ic035317p
Return to citation in text: [1] -
Zhang, S.; Moudgil, K.; Jucov, E.; Risko, C.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Inorg. Chim. Acta 2019, 489, 67–77. doi:10.1016/j.ica.2019.02.003
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Wright, A. G.; Weissbach, T.; Holdcroft, S. Angew. Chem., Int. Ed. 2016, 55, 4818–4821. doi:10.1002/anie.201511184
Return to citation in text: [1] -
Mehrotra, S.; Raje, S.; Jain, A. K.; Angamuthu, R. ACS Sustainable Chem. Eng. 2017, 5, 6322–6328. doi:10.1021/acssuschemeng.7b01495
Return to citation in text: [1] -
Li, X.; Hao, P.; Shen, J.; Fu, Y. Dalton Trans. 2018, 47, 6031–6035. doi:10.1039/c8dt00829a
Return to citation in text: [1] -
Moudgil, K.; Mann, M. A.; Risko, C.; Bottomley, L. A.; Marder, S. R.; Barlow, S. Organometallics 2015, 34, 3706–3712. doi:10.1021/acs.organomet.5b00327
Return to citation in text: [1] -
Uebe, M.; Yoshihashi, Y.; Noda, K.; Matsubara, M.; Ito, A. J. Mater. Chem. C 2018, 6, 6429–6439. doi:10.1039/c8tc01280f
Return to citation in text: [1] [2] -
Kwon, O.; Barlow, S.; Odom, S. A.; Beverina, L.; Thompson, N. J.; Zojer, E.; Brédas, J.-L.; Marder, S. R. J. Phys. Chem. A 2005, 109, 9346–9352. doi:10.1021/jp054334s
Return to citation in text: [1] [2] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] [2] [3] -
Tanner, D. D.; Chen, J. J. J. Org. Chem. 1989, 54, 3842–3846. doi:10.1021/jo00277a020
Return to citation in text: [1] -
Riera-Galindo, S.; Orbelli Biroli, A.; Forni, A.; Puttisong, Y.; Tessore, F.; Pizzotti, M.; Pavlopoulou, E.; Solano, E.; Wang, S.; Wang, G.; Ruoko, T.-P.; Chen, W. M.; Kemerink, M.; Berggren, M.; di Carlo, G.; Fabiano, S. ACS Appl. Mater. Interfaces 2019, 11, 37981–37990. doi:10.1021/acsami.9b12441
Return to citation in text: [1] -
Zeng, Y.; Zheng, W.; Guo, Y.; Han, G.; Yi, Y. J. Mater. Chem. A 2020, 8, 8323–8328. doi:10.1039/d0ta01087a
Return to citation in text: [1] -
Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Bennett, R. W.; Wharry, D. L.; Koch, T. H. J. Am. Chem. Soc. 1980, 102, 2345–2349. doi:10.1021/ja00527a036
Return to citation in text: [1] -
Colter, A. K.; Lai, C. C.; Parsons, A. G.; Ramsey, N. B.; Saito, G. Can. J. Chem. 1985, 63, 445–451. doi:10.1139/v85-073
Return to citation in text: [1] -
Al Kurdi, K. Investigating Charge Transport in Conjugated Organic Materials. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2021.
Return to citation in text: [1]
| 27. | Pallini, F.; Mattiello, S.; Cassinelli, M.; Rossi, P.; Mecca, S.; Tan, W. L.; Sassi, M.; Lanzani, G.; McNeill, C. R.; Caironi, M.; Beverina, L. ACS Appl. Energy Mater. 2022, 5, 2421–2429. doi:10.1021/acsaem.1c03893 |
| 34. | Bardagot, O.; Aumaître, C.; Monmagnon, A.; Pécaut, J.; Bayle, P.-A.; Demadrille, R. Appl. Phys. Lett. 2021, 118, 203904. doi:10.1063/5.0047637 |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 26. | Pham, P. H.; Barlow, S.; Marder, S. R.; Luca, O. R. Chem Catal. 2023, 3, 100675. doi:10.1016/j.checat.2023.100675 |
| 8. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 50. | Zhang, S.; Moudgil, K.; Jucov, E.; Risko, C.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Inorg. Chim. Acta 2019, 489, 67–77. doi:10.1016/j.ica.2019.02.003 |
| 61. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 61. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 35. | Miler-Srenger, E. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1973, 29, 1119–1124. doi:10.1107/s0567740873003973 |
| 37. | Preuss, J.; Zanker, V.; Gieren, A. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1977, 33, 2317–2319. doi:10.1107/s0567740877008346 |
| 38. | Haltiwanger, R. C.; Koch, T. H.; Olesen, J. A.; Kim, C. S.; Kim, N. K. J. Am. Chem. Soc. 1977, 99, 6327–6331. doi:10.1021/ja00461a026 |
| 35. | Miler-Srenger, E. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1973, 29, 1119–1124. doi:10.1107/s0567740873003973 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 46. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 61. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 35. | Miler-Srenger, E. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1973, 29, 1119–1124. doi:10.1107/s0567740873003973 |
| 36. | Ramirez-Montes, P. I.; Ochoa, M. E.; Rodríguez, V.; Santillan, R.; García-Ortega, H.; Rodríguez, P.; Farfán, N. Tetrahedron Lett. 2012, 53, 5887–5890. doi:10.1016/j.tetlet.2012.08.094 |
| 37. | Preuss, J.; Zanker, V.; Gieren, A. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1977, 33, 2317–2319. doi:10.1107/s0567740877008346 |
| 38. | Haltiwanger, R. C.; Koch, T. H.; Olesen, J. A.; Kim, C. S.; Kim, N. K. J. Am. Chem. Soc. 1977, 99, 6327–6331. doi:10.1021/ja00461a026 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 46. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 61. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 62. | Bennett, R. W.; Wharry, D. L.; Koch, T. H. J. Am. Chem. Soc. 1980, 102, 2345–2349. doi:10.1021/ja00527a036 |
| 34. | Bardagot, O.; Aumaître, C.; Monmagnon, A.; Pécaut, J.; Bayle, P.-A.; Demadrille, R. Appl. Phys. Lett. 2021, 118, 203904. doi:10.1063/5.0047637 |
| 56. | Kwon, O.; Barlow, S.; Odom, S. A.; Beverina, L.; Thompson, N. J.; Zojer, E.; Brédas, J.-L.; Marder, S. R. J. Phys. Chem. A 2005, 109, 9346–9352. doi:10.1021/jp054334s |
| 57. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 57. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 22. | Mohapatra, S. K.; Fonari, A.; Risko, C.; Yesudas, K.; Moudgil, K.; Delcamp, J. H.; Timofeeva, T. V.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2014, 20, 15385–15394. doi:10.1002/chem.201404007 |
| 39. | Andrianov, V. G.; Struchkov, Y. T.; Petrakova, V. A.; Vol'kenau, N. A. Koord. Khim. 1986, 12, 978–980. |
| 40. | Gaudet, M. V.; Hanson, A. W.; White, P. S.; Zaworotko, M. J. Organometallics 1989, 8, 286–293. doi:10.1021/om00104a004 |
| 41. | Lee, S.; Lovelace, S. R.; Arford, D. J.; Geib, S. J.; Weber, S. G.; Cooper, N. J. J. Am. Chem. Soc. 1996, 118, 4190–4191. doi:10.1021/ja952217c |
| 42. | Hsu, S. C. N.; Yeh, W.-Y.; Lee, G.-H.; Peng, S.-M. J. Am. Chem. Soc. 1998, 120, 13250–13251. doi:10.1021/ja982773h |
| 43. | Hitchcock, P. B.; Lappert, M. F.; Protchenko, A. V. J. Am. Chem. Soc. 2001, 123, 189–190. doi:10.1021/ja005580e |
| 44. | Shao, L.; Geib, S. J.; Badger, P. D.; Cooper, N. J. J. Am. Chem. Soc. 2002, 124, 14812–14813. doi:10.1021/ja027081e |
| 45. | Tamm, M.; Bannenberg, T.; Fröhlich, R.; Grimme, S.; Gerenkamp, M. Dalton Trans. 2004, 482–491. doi:10.1039/b314347c |
| 46. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 64. | Al Kurdi, K. Investigating Charge Transport in Conjugated Organic Materials. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2021. |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 46. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 61. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 61. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 63. | Colter, A. K.; Lai, C. C.; Parsons, A. G.; Ramsey, N. B.; Saito, G. Can. J. Chem. 1985, 63, 445–451. doi:10.1139/v85-073 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 46. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 61. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 8. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 36. | Ramirez-Montes, P. I.; Ochoa, M. E.; Rodríguez, V.; Santillan, R.; García-Ortega, H.; Rodríguez, P.; Farfán, N. Tetrahedron Lett. 2012, 53, 5887–5890. doi:10.1016/j.tetlet.2012.08.094 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 47. | Kratt, G.; Beckhaus, H.-D.; Lindner, H. J.; Rüchardt, C. Chem. Ber. 1983, 116, 3235–3263. doi:10.1002/cber.19831160921 |
| 37. | Preuss, J.; Zanker, V.; Gieren, A. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1977, 33, 2317–2319. doi:10.1107/s0567740877008346 |
| 35. | Miler-Srenger, E. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1973, 29, 1119–1124. doi:10.1107/s0567740873003973 |
| 38. | Haltiwanger, R. C.; Koch, T. H.; Olesen, J. A.; Kim, C. S.; Kim, N. K. J. Am. Chem. Soc. 1977, 99, 6327–6331. doi:10.1021/ja00461a026 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 22. | Mohapatra, S. K.; Fonari, A.; Risko, C.; Yesudas, K.; Moudgil, K.; Delcamp, J. H.; Timofeeva, T. V.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2014, 20, 15385–15394. doi:10.1002/chem.201404007 |
| 46. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 34. | Bardagot, O.; Aumaître, C.; Monmagnon, A.; Pécaut, J.; Bayle, P.-A.; Demadrille, R. Appl. Phys. Lett. 2021, 118, 203904. doi:10.1063/5.0047637 |
| 48. | Beauchamp, A. L.; Montgrain, F.; Wuest, J. D. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1987, 43, 1557–1560. doi:10.1107/s0108270187091108 |
| 49. | Mas-Marzá, E.; Poyatos, M.; Sanaú, M.; Peris, E. Inorg. Chem. 2004, 43, 2213–2219. doi:10.1021/ic035317p |
| 50. | Zhang, S.; Moudgil, K.; Jucov, E.; Risko, C.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Inorg. Chim. Acta 2019, 489, 67–77. doi:10.1016/j.ica.2019.02.003 |
| 22. | Mohapatra, S. K.; Fonari, A.; Risko, C.; Yesudas, K.; Moudgil, K.; Delcamp, J. H.; Timofeeva, T. V.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2014, 20, 15385–15394. doi:10.1002/chem.201404007 |
| 46. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 1. | Walzer, K.; Maennig, B.; Pfeiffer, M.; Leo, K. Chem. Rev. 2007, 107, 1233–1271. doi:10.1021/cr050156n |
| 2. | Russ, B.; Glaudell, A.; Urban, J. J.; Chabinyc, M. L.; Segalman, R. A. Nat. Rev. Mater. 2016, 1, 16050. doi:10.1038/natrevmats.2016.50 |
| 3. | Lüssem, B.; Keum, C.-M.; Kasemann, D.; Naab, B.; Bao, Z.; Leo, K. Chem. Rev. 2016, 116, 13714–13751. doi:10.1021/acs.chemrev.6b00329 |
| 4. | Wang, Z.-K.; Liao, L.-S. Adv. Opt. Mater. 2018, 6, 1800276. doi:10.1002/adom.201800276 |
| 5. | Barlow, S.; Marder, S. R.; Lin, X.; Zhang, F.; Kahn, A. Electrial Doping of Organic Semiconductors with Molecular Oxidants and Reductants. In Conjugated Polymers, 4th ed.; Skotheim, T. A.; Reynolds, J.; Thompson, B. C., Eds.; CRC Press: Boca Raton, FL, USA, 2019. doi:10.1201/9780429190520-2 |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 12. | Ludvík, J.; Pragst, F.; Volke, J. J. Electroanal. Chem. Interfacial Electrochem. 1984, 180, 141–156. doi:10.1016/0368-1874(84)83576-5 |
| 13. | Naab, B. D.; Zhang, S.; Vandewal, K.; Salleo, A.; Barlow, S.; Marder, S. R.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2014, 26, 4268–4272. doi:10.1002/adma.201400668 |
| 16. | Yuan, D.; Huang, D.; Zhang, C.; Zou, Y.; Di, C.-a.; Zhu, X.; Zhu, D. ACS Appl. Mater. Interfaces 2017, 9, 28795–28801. doi:10.1021/acsami.7b07282 |
| 19. | Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817 |
| 26. | Pham, P. H.; Barlow, S.; Marder, S. R.; Luca, O. R. Chem Catal. 2023, 3, 100675. doi:10.1016/j.checat.2023.100675 |
| 54. | Moudgil, K.; Mann, M. A.; Risko, C.; Bottomley, L. A.; Marder, S. R.; Barlow, S. Organometallics 2015, 34, 3706–3712. doi:10.1021/acs.organomet.5b00327 |
| 8. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 24. | Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m |
| 27. | Pallini, F.; Mattiello, S.; Cassinelli, M.; Rossi, P.; Mecca, S.; Tan, W. L.; Sassi, M.; Lanzani, G.; McNeill, C. R.; Caironi, M.; Beverina, L. ACS Appl. Energy Mater. 2022, 5, 2421–2429. doi:10.1021/acsaem.1c03893 |
| 7. | Wei, P.; Oh, J. H.; Dong, G.; Bao, Z. J. Am. Chem. Soc. 2010, 132, 8852–8853. doi:10.1021/ja103173m |
| 16. | Yuan, D.; Huang, D.; Zhang, C.; Zou, Y.; Di, C.-a.; Zhu, X.; Zhu, D. ACS Appl. Mater. Interfaces 2017, 9, 28795–28801. doi:10.1021/acsami.7b07282 |
| 24. | Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m |
| 8. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 25. | Küçükbay, H.; Çetinkaya, E.; Çetinkaya, B.; Lappert, M. F. Synth. Commun. 1997, 27, 4059–4066. doi:10.1080/00397919708005451 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 21. | Guo, S.; Kim, S. B.; Mohapatra, S. K.; Qi, Y.; Sajoto, T.; Kahn, A.; Marder, S. R.; Barlow, S. Adv. Mater. (Weinheim, Ger.) 2012, 24, 699–703. doi:10.1002/adma.201103238 |
| 22. | Mohapatra, S. K.; Fonari, A.; Risko, C.; Yesudas, K.; Moudgil, K.; Delcamp, J. H.; Timofeeva, T. V.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2014, 20, 15385–15394. doi:10.1002/chem.201404007 |
| 23. | Mohapatra, S. K.; Marder, S. R.; Barlow, S. Acc. Chem. Res. 2022, 55, 319–332. doi:10.1021/acs.accounts.1c00612 |
| 19. | Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817 |
| 50. | Zhang, S.; Moudgil, K.; Jucov, E.; Risko, C.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Inorg. Chim. Acta 2019, 489, 67–77. doi:10.1016/j.ica.2019.02.003 |
| 13. | Naab, B. D.; Zhang, S.; Vandewal, K.; Salleo, A.; Barlow, S.; Marder, S. R.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2014, 26, 4268–4272. doi:10.1002/adma.201400668 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 15. | Naab, B. D.; Gu, X.; Kurosawa, T.; To, J. W. F.; Salleo, A.; Bao, Z. Adv. Electron. Mater. 2016, 2, 1600004. doi:10.1002/aelm.201600004 |
| 16. | Yuan, D.; Huang, D.; Zhang, C.; Zou, Y.; Di, C.-a.; Zhu, X.; Zhu, D. ACS Appl. Mater. Interfaces 2017, 9, 28795–28801. doi:10.1021/acsami.7b07282 |
| 17. | Schwarze, M.; Gaul, C.; Scholz, R.; Bussolotti, F.; Hofacker, A.; Schellhammer, K. S.; Nell, B.; Naab, B. D.; Bao, Z.; Spoltore, D.; Vandewal, K.; Widmer, J.; Kera, S.; Ueno, N.; Ortmann, F.; Leo, K. Nat. Mater. 2019, 18, 242–248. doi:10.1038/s41563-018-0277-0 |
| 18. | Al Kurdi, K.; Gregory, S. A.; Jhulki, S.; Conte, M.; Barlow, S.; Yee, S. K.; Marder, S. R. Mater. Adv. 2020, 1, 1829–1834. doi:10.1039/d0ma00406e |
| 19. | Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817 |
| 20. | Lungwitz, D.; Joy, S.; Mansour, A. E.; Opitz, A.; Karunasena, C.; Li, H.; Panjwani, N. A.; Moudgil, K.; Tang, K.; Behrends, J.; Barlow, S.; Marder, S. R.; Brédas, J.-L.; Graham, K.; Koch, N.; Kahn, A. J. Phys. Chem. Lett. 2023, 14, 5633–5640. doi:10.1021/acs.jpclett.3c01022 |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 24. | Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m |
| 9. | Naab, B. D.; Guo, S.; Olthof, S.; Evans, E. G. B.; Wei, P.; Millhauser, G. L.; Kahn, A.; Barlow, S.; Marder, S. R.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 15018–15025. doi:10.1021/ja403906d |
| 13. | Naab, B. D.; Zhang, S.; Vandewal, K.; Salleo, A.; Barlow, S.; Marder, S. R.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2014, 26, 4268–4272. doi:10.1002/adma.201400668 |
| 19. | Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817 |
| 24. | Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m |
| 12. | Ludvík, J.; Pragst, F.; Volke, J. J. Electroanal. Chem. Interfacial Electrochem. 1984, 180, 141–156. doi:10.1016/0368-1874(84)83576-5 |
| 19. | Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817 |
| 34. | Bardagot, O.; Aumaître, C.; Monmagnon, A.; Pécaut, J.; Bayle, P.-A.; Demadrille, R. Appl. Phys. Lett. 2021, 118, 203904. doi:10.1063/5.0047637 |
| 8. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 10. | Guo, H.; Yang, C.-Y.; Zhang, X.; Motta, A.; Feng, K.; Xia, Y.; Shi, Y.; Wu, Z.; Yang, K.; Chen, J.; Liao, Q.; Tang, Y.; Sun, H.; Woo, H. Y.; Fabiano, S.; Facchetti, A.; Guo, X. Nature 2021, 599, 67–73. doi:10.1038/s41586-021-03942-0 |
| 11. | Pallini, F.; Mattiello, S.; Manfredi, N.; Mecca, S.; Fedorov, A.; Sassi, M.; Al Kurdi, K.; Ding, Y.-F.; Pan, C.-K.; Pei, J.; Barlow, S.; Marder, S. R.; Nguyen, T.-Q.; Beverina, L. J. Mater. Chem. A 2023, 11, 8192–8201. doi:10.1039/d3ta00231d |
| 8. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 51. | Wright, A. G.; Weissbach, T.; Holdcroft, S. Angew. Chem., Int. Ed. 2016, 55, 4818–4821. doi:10.1002/anie.201511184 |
| 52. | Mehrotra, S.; Raje, S.; Jain, A. K.; Angamuthu, R. ACS Sustainable Chem. Eng. 2017, 5, 6322–6328. doi:10.1021/acssuschemeng.7b01495 |
| 53. | Li, X.; Hao, P.; Shen, J.; Fu, Y. Dalton Trans. 2018, 47, 6031–6035. doi:10.1039/c8dt00829a |
| 24. | Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m |
| 24. | Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m |
| 13. | Naab, B. D.; Zhang, S.; Vandewal, K.; Salleo, A.; Barlow, S.; Marder, S. R.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2014, 26, 4268–4272. doi:10.1002/adma.201400668 |
| 19. | Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817 |
| 8. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 50. | Zhang, S.; Moudgil, K.; Jucov, E.; Risko, C.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Inorg. Chim. Acta 2019, 489, 67–77. doi:10.1016/j.ica.2019.02.003 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 55. | Uebe, M.; Yoshihashi, Y.; Noda, K.; Matsubara, M.; Ito, A. J. Mater. Chem. C 2018, 6, 6429–6439. doi:10.1039/c8tc01280f |
| 33. | Reddy, A. P. R.; Veeranagaiah, V.; Ratnam, C. V. Indian J. Chem. 1985, B24, 367–371. |
| 27. | Pallini, F.; Mattiello, S.; Cassinelli, M.; Rossi, P.; Mecca, S.; Tan, W. L.; Sassi, M.; Lanzani, G.; McNeill, C. R.; Caironi, M.; Beverina, L. ACS Appl. Energy Mater. 2022, 5, 2421–2429. doi:10.1021/acsaem.1c03893 |
| 34. | Bardagot, O.; Aumaître, C.; Monmagnon, A.; Pécaut, J.; Bayle, P.-A.; Demadrille, R. Appl. Phys. Lett. 2021, 118, 203904. doi:10.1063/5.0047637 |
| 30. | Crippa, G. B.; Maffei, S. Gazz. Chim. Ital. 1941, 71, 194–200. |
| 31. | Balachandran, K. S.; George, M. V. Indian J. Chem. 1973, 11, 1267–1271. |
| 32. | Speier, G.; Párkányi, L. J. Org. Chem. 1986, 51, 218–221. doi:10.1021/jo00352a016 |
| 58. | Tanner, D. D.; Chen, J. J. J. Org. Chem. 1989, 54, 3842–3846. doi:10.1021/jo00277a020 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 50. | Zhang, S.; Moudgil, K.; Jucov, E.; Risko, C.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Inorg. Chim. Acta 2019, 489, 67–77. doi:10.1016/j.ica.2019.02.003 |
| 55. | Uebe, M.; Yoshihashi, Y.; Noda, K.; Matsubara, M.; Ito, A. J. Mater. Chem. C 2018, 6, 6429–6439. doi:10.1039/c8tc01280f |
| 59. | Riera-Galindo, S.; Orbelli Biroli, A.; Forni, A.; Puttisong, Y.; Tessore, F.; Pizzotti, M.; Pavlopoulou, E.; Solano, E.; Wang, S.; Wang, G.; Ruoko, T.-P.; Chen, W. M.; Kemerink, M.; Berggren, M.; di Carlo, G.; Fabiano, S. ACS Appl. Mater. Interfaces 2019, 11, 37981–37990. doi:10.1021/acsami.9b12441 |
| 60. | Zeng, Y.; Zheng, W.; Guo, Y.; Han, G.; Yi, Y. J. Mater. Chem. A 2020, 8, 8323–8328. doi:10.1039/d0ta01087a |
| 24. | Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. J. Am. Chem. Soc. 2008, 130, 2501–2516. doi:10.1021/ja075523m |
| 56. | Kwon, O.; Barlow, S.; Odom, S. A.; Beverina, L.; Thompson, N. J.; Zojer, E.; Brédas, J.-L.; Marder, S. R. J. Phys. Chem. A 2005, 109, 9346–9352. doi:10.1021/jp054334s |
| 29. | Ghosh, R.; Kushwaha, A.; Das, D. J. Phys. Chem. B 2017, 121, 8786–8794. doi:10.1021/acs.jpcb.7b05947 |
| 57. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 28. | Lim, C.-H.; Ilic, S.; Alherz, A.; Worrell, B. T.; Bacon, S. S.; Hynes, J. T.; Glusac, K. D.; Musgrave, C. B. J. Am. Chem. Soc. 2019, 141, 272–280. doi:10.1021/jacs.8b09653 |
| 14. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 29. | Ghosh, R.; Kushwaha, A.; Das, D. J. Phys. Chem. B 2017, 121, 8786–8794. doi:10.1021/acs.jpcb.7b05947 |
| 50. | Zhang, S.; Moudgil, K.; Jucov, E.; Risko, C.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Inorg. Chim. Acta 2019, 489, 67–77. doi:10.1016/j.ica.2019.02.003 |
© 2023 Mohapatra et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.