Abstract
A practical NaI/PPh3-catalyzed decarboxylative radical cascade cyclization of N-arylacrylamides with redox-active esters is described, which is mediated by visible light irradiation. A wide range of substrates bearing different substituents and derived from ubiquitous carboxylic acids, including α-amino acids, were synthesized and examined under this very mild, efficient, and cost effective transition-metal-free synthetic method. These afforded various functionalized oxindoles featuring a C3 quaternary stereogenic center. Mechanistic experiments suggest a radical mechanism.

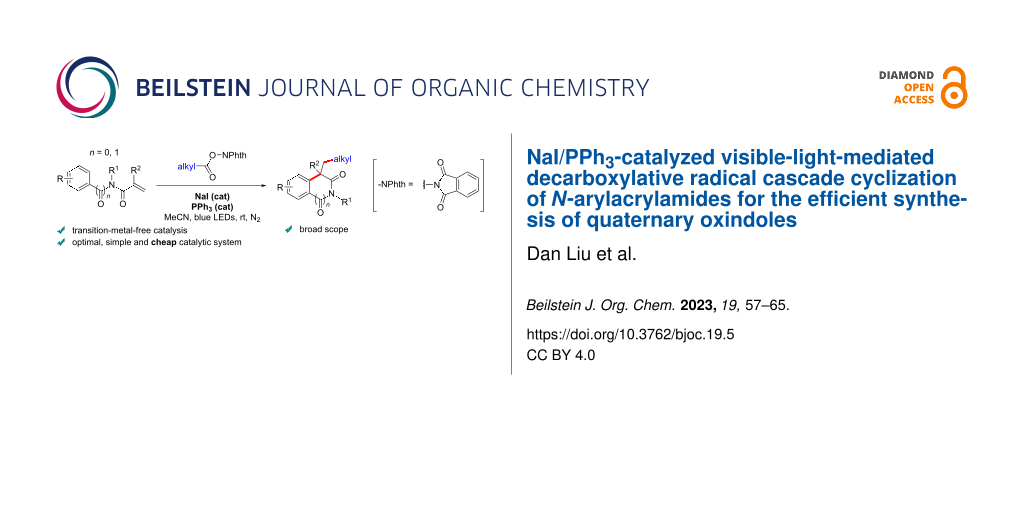
Graphical Abstract
Introduction
Radical-initiated cascade reactions constitute a powerful synthetic approach to construct multiple C–C or C–X bonds in one pot. As such, these tend to allow facile access to many complex natural molecules and drugs [1-6]. Recently, radical-initiated cascade cyclizations involving acrylamides have attracted considerable attention due to their propensity to build important oxindole scaffolds. These are broadly found in natural products, pharmaceuticals, and bioactive molecules (Figure 1) [7-13]. Although a number of synthetic approaches have already been explored [14-20], these existing methods generally require stoichiometric, often onerous reagents [21-28], and/or high temperatures [29-38].
![[1860-5397-19-5-1]](/bjoc/content/figures/1860-5397-19-5-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative natural products and biologically active molecules containing an oxindole moiety [7-13].
Figure 1: Representative natural products and biologically active molecules containing an oxindole moiety [7-13].
In the past few years, photocatalytic processes have become one of the most powerful tools in developing radical-initiated addition/cyclization cascades from acrylamides for the synthesis of oxindoles [39-41]. The radicals are typically generated from alkyl halides [42-44], carboxylic acids [45-47], simple alkanes [48], alkylboronic acids [49], isocyanides [50], or other [51-53]. In this context, the group of Fu reported a Ru(bpy)3Cl2-catalyzed synthesis of N-Boc proline oxindole derivatives under visible-light assistance [47]. Therein, N-hydroxyphthalimide (NPhth) esters were utilized as alkyl radical precursors, which can be readily prepared from highly available carboxylic acids. In 2015, Cheng and co-workers disclosed a visible light-mediated radical tandem cyclization of N-arylacrylamides with N-(acyloxy)phthalimides to access 3,3-dialkylated oxindoles in the presence of [Ru(bpy)3Cl2]·6H2O [46]. However, these seminal methods remain limited by the need of noble-metal-based photocatalysts, excess additives and limited substrate scopes (Scheme 1a).
![[1860-5397-19-5-i1]](/bjoc/content/inline/1860-5397-19-5-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Selected photocatalytic decarboxylative radical cascade reactions of N-arylamides.
Scheme 1: Selected photocatalytic decarboxylative radical cascade reactions of N-arylamides.
With the rapid development of sustainable chemistry, developing low-cost and transition-metal-free photocatalytic methods has become a strategic priority. In 2019 [54], the groups of Fu and Shang pioneered the photocatalytic decarboxylative alkylation of silyl enol ethers and N-heteroarenes by using a novel catalytic system based on sodium iodide (NaI) and triphenylphosphine (PPh3), suggested to function as an electron donor–acceptor (EDA) complex [55-60]. Compared to previously reported radical reactions, this novel catalytic system has the key advantage of circumventing the need for external redox additives and/or noble metals, using readily available and cost-effective NaI and PPh3 under mild reaction conditions. In a broader context, phosphine organocatalysis is probably still underappreciated in organic synthesis, and could lead to important future synthetic developments [61-67]. The NaI/PPh3 system has been further broadly applied to the functionalization of alkenes [68-70], as well as to decarboxylative C(sp3)–X bond formation [71], cyclization of 1,7-enynes [72,73] and other reactions [74-77]. Inspired by these advances, we developed here a visible light-mediated decarboxylative radical cascade cyclization of N-arylacrylamides under NaI/PPh3 catalysis, for the most efficient and practical synthesis of quaternary oxindoles (Scheme 1b and 1c). It should be noted that during the finalization of this work, a similar, however stoichiometric CsI/PPh2Cy-mediated method appeared from the Yang and Li groups (Scheme 1b) [28]. In contrast, the method we present here is 1) catalytic, 2) it employs the far less onerous NaI/PPh3 system, and 3) it displays a considerably broader substrate scope.
Results and Discussion
Key elements of reaction optimization are summarized in Table 1. With NaI (20 mol %) and PPh3 (20 mol %), acrylamide 1a and redox-active ester 2a were used as model substrates to react for 36 h in acetonitrile (MeCN) under blue LEDs irradiation and N2 atmosphere, delivering the desired oxindole derivative 3aa with 72% isolated yield (Table 1, entry 1). Other iodide sources, such as LiI, KI, RbI, CsI, CaI2, and a quaternary ammonium iodide, while also effective, provided slightly lower yields (Table 1, entries 2–7). It should be noted that all tested iodide sources were found soluble under those conditions. Some diverse phosphines were then screened. Aromatic phosphines performed best (Table 1, entries 8 and 9), the cheapest PPh3 remaining however optimal. In contrast, tricyclohexylphosphine PCy3 performed poorly (Table 1, entry 10), and bulky tri-o-tolylphosphine almost shut down the reaction (Table 1, entry 11). These results indicate that the accessibility of the phosphorus center is important. Next, the solvent was investigated. Replacing acetonitrile with dimethyl sulfoxide (DMSO), or dimethylacetamide (DMA), or acetone, or ethyl acetate (EA), resulted in inferior yields (Table 1, entries 12–15), and no product was detected when using 1,4-dioxane or dichloromethane (DCM) as reaction solvent (Table 1, entries 16 and 17). Although the reaction also proceeded without NaI, only a low yield of 3aa was then obtained (Table 1, entry 18). PPh3 and irradiation are however both essential for this decarboxylative cascade cyclization process (Table 1, entries 19 and 20).
Table 1: Optimization tablea.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-5-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Variation from standard conditions | 3aa, Yield (%)b |
|---|---|---|
| 1 | none | 76 (72)c |
| 2 | LiI instead of NaI | 70 |
| 3 | KI instead of NaI | 62 |
| 4 | RbI instead of NaI | 64 |
| 5 | CsI instead of NaI | 39 |
| 6 | CaI2 instead of NaI | 56 |
| 7 | n-Bu4NI instead of NaI | 57 |
| 8 | P(4-F-C6H4)3 instead of PPh3 | 73 |
| 9 | P(4-OMe-C6H4)3 instead of PPh3 | 60 |
| 10 | PCy3 instead of PPh3 | 23 |
| 11 | P(2-Me-C6H4)3 instead of PPh3 | trace |
| 12 | DMSO instead of MeCN | 60 |
| 13 | DMA instead of MeCN | 44 |
| 14 | acetone instead of MeCN | 52 |
| 15 | EA instead of MeCN | 57 |
| 16 | DCM instead of MeCN | nr |
| 17 | 1,4-dioxane instead MeCN | nr |
| 18 | without NaI | 14 |
| 19 | without PPh3 | 0 |
| 20 | without blue LED | 0 |
aUnless otherwise noted, the standard reaction conditions were as follows: 1a (0.3 mmol), 2a (0.2 mmol), solvent (2 mL); bthe yield was determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard; cisolated yield.
With the optimized conditions in hand, we then explored the scope of N-arylacrylamides with different substituents. A series of acrylamides showed good compatibility under standard conditions, offering the desired oxindoles in moderate to good yields (Scheme 2). Electron-donating groups at the para-position of the phenyl ring, such as methyl or methoxy groups, decreased slightly the yield, to 68% and 66%, respectively (3ba and 3ca). When these substituents were replaced by common halogens or electron-withdrawing groups, good yields of the corresponding oxindoles (3da–ga) were achieved. A trifluoromethyl-substituted acrylamide afforded the product 3fa in very high 85% yield. In addition, ortho-substitution at the N-aryl moiety was also well tolerated, albeit with slightly decreased yields (3ha–ka, 50–63%).
![[1860-5397-19-5-i2]](/bjoc/content/inline/1860-5397-19-5-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Arylamide substrate scope with isolated yields of products.
Scheme 2: Arylamide substrate scope with isolated yields of products.
Interestingly, a cyclic N-arylamide derivative was also well tolerated, furnishing polycyclic structure 3la in 67% yield. In addition, substrates with different N-substituents, such as ethyl, benzyl, and phenyl, could be converted into the expected products 3ma–oa in good yields. It should be noted that replacing the methyl with a phenyl group at the N-arylacrylamide core significantly affected the reaction efficiency from 72% to 34% yield (3pa). Satisfyingly, substrate 1q could successfully undergo decarboxylative cascade cyclization to afford 3qa with 70% yield, which is used as a key intermediate in the synthesis of (±)-physovenine and (±)-physostigmine alkyl analogues exhibiting inhibitory activity against acetylcholinesterase and butyrylcholinesterase [30,78-84]. Subsequently, we expanded the scope of this protocol to include a benzamide derived acrylamide 1r. The expected six-membered ring structure 3ra could be successfully isolated with a good yield (66%).
A number of alkyl radical precursors were then synthesized and evaluated in the reaction (Scheme 3). We found that redox-active esters derived from primary, secondary, and tertiary aliphatic carboxylic acids were all compatible with the method. Cyclic substrates bearing cyclobutyl, cyclopentyl, and indenyl groups could deliver the corresponding desired products with good yields (3ab–ad, 63–74%), while an adamantyl-derived substituent proved more challenging (3ae, 40%). The use of other cyclic substituents such as oxygen-containing and nitrogen-containing rings gave good yields of the target oxindoles (3af–ah, 65–76%). In addition, a symmetrically α-substituted redox-active esters furnished the corresponding quaternary oxindole 3ai with 69% yield. Moreover, an asymmetrically α-branched starting material could react with similar efficiency, affording oxindole 3aj as a 1:1.1 mixture of diastereomers. Interestingly, this method also enabled the synthesis of the highly sterically demanding oxindole 3ak in good yield when using a tert-butyl N-hydroxyphthalimide ester as the tert-butyl radical precursor. Importantly, a redox-active ester derived from methionine could be converted effectively to α-aminoalkylation product 3al in overall 70% yield, which thus provides a mild method for the functionalization and derivation of abundant natural or unnatural amino acids. Some functional groups such as a terminal alkene in 3am, a terminal alkyne in 3an, and an alkyl chloride in 3ao proved compatible, associated with encouraging yields. In order to further demonstrate the utility of our protocol, a complex scaffold derived from lithocholic acid was tested, and was found to smoothly undergo the decarboxylative cyclization towards oxindole 3ap in 63% yield.
![[1860-5397-19-5-i3]](/bjoc/content/inline/1860-5397-19-5-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Alkyl radical precursor scope with isolated yields of products.
Scheme 3: Alkyl radical precursor scope with isolated yields of products.
In order to gain insight into the reaction mechanism, some control experiments were further performed. When a radical scavenger such as 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) was added to the catalytic system under standard conditions, the reaction was fully inhibited, and a TEMPO-trapped adduct (4) was detected by HRMS (Scheme 4a). Moreover, the radical-mediated ring-opening product 3am could be obtained with 66% yield in a radical clock experiment when redox-active ester 5 was engaged to react with acrylamide 1a under standard conditions (Scheme 4b). Finally, it should be noted that benzoyl ester substrate 6a did not deliver the corresponding cyclized product 7aa (Scheme 4c). All of these outcomes indicate that a radical species should be involved in this decarboxylative cascade cyclization towards oxindoles under NaI/PPh3 catalysis. Thus, the mechanism should run in a similar fashion to related well-documented previous reports [54,68-77], through a light-induced, phosphine-assisted, intermolecular electron transfer from sodium iodide to the redox-active ester.
![[1860-5397-19-5-i4]](/bjoc/content/inline/1860-5397-19-5-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Selected mechanistic experiments.
Scheme 4: Selected mechanistic experiments.
Conclusion
In summary, we developed an effective photocatalytic decarboxylative radical cascade cyclization of N-arylacrylamides with various redox-active esters derived from common and/or important carboxylic acids under mild conditions. Complementary to traditional transition metal photocatalysis and organo-photocatalysis [85], the readily available and inexpensive NaI/PPh3 can operate as an efficient photoredox catalyst, providing an economical access to construct important oxindole scaffolds containing a quaternary carbon center. This synthetic method features a broad substrate scope, good functional group tolerance and operational simplicity. Mechanistic investigations revealed that this cyclization reaction proceeds via a cascade radical pathway. We expect these results to encourage the further development of NaI/PPh3-catalyzed and related synthetic methods.
Supporting Information
| Supporting Information File 1: Experimental section and characterization of synthesized compounds. | ||
| Format: PDF | Size: 8.3 MB | Download |
References
-
Molander, G. A.; Harris, C. R. Chem. Rev. 1996, 96, 307–338. doi:10.1021/cr950019y
Return to citation in text: [1] -
Snider, B. B. Chem. Rev. 1996, 96, 339–364. doi:10.1021/cr950026m
Return to citation in text: [1] -
Nair, V.; Mathew, J.; Prabhakaran, J. Chem. Soc. Rev. 1997, 26, 127–132. doi:10.1039/cs9972600127
Return to citation in text: [1] -
McCarroll, A. J.; Walton, J. C. Angew. Chem., Int. Ed. 2001, 40, 2224–2248. doi:10.1002/1521-3773(20010618)40:12<2224::aid-anie2224>3.0.co;2-f
Return to citation in text: [1] -
Albert, M.; Fensterbank, L.; Lacote, E.; Malacria, M. Tandem Radical Reactions. In Radicals in Synthesis II; Gansäuer, A., Ed.; Springer: Berlin, 2006; pp 1–62. doi:10.1007/128_026
Return to citation in text: [1] -
Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d
Return to citation in text: [1] -
Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Wang, S. J. Med. Chem. 2006, 49, 3432–3435. doi:10.1021/jm051122a
Return to citation in text: [1] [2] -
Christensen, M. K.; Erichsen, K. D.; Trojel-Hansen, C.; Tjørnelund, J.; Nielsen, S. J.; Frydenvang, K.; Johansen, T. N.; Nielsen, B.; Sehested, M.; Jensen, P. B.; Ikaunieks, M.; Zaichenko, A.; Loza, E.; Kalvinsh, I.; Björkling, F. J. Med. Chem. 2010, 53, 7140–7145. doi:10.1021/jm100763j
Return to citation in text: [1] [2] -
Millemaggi, A.; Taylor, R. J. K. Eur. J. Org. Chem. 2010, 4527–4547. doi:10.1002/ejoc.201000643
Return to citation in text: [1] [2] -
Rudrangi, S. R. S.; Bontha, V. K.; Manda, V. R.; Bethi, S. Asian J. Res. Chem. 2011, 4, 335.
Return to citation in text: [1] [2] -
Yu, B.; Yu, D. Q.; Liu, H. M. Eur. J. Med. Chem. 2015, 97, 763. doi:10.1016/j.ejmech.2014.06.056
Return to citation in text: [1] [2] -
Kaur, M.; Singh, M.; Chadha, N.; Silakari, O. Eur. J. Med. Chem. 2016, 123, 858–894. doi:10.1016/j.ejmech.2016.08.011
Return to citation in text: [1] [2] -
Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041
Return to citation in text: [1] [2] -
Klein, J. E. M. N.; Taylor, R. J. K. Eur. J. Org. Chem. 2011, 6821–6841. doi:10.1002/ejoc.201100836
Return to citation in text: [1] -
Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247. doi:10.1039/c2cs35100e
Return to citation in text: [1] -
Mai, W.; Wang, J.; Yang, L.; Yuan, J.; Mao, P.; Xiao, Y.; Qu, L. Chin. J. Org. Chem. 2014, 34, 1958. doi:10.6023/cjoc201405006
Return to citation in text: [1] -
Li, C.-C.; Yang, S.-D. Org. Biomol. Chem. 2016, 14, 4365–4377. doi:10.1039/c6ob00554c
Return to citation in text: [1] -
Cao, Z.-Y.; Zhou, F.; Zhou, J. Acc. Chem. Res. 2018, 51, 1443–1454. doi:10.1021/acs.accounts.8b00097
Return to citation in text: [1] -
Marchese, A. D.; Larin, E. M.; Mirabi, B.; Lautens, M. Acc. Chem. Res. 2020, 53, 1605–1619. doi:10.1021/acs.accounts.0c00297
Return to citation in text: [1] -
Boddy, A. J.; Bull, J. A. Org. Chem. Front. 2021, 8, 1026–1084. doi:10.1039/d0qo01085e
Return to citation in text: [1] -
Xu, Z.; Yan, C.; Liu, Z.-Q. Org. Lett. 2014, 16, 5670–5673. doi:10.1021/ol502738a
Return to citation in text: [1] -
Dai, Q.; Yu, J.; Jiang, Y.; Guo, S.; Yang, H.; Cheng, J. Chem. Commun. 2014, 50, 3865. doi:10.1039/c4cc01053a
Return to citation in text: [1] -
Zhou, D.; Li, Z.-H.; Li, J.; Li, S.-H.; Wang, M.-W.; Luo, X.-L.; Ding, G.-L.; Sheng, R.-L.; Fu, M.-J.; Tang, S. Eur. J. Org. Chem. 2015, 1606–1612. doi:10.1002/ejoc.201403499
Return to citation in text: [1] -
He, Z.-Y.; Guo, J.-Y.; Tian, S.-K. Adv. Synth. Catal. 2018, 360, 1544–1548. doi:10.1002/adsc.201800012
Return to citation in text: [1] -
Shi, Y.; Xiao, H.; Xu, X.-H.; Huang, Y. Org. Biomol. Chem. 2018, 16, 8472–8476. doi:10.1039/c8ob02457j
Return to citation in text: [1] -
Wang, X.-Y.; Zhong, Y.-F.; Mo, Z.-Y.; Wu, S.-H.; Xu, Y.-L.; Tang, H.-T.; Pan, Y.-M. Adv. Synth. Catal. 2021, 363, 208–214. doi:10.1002/adsc.202001192
Return to citation in text: [1] -
Wu, H.; Zhou, M.; Li, W.; Zhang, P. Catal. Commun. 2020, 133, 105832. doi:10.1016/j.catcom.2019.105832
Return to citation in text: [1] -
Fan, X.; Liu, H.; Ma, S.; Wang, F.; Yang, J.; Li, D. Tetrahedron 2022, 117-118, 132849. doi:10.1016/j.tet.2022.132849
Return to citation in text: [1] [2] -
Fan, J.-H.; Wei, W.-T.; Zhou, M.-B.; Song, R.-J.; Li, J.-H. Angew. Chem., Int. Ed. 2014, 53, 6650–6654. doi:10.1002/anie.201402893
Return to citation in text: [1] -
Biswas, P.; Paul, S.; Guin, J. Angew. Chem., Int. Ed. 2016, 55, 7756–7760. doi:10.1002/anie.201603809
Return to citation in text: [1] [2] -
Tang, S.; Zhou, D.; Li, Z.-H.; Fu, M.-J.; Jie, L.; Sheng, R.-L.; Li, S.-H. Synthesis 2015, 47, 1567–1580. doi:10.1055/s-0034-1379902
Return to citation in text: [1] -
Wang, H.; Guo, L.; Duan, X.-H. J. Org. Chem. 2016, 81, 860–867. doi:10.1021/acs.joc.5b02433
Return to citation in text: [1] -
Yang, Z.; Cheng, Y.; Long, J.; Feng, X.; Tang, R.; Wei, J. New J. Chem. 2019, 43, 18760–18766. doi:10.1039/c9nj04458b
Return to citation in text: [1] -
Che, F.; Zhong, J.; Yu, L.; Ma, C.; Yu, C.; Wang, M.; Hou, Z.; Zhang, Y. Adv. Synth. Catal. 2020, 362, 5020–5025. doi:10.1002/adsc.202000600
Return to citation in text: [1] -
Zhang, L.; Zhou, H.; Bai, S.; Li, S. Dalton Trans. 2021, 50, 3201–3206. doi:10.1039/d0dt04295a
Return to citation in text: [1] -
Zhang, L.; Wang, Y.; Yang, Y.; Zhang, P.; Wang, C. Org. Chem. Front. 2020, 7, 3234–3241. doi:10.1039/d0qo00953a
Return to citation in text: [1] -
Wang, C.; Liu, L. Org. Chem. Front. 2021, 8, 1454–1460. doi:10.1039/d0qo01508c
Return to citation in text: [1] -
Su, L.; Sun, H.; Liu, J.; Wang, C. Org. Lett. 2021, 23, 4662–4666. doi:10.1021/acs.orglett.1c01400
Return to citation in text: [1] -
Festa, A. A.; Voskressensky, L. G.; Van der Eycken, E. V. Chem. Soc. Rev. 2019, 48, 4401–4423. doi:10.1039/c8cs00790j
Return to citation in text: [1] -
Singh, J.; Sharma, A. Adv. Synth. Catal. 2021, 363, 4284–4308. doi:10.1002/adsc.202100515
Return to citation in text: [1] -
Ghosh, S.; Qu, Z.-W.; Pradhan, S.; Ghosh, A.; Grimme, S.; Chatterjee, I. Angew. Chem., Int. Ed. 2022, 61, 10.1002/anie.202115272. doi:10.1002/anie.202115272
Return to citation in text: [1] -
An, Y.; Li, Y.; Wu, J. Org. Chem. Front. 2016, 3, 570. doi:10.1039/c6qo00055j
Return to citation in text: [1] -
Muralirajan, K.; Kancherla, R.; Gimnkhan, A.; Rueping, M. Org. Lett. 2021, 23, 6905–6910. doi:10.1021/acs.orglett.1c02467
Return to citation in text: [1] -
Du, J.; Wang, X.; Wang, H.; Wei, J.; Huang, X.; Song, J.; Zhang, J. Org. Lett. 2021, 23, 5631–5635. doi:10.1021/acs.orglett.1c01698
Return to citation in text: [1] -
Xie, J.; Xu, P.; Li, H.; Xue, Q.; Jin, H.; Cheng, Y.; Zhu, C. Chem. Commun. 2013, 49, 5672. doi:10.1039/c3cc42672f
Return to citation in text: [1] -
Tang, Q.; Liu, X.; Liu, S.; Xie, H.; Liu, W.; Zeng, J.; Cheng, P. RSC Adv. 2015, 5, 89009–89014. doi:10.1039/c5ra17292f
Return to citation in text: [1] [2] -
Jin, Y.; Jiang, M.; Wang, H.; Fu, H. Sci. Rep. 2016, 6, 20068. doi:10.1038/srep20068
Return to citation in text: [1] [2] -
Li, Z.; Zhang, Y.; Zhang, L.; Liu, Z.-Q. Org. Lett. 2014, 16, 382–385. doi:10.1021/ol4032478
Return to citation in text: [1] -
Li, X.; Han, M.-Y.; Wang, B.; Wang, L.; Wang, M. Org. Biomol. Chem. 2019, 17, 6612–6619. doi:10.1039/c9ob01023h
Return to citation in text: [1] -
Zhao, Y.; Li, Z.; Sharma, U. K.; Sharma, N.; Song, G.; Van der Eycken, E. V. Chem. Commun. 2016, 52, 6395–6398. doi:10.1039/c6cc02024k
Return to citation in text: [1] -
Xu, P.; Xie, J.; Xue, Q.; Pan, C.; Cheng, Y.; Zhu, C. Chem. – Eur. J. 2013, 19, 14039–14042. doi:10.1002/chem.201302407
Return to citation in text: [1] -
Chen, J.-Q.; Wei, Y.-L.; Xu, G.-Q.; Liang, Y.-M.; Xu, P.-F. Chem. Commun. 2016, 52, 6455–6458. doi:10.1039/c6cc02007k
Return to citation in text: [1] -
Wang, Y.-Z.; Lin, W.-J.; Zou, J.-Y.; Yu, W.; Liu, X.-Y. Adv. Synth. Catal. 2020, 362, 3116–3120. doi:10.1002/adsc.202000609
Return to citation in text: [1] -
Fu, M.-C.; Shang, R.; Zhao, B.; Wang, B.; Fu, Y. Science 2019, 363, 1429–1434. doi:10.1126/science.aav3200
Return to citation in text: [1] [2] -
Rosokha, S. V.; Kochi, J. K. Acc. Chem. Res. 2008, 41, 641–653. doi:10.1021/ar700256a
Return to citation in text: [1] -
Lima, C. G. S.; de M. Lima, T.; Duarte, M.; Jurberg, I. D.; Paixão, M. W. ACS Catal. 2016, 6, 1389–1407. doi:10.1021/acscatal.5b02386
Return to citation in text: [1] -
Yuan, Y.-q.; Majumder, S.; Yang, M.-h.; Guo, S.-r. Tetrahedron Lett. 2020, 61, 151506. doi:10.1016/j.tetlet.2019.151506
Return to citation in text: [1] -
Crisenza, G. E. M.; Mazzarella, D.; Melchiorre, P. J. Am. Chem. Soc. 2020, 142, 5461–5476. doi:10.1021/jacs.0c01416
Return to citation in text: [1] -
Yang, Z.; Liu, Y.; Cao, K.; Zhang, X.; Jiang, H.; Li, J. Beilstein J. Org. Chem. 2021, 17, 771–799. doi:10.3762/bjoc.17.67
Return to citation in text: [1] -
Sumida, Y.; Ohmiya, H. Chem. Soc. Rev. 2021, 50, 6320–6332. doi:10.1039/d1cs00262g
Return to citation in text: [1] -
Denmark, S. E.; Beutner, G. L. Angew. Chem., Int. Ed. 2008, 47, 1560–1638. doi:10.1002/anie.200604943
Return to citation in text: [1] -
Ye, L.-W.; Zhou, J.; Tang, Y. Chem. Soc. Rev. 2008, 37, 1140. doi:10.1039/b717758e
Return to citation in text: [1] -
Guo, H.; Fan, Y. C.; Sun, Z.; Wu, Y.; Kwon, O. Chem. Rev. 2018, 118, 10049–10293. doi:10.1021/acs.chemrev.8b00081
Return to citation in text: [1] -
Huang, Y.; Liao, J.; Wang, W.; Liu, H.; Guo, H. Chem. Commun. 2020, 56, 15235–15281. doi:10.1039/d0cc05699e
Return to citation in text: [1] -
Xie, C.; Smaligo, A. J.; Song, X.-R.; Kwon, O. ACS Cent. Sci. 2021, 7, 536–558. doi:10.1021/acscentsci.0c01493
Return to citation in text: [1] -
Khong, S.; Venkatesh, T.; Kwon, O. Asian J. Org. Chem. 2021, 10, 2699–2708. doi:10.1002/ajoc.202100496
Return to citation in text: [1] -
Wang, X.; Yu, C.; Atodiresei, I. L.; Patureau, F. W. Org. Lett. 2022, 24, 1127–1131. doi:10.1021/acs.orglett.1c04045
Return to citation in text: [1] -
Wang, Y.-T.; Fu, M.-C.; Zhao, B.; Shang, R.; Fu, Y. Chem. Commun. 2020, 56, 2495–2498. doi:10.1039/c9cc09654j
Return to citation in text: [1] [2] -
Wang, H.-Y.; Zhong, L.-J.; Lv, G.-F.; Li, Y.; Li, J.-H. Org. Biomol. Chem. 2020, 18, 5589–5593. doi:10.1039/d0ob01242d
Return to citation in text: [1] [2] -
Wang, J.-X.; Wang, Y.-T.; Zhang, H.; Fu, M.-C. Org. Chem. Front. 2021, 8, 4466–4472. doi:10.1039/d1qo00660f
Return to citation in text: [1] [2] -
Chen, K.-Q.; Wang, Z.-X.; Chen, X.-Y. Org. Lett. 2020, 22, 8059–8064. doi:10.1021/acs.orglett.0c03006
Return to citation in text: [1] [2] -
Liu, H.-Y.; Lu, Y.; Li, Y.; Li, J.-H. Org. Lett. 2020, 22, 8819–8823. doi:10.1021/acs.orglett.0c03182
Return to citation in text: [1] [2] -
Liu, X.-J.; Zhou, S.-Y.; Xiao, Y.; Sun, Q.; Lu, X.; Li, Y.; Li, J.-H. Org. Lett. 2021, 23, 7839–7844. doi:10.1021/acs.orglett.1c02858
Return to citation in text: [1] [2] -
Wadekar, K.; Aswale, S.; Yatham, V. R. RSC Adv. 2020, 10, 16510–16514. doi:10.1039/d0ra03211e
Return to citation in text: [1] [2] -
Hou, T.; Peng, H.; Xin, Y.; Wang, S.; Zhu, W.; Chen, L.; Yao, Y.; Zhang, W.; Liang, S.; Wang, L. ACS Catal. 2020, 10, 5502–5510. doi:10.1021/acscatal.0c00920
Return to citation in text: [1] [2] -
Qu, Z.; Chen, X.; Zhong, S.; Deng, G.-J.; Huang, H. Org. Lett. 2021, 23, 5349–5353. doi:10.1021/acs.orglett.1c01654
Return to citation in text: [1] [2] -
Zhang, W.-K.; Li, J.-Z.; Zhang, C.-C.; Zhang, J.; Zheng, Y.-N.; Hu, Y.; Li, T.; Wei, W.-T. Eur. J. Org. Chem. 2022, e202200523. doi:10.1002/ejoc.202200523
Return to citation in text: [1] [2] -
Takano, S.; Moriya, M.; Ogasawara, K. J. Org. Chem. 1991, 56, 5982–5984. doi:10.1021/jo00021a006
Return to citation in text: [1] -
Greig, N. H.; Pei, X.-F.; Soncrant, T. T.; Ingram, D. K.; Brossi, A. Med. Res. Rev. 1995, 15, 3–31. doi:10.1002/med.2610150103
Return to citation in text: [1] -
Yu, Q.-s.; Pei, X.-F.; Holloway, H. W.; Greig, N. H.; Brossi, A. J. Med. Chem. 1997, 40, 2895–2901. doi:10.1021/jm970210v
Return to citation in text: [1] -
Nigel, H. G.; Kumar, S.; Qiansheng, Y.; Arnold, B.; Gosse, B. B.; Debomoy, K. L. Curr. Alzheimer Res. 2005, 2, 281. doi:10.2174/1567205054367829
Return to citation in text: [1] -
Shafferman, A.; Barak, D.; Stein, D.; Kronman, C.; Velan, B.; Greig, N. H.; Ordentlich, A. Chem.-Biol. Interact. 2008, 175, 166–172. doi:10.1016/j.cbi.2008.03.013
Return to citation in text: [1] -
Becker, R. E.; Greig, N. H. Curr. Alzheimer Res. 2010, 7, 642–651. doi:10.2174/156720510793499075
Return to citation in text: [1] -
Suzuki, T.; Choi, J.-H.; Kawaguchi, T.; Yamashita, K.; Morita, A.; Hirai, H.; Nagai, K.; Hirose, T.; Ōmura, S.; Sunazuka, T.; Kawagishi, H. Bioorg. Med. Chem. Lett. 2012, 22, 4246–4248. doi:10.1016/j.bmcl.2012.05.021
Return to citation in text: [1] -
Sun, Z.; Huang, H.; Wang, Q.; Huang, C.; Mao, G.; Deng, G.-J. Org. Chem. Front. 2022, 9, 3506–3514. doi:10.1039/d2qo00319h
Return to citation in text: [1]
| 1. | Molander, G. A.; Harris, C. R. Chem. Rev. 1996, 96, 307–338. doi:10.1021/cr950019y |
| 2. | Snider, B. B. Chem. Rev. 1996, 96, 339–364. doi:10.1021/cr950026m |
| 3. | Nair, V.; Mathew, J.; Prabhakaran, J. Chem. Soc. Rev. 1997, 26, 127–132. doi:10.1039/cs9972600127 |
| 4. | McCarroll, A. J.; Walton, J. C. Angew. Chem., Int. Ed. 2001, 40, 2224–2248. doi:10.1002/1521-3773(20010618)40:12<2224::aid-anie2224>3.0.co;2-f |
| 5. | Albert, M.; Fensterbank, L.; Lacote, E.; Malacria, M. Tandem Radical Reactions. In Radicals in Synthesis II; Gansäuer, A., Ed.; Springer: Berlin, 2006; pp 1–62. doi:10.1007/128_026 |
| 6. | Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d |
| 29. | Fan, J.-H.; Wei, W.-T.; Zhou, M.-B.; Song, R.-J.; Li, J.-H. Angew. Chem., Int. Ed. 2014, 53, 6650–6654. doi:10.1002/anie.201402893 |
| 30. | Biswas, P.; Paul, S.; Guin, J. Angew. Chem., Int. Ed. 2016, 55, 7756–7760. doi:10.1002/anie.201603809 |
| 31. | Tang, S.; Zhou, D.; Li, Z.-H.; Fu, M.-J.; Jie, L.; Sheng, R.-L.; Li, S.-H. Synthesis 2015, 47, 1567–1580. doi:10.1055/s-0034-1379902 |
| 32. | Wang, H.; Guo, L.; Duan, X.-H. J. Org. Chem. 2016, 81, 860–867. doi:10.1021/acs.joc.5b02433 |
| 33. | Yang, Z.; Cheng, Y.; Long, J.; Feng, X.; Tang, R.; Wei, J. New J. Chem. 2019, 43, 18760–18766. doi:10.1039/c9nj04458b |
| 34. | Che, F.; Zhong, J.; Yu, L.; Ma, C.; Yu, C.; Wang, M.; Hou, Z.; Zhang, Y. Adv. Synth. Catal. 2020, 362, 5020–5025. doi:10.1002/adsc.202000600 |
| 35. | Zhang, L.; Zhou, H.; Bai, S.; Li, S. Dalton Trans. 2021, 50, 3201–3206. doi:10.1039/d0dt04295a |
| 36. | Zhang, L.; Wang, Y.; Yang, Y.; Zhang, P.; Wang, C. Org. Chem. Front. 2020, 7, 3234–3241. doi:10.1039/d0qo00953a |
| 37. | Wang, C.; Liu, L. Org. Chem. Front. 2021, 8, 1454–1460. doi:10.1039/d0qo01508c |
| 38. | Su, L.; Sun, H.; Liu, J.; Wang, C. Org. Lett. 2021, 23, 4662–4666. doi:10.1021/acs.orglett.1c01400 |
| 46. | Tang, Q.; Liu, X.; Liu, S.; Xie, H.; Liu, W.; Zeng, J.; Cheng, P. RSC Adv. 2015, 5, 89009–89014. doi:10.1039/c5ra17292f |
| 21. | Xu, Z.; Yan, C.; Liu, Z.-Q. Org. Lett. 2014, 16, 5670–5673. doi:10.1021/ol502738a |
| 22. | Dai, Q.; Yu, J.; Jiang, Y.; Guo, S.; Yang, H.; Cheng, J. Chem. Commun. 2014, 50, 3865. doi:10.1039/c4cc01053a |
| 23. | Zhou, D.; Li, Z.-H.; Li, J.; Li, S.-H.; Wang, M.-W.; Luo, X.-L.; Ding, G.-L.; Sheng, R.-L.; Fu, M.-J.; Tang, S. Eur. J. Org. Chem. 2015, 1606–1612. doi:10.1002/ejoc.201403499 |
| 24. | He, Z.-Y.; Guo, J.-Y.; Tian, S.-K. Adv. Synth. Catal. 2018, 360, 1544–1548. doi:10.1002/adsc.201800012 |
| 25. | Shi, Y.; Xiao, H.; Xu, X.-H.; Huang, Y. Org. Biomol. Chem. 2018, 16, 8472–8476. doi:10.1039/c8ob02457j |
| 26. | Wang, X.-Y.; Zhong, Y.-F.; Mo, Z.-Y.; Wu, S.-H.; Xu, Y.-L.; Tang, H.-T.; Pan, Y.-M. Adv. Synth. Catal. 2021, 363, 208–214. doi:10.1002/adsc.202001192 |
| 27. | Wu, H.; Zhou, M.; Li, W.; Zhang, P. Catal. Commun. 2020, 133, 105832. doi:10.1016/j.catcom.2019.105832 |
| 28. | Fan, X.; Liu, H.; Ma, S.; Wang, F.; Yang, J.; Li, D. Tetrahedron 2022, 117-118, 132849. doi:10.1016/j.tet.2022.132849 |
| 54. | Fu, M.-C.; Shang, R.; Zhao, B.; Wang, B.; Fu, Y. Science 2019, 363, 1429–1434. doi:10.1126/science.aav3200 |
| 14. | Klein, J. E. M. N.; Taylor, R. J. K. Eur. J. Org. Chem. 2011, 6821–6841. doi:10.1002/ejoc.201100836 |
| 15. | Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247. doi:10.1039/c2cs35100e |
| 16. | Mai, W.; Wang, J.; Yang, L.; Yuan, J.; Mao, P.; Xiao, Y.; Qu, L. Chin. J. Org. Chem. 2014, 34, 1958. doi:10.6023/cjoc201405006 |
| 17. | Li, C.-C.; Yang, S.-D. Org. Biomol. Chem. 2016, 14, 4365–4377. doi:10.1039/c6ob00554c |
| 18. | Cao, Z.-Y.; Zhou, F.; Zhou, J. Acc. Chem. Res. 2018, 51, 1443–1454. doi:10.1021/acs.accounts.8b00097 |
| 19. | Marchese, A. D.; Larin, E. M.; Mirabi, B.; Lautens, M. Acc. Chem. Res. 2020, 53, 1605–1619. doi:10.1021/acs.accounts.0c00297 |
| 20. | Boddy, A. J.; Bull, J. A. Org. Chem. Front. 2021, 8, 1026–1084. doi:10.1039/d0qo01085e |
| 51. | Xu, P.; Xie, J.; Xue, Q.; Pan, C.; Cheng, Y.; Zhu, C. Chem. – Eur. J. 2013, 19, 14039–14042. doi:10.1002/chem.201302407 |
| 52. | Chen, J.-Q.; Wei, Y.-L.; Xu, G.-Q.; Liang, Y.-M.; Xu, P.-F. Chem. Commun. 2016, 52, 6455–6458. doi:10.1039/c6cc02007k |
| 53. | Wang, Y.-Z.; Lin, W.-J.; Zou, J.-Y.; Yu, W.; Liu, X.-Y. Adv. Synth. Catal. 2020, 362, 3116–3120. doi:10.1002/adsc.202000609 |
| 7. | Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Wang, S. J. Med. Chem. 2006, 49, 3432–3435. doi:10.1021/jm051122a |
| 8. | Christensen, M. K.; Erichsen, K. D.; Trojel-Hansen, C.; Tjørnelund, J.; Nielsen, S. J.; Frydenvang, K.; Johansen, T. N.; Nielsen, B.; Sehested, M.; Jensen, P. B.; Ikaunieks, M.; Zaichenko, A.; Loza, E.; Kalvinsh, I.; Björkling, F. J. Med. Chem. 2010, 53, 7140–7145. doi:10.1021/jm100763j |
| 9. | Millemaggi, A.; Taylor, R. J. K. Eur. J. Org. Chem. 2010, 4527–4547. doi:10.1002/ejoc.201000643 |
| 10. | Rudrangi, S. R. S.; Bontha, V. K.; Manda, V. R.; Bethi, S. Asian J. Res. Chem. 2011, 4, 335. |
| 11. | Yu, B.; Yu, D. Q.; Liu, H. M. Eur. J. Med. Chem. 2015, 97, 763. doi:10.1016/j.ejmech.2014.06.056 |
| 12. | Kaur, M.; Singh, M.; Chadha, N.; Silakari, O. Eur. J. Med. Chem. 2016, 123, 858–894. doi:10.1016/j.ejmech.2016.08.011 |
| 13. | Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041 |
| 47. | Jin, Y.; Jiang, M.; Wang, H.; Fu, H. Sci. Rep. 2016, 6, 20068. doi:10.1038/srep20068 |
| 45. | Xie, J.; Xu, P.; Li, H.; Xue, Q.; Jin, H.; Cheng, Y.; Zhu, C. Chem. Commun. 2013, 49, 5672. doi:10.1039/c3cc42672f |
| 46. | Tang, Q.; Liu, X.; Liu, S.; Xie, H.; Liu, W.; Zeng, J.; Cheng, P. RSC Adv. 2015, 5, 89009–89014. doi:10.1039/c5ra17292f |
| 47. | Jin, Y.; Jiang, M.; Wang, H.; Fu, H. Sci. Rep. 2016, 6, 20068. doi:10.1038/srep20068 |
| 49. | Li, X.; Han, M.-Y.; Wang, B.; Wang, L.; Wang, M. Org. Biomol. Chem. 2019, 17, 6612–6619. doi:10.1039/c9ob01023h |
| 42. | An, Y.; Li, Y.; Wu, J. Org. Chem. Front. 2016, 3, 570. doi:10.1039/c6qo00055j |
| 43. | Muralirajan, K.; Kancherla, R.; Gimnkhan, A.; Rueping, M. Org. Lett. 2021, 23, 6905–6910. doi:10.1021/acs.orglett.1c02467 |
| 44. | Du, J.; Wang, X.; Wang, H.; Wei, J.; Huang, X.; Song, J.; Zhang, J. Org. Lett. 2021, 23, 5631–5635. doi:10.1021/acs.orglett.1c01698 |
| 50. | Zhao, Y.; Li, Z.; Sharma, U. K.; Sharma, N.; Song, G.; Van der Eycken, E. V. Chem. Commun. 2016, 52, 6395–6398. doi:10.1039/c6cc02024k |
| 39. | Festa, A. A.; Voskressensky, L. G.; Van der Eycken, E. V. Chem. Soc. Rev. 2019, 48, 4401–4423. doi:10.1039/c8cs00790j |
| 40. | Singh, J.; Sharma, A. Adv. Synth. Catal. 2021, 363, 4284–4308. doi:10.1002/adsc.202100515 |
| 41. | Ghosh, S.; Qu, Z.-W.; Pradhan, S.; Ghosh, A.; Grimme, S.; Chatterjee, I. Angew. Chem., Int. Ed. 2022, 61, 10.1002/anie.202115272. doi:10.1002/anie.202115272 |
| 7. | Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Wang, S. J. Med. Chem. 2006, 49, 3432–3435. doi:10.1021/jm051122a |
| 8. | Christensen, M. K.; Erichsen, K. D.; Trojel-Hansen, C.; Tjørnelund, J.; Nielsen, S. J.; Frydenvang, K.; Johansen, T. N.; Nielsen, B.; Sehested, M.; Jensen, P. B.; Ikaunieks, M.; Zaichenko, A.; Loza, E.; Kalvinsh, I.; Björkling, F. J. Med. Chem. 2010, 53, 7140–7145. doi:10.1021/jm100763j |
| 9. | Millemaggi, A.; Taylor, R. J. K. Eur. J. Org. Chem. 2010, 4527–4547. doi:10.1002/ejoc.201000643 |
| 10. | Rudrangi, S. R. S.; Bontha, V. K.; Manda, V. R.; Bethi, S. Asian J. Res. Chem. 2011, 4, 335. |
| 11. | Yu, B.; Yu, D. Q.; Liu, H. M. Eur. J. Med. Chem. 2015, 97, 763. doi:10.1016/j.ejmech.2014.06.056 |
| 12. | Kaur, M.; Singh, M.; Chadha, N.; Silakari, O. Eur. J. Med. Chem. 2016, 123, 858–894. doi:10.1016/j.ejmech.2016.08.011 |
| 13. | Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041 |
| 48. | Li, Z.; Zhang, Y.; Zhang, L.; Liu, Z.-Q. Org. Lett. 2014, 16, 382–385. doi:10.1021/ol4032478 |
| 68. | Wang, Y.-T.; Fu, M.-C.; Zhao, B.; Shang, R.; Fu, Y. Chem. Commun. 2020, 56, 2495–2498. doi:10.1039/c9cc09654j |
| 69. | Wang, H.-Y.; Zhong, L.-J.; Lv, G.-F.; Li, Y.; Li, J.-H. Org. Biomol. Chem. 2020, 18, 5589–5593. doi:10.1039/d0ob01242d |
| 70. | Wang, J.-X.; Wang, Y.-T.; Zhang, H.; Fu, M.-C. Org. Chem. Front. 2021, 8, 4466–4472. doi:10.1039/d1qo00660f |
| 55. | Rosokha, S. V.; Kochi, J. K. Acc. Chem. Res. 2008, 41, 641–653. doi:10.1021/ar700256a |
| 56. | Lima, C. G. S.; de M. Lima, T.; Duarte, M.; Jurberg, I. D.; Paixão, M. W. ACS Catal. 2016, 6, 1389–1407. doi:10.1021/acscatal.5b02386 |
| 57. | Yuan, Y.-q.; Majumder, S.; Yang, M.-h.; Guo, S.-r. Tetrahedron Lett. 2020, 61, 151506. doi:10.1016/j.tetlet.2019.151506 |
| 58. | Crisenza, G. E. M.; Mazzarella, D.; Melchiorre, P. J. Am. Chem. Soc. 2020, 142, 5461–5476. doi:10.1021/jacs.0c01416 |
| 59. | Yang, Z.; Liu, Y.; Cao, K.; Zhang, X.; Jiang, H.; Li, J. Beilstein J. Org. Chem. 2021, 17, 771–799. doi:10.3762/bjoc.17.67 |
| 60. | Sumida, Y.; Ohmiya, H. Chem. Soc. Rev. 2021, 50, 6320–6332. doi:10.1039/d1cs00262g |
| 61. | Denmark, S. E.; Beutner, G. L. Angew. Chem., Int. Ed. 2008, 47, 1560–1638. doi:10.1002/anie.200604943 |
| 62. | Ye, L.-W.; Zhou, J.; Tang, Y. Chem. Soc. Rev. 2008, 37, 1140. doi:10.1039/b717758e |
| 63. | Guo, H.; Fan, Y. C.; Sun, Z.; Wu, Y.; Kwon, O. Chem. Rev. 2018, 118, 10049–10293. doi:10.1021/acs.chemrev.8b00081 |
| 64. | Huang, Y.; Liao, J.; Wang, W.; Liu, H.; Guo, H. Chem. Commun. 2020, 56, 15235–15281. doi:10.1039/d0cc05699e |
| 65. | Xie, C.; Smaligo, A. J.; Song, X.-R.; Kwon, O. ACS Cent. Sci. 2021, 7, 536–558. doi:10.1021/acscentsci.0c01493 |
| 66. | Khong, S.; Venkatesh, T.; Kwon, O. Asian J. Org. Chem. 2021, 10, 2699–2708. doi:10.1002/ajoc.202100496 |
| 67. | Wang, X.; Yu, C.; Atodiresei, I. L.; Patureau, F. W. Org. Lett. 2022, 24, 1127–1131. doi:10.1021/acs.orglett.1c04045 |
| 85. | Sun, Z.; Huang, H.; Wang, Q.; Huang, C.; Mao, G.; Deng, G.-J. Org. Chem. Front. 2022, 9, 3506–3514. doi:10.1039/d2qo00319h |
| 30. | Biswas, P.; Paul, S.; Guin, J. Angew. Chem., Int. Ed. 2016, 55, 7756–7760. doi:10.1002/anie.201603809 |
| 78. | Takano, S.; Moriya, M.; Ogasawara, K. J. Org. Chem. 1991, 56, 5982–5984. doi:10.1021/jo00021a006 |
| 79. | Greig, N. H.; Pei, X.-F.; Soncrant, T. T.; Ingram, D. K.; Brossi, A. Med. Res. Rev. 1995, 15, 3–31. doi:10.1002/med.2610150103 |
| 80. | Yu, Q.-s.; Pei, X.-F.; Holloway, H. W.; Greig, N. H.; Brossi, A. J. Med. Chem. 1997, 40, 2895–2901. doi:10.1021/jm970210v |
| 81. | Nigel, H. G.; Kumar, S.; Qiansheng, Y.; Arnold, B.; Gosse, B. B.; Debomoy, K. L. Curr. Alzheimer Res. 2005, 2, 281. doi:10.2174/1567205054367829 |
| 82. | Shafferman, A.; Barak, D.; Stein, D.; Kronman, C.; Velan, B.; Greig, N. H.; Ordentlich, A. Chem.-Biol. Interact. 2008, 175, 166–172. doi:10.1016/j.cbi.2008.03.013 |
| 83. | Becker, R. E.; Greig, N. H. Curr. Alzheimer Res. 2010, 7, 642–651. doi:10.2174/156720510793499075 |
| 84. | Suzuki, T.; Choi, J.-H.; Kawaguchi, T.; Yamashita, K.; Morita, A.; Hirai, H.; Nagai, K.; Hirose, T.; Ōmura, S.; Sunazuka, T.; Kawagishi, H. Bioorg. Med. Chem. Lett. 2012, 22, 4246–4248. doi:10.1016/j.bmcl.2012.05.021 |
| 54. | Fu, M.-C.; Shang, R.; Zhao, B.; Wang, B.; Fu, Y. Science 2019, 363, 1429–1434. doi:10.1126/science.aav3200 |
| 68. | Wang, Y.-T.; Fu, M.-C.; Zhao, B.; Shang, R.; Fu, Y. Chem. Commun. 2020, 56, 2495–2498. doi:10.1039/c9cc09654j |
| 69. | Wang, H.-Y.; Zhong, L.-J.; Lv, G.-F.; Li, Y.; Li, J.-H. Org. Biomol. Chem. 2020, 18, 5589–5593. doi:10.1039/d0ob01242d |
| 70. | Wang, J.-X.; Wang, Y.-T.; Zhang, H.; Fu, M.-C. Org. Chem. Front. 2021, 8, 4466–4472. doi:10.1039/d1qo00660f |
| 71. | Chen, K.-Q.; Wang, Z.-X.; Chen, X.-Y. Org. Lett. 2020, 22, 8059–8064. doi:10.1021/acs.orglett.0c03006 |
| 72. | Liu, H.-Y.; Lu, Y.; Li, Y.; Li, J.-H. Org. Lett. 2020, 22, 8819–8823. doi:10.1021/acs.orglett.0c03182 |
| 73. | Liu, X.-J.; Zhou, S.-Y.; Xiao, Y.; Sun, Q.; Lu, X.; Li, Y.; Li, J.-H. Org. Lett. 2021, 23, 7839–7844. doi:10.1021/acs.orglett.1c02858 |
| 74. | Wadekar, K.; Aswale, S.; Yatham, V. R. RSC Adv. 2020, 10, 16510–16514. doi:10.1039/d0ra03211e |
| 75. | Hou, T.; Peng, H.; Xin, Y.; Wang, S.; Zhu, W.; Chen, L.; Yao, Y.; Zhang, W.; Liang, S.; Wang, L. ACS Catal. 2020, 10, 5502–5510. doi:10.1021/acscatal.0c00920 |
| 76. | Qu, Z.; Chen, X.; Zhong, S.; Deng, G.-J.; Huang, H. Org. Lett. 2021, 23, 5349–5353. doi:10.1021/acs.orglett.1c01654 |
| 77. | Zhang, W.-K.; Li, J.-Z.; Zhang, C.-C.; Zhang, J.; Zheng, Y.-N.; Hu, Y.; Li, T.; Wei, W.-T. Eur. J. Org. Chem. 2022, e202200523. doi:10.1002/ejoc.202200523 |
| 74. | Wadekar, K.; Aswale, S.; Yatham, V. R. RSC Adv. 2020, 10, 16510–16514. doi:10.1039/d0ra03211e |
| 75. | Hou, T.; Peng, H.; Xin, Y.; Wang, S.; Zhu, W.; Chen, L.; Yao, Y.; Zhang, W.; Liang, S.; Wang, L. ACS Catal. 2020, 10, 5502–5510. doi:10.1021/acscatal.0c00920 |
| 76. | Qu, Z.; Chen, X.; Zhong, S.; Deng, G.-J.; Huang, H. Org. Lett. 2021, 23, 5349–5353. doi:10.1021/acs.orglett.1c01654 |
| 77. | Zhang, W.-K.; Li, J.-Z.; Zhang, C.-C.; Zhang, J.; Zheng, Y.-N.; Hu, Y.; Li, T.; Wei, W.-T. Eur. J. Org. Chem. 2022, e202200523. doi:10.1002/ejoc.202200523 |
| 28. | Fan, X.; Liu, H.; Ma, S.; Wang, F.; Yang, J.; Li, D. Tetrahedron 2022, 117-118, 132849. doi:10.1016/j.tet.2022.132849 |
| 71. | Chen, K.-Q.; Wang, Z.-X.; Chen, X.-Y. Org. Lett. 2020, 22, 8059–8064. doi:10.1021/acs.orglett.0c03006 |
| 72. | Liu, H.-Y.; Lu, Y.; Li, Y.; Li, J.-H. Org. Lett. 2020, 22, 8819–8823. doi:10.1021/acs.orglett.0c03182 |
| 73. | Liu, X.-J.; Zhou, S.-Y.; Xiao, Y.; Sun, Q.; Lu, X.; Li, Y.; Li, J.-H. Org. Lett. 2021, 23, 7839–7844. doi:10.1021/acs.orglett.1c02858 |
© 2023 Liu et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.