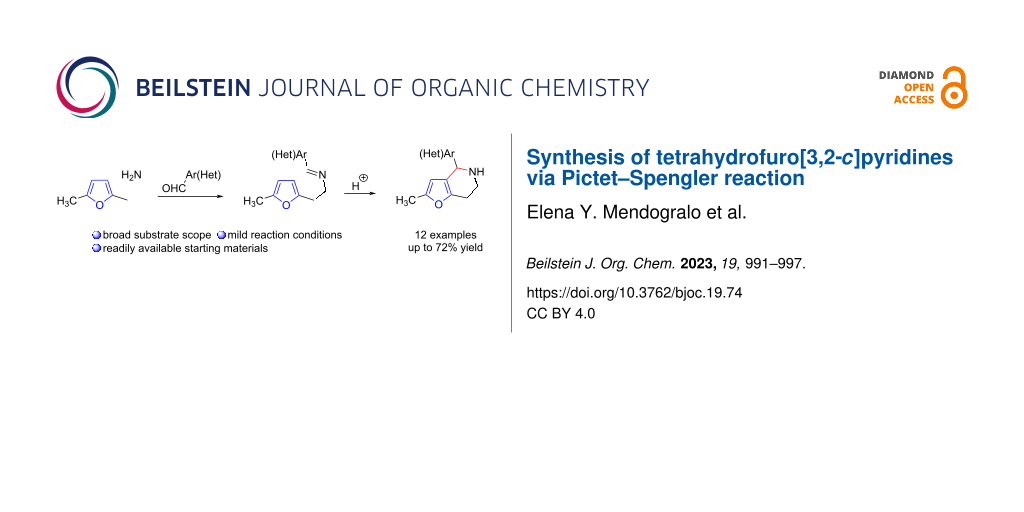
Abstract
A semi-one-pot method for the synthesis of 4-substituted tetrahydrofuro[3,2-c]pyridines by the Pictet–Spengler reaction was developed. The method is based on the condensation of easily accessibly 2-(5-methylfuran-2-yl)ethanamine with commercially available aromatic aldehydes followed by acid-catalyzed Pictet–Spengler cyclization. Using this approach, we synthesized a range of 4-substituted tetrahydrofuro[3,2-c]pyridines in reasonable yields. The reactivity of some of the products was investigated and selected synthetic transformations of the obtained tetrahydrofuro[3,2-c]pyridines were shown.

Graphical Abstract
Introduction
Hydrogenated furo[3,2-c]pyridines represent an important class of heterocyclic compounds, which skeleton is the key frame of many bioactive and natural compounds. For example, tetrahydrofuro[3,2-c]pyridine A demonstrates excellent in vitro JAK2 inhibitory activity superior to tofacitinib (Figure 1) [1]. Furan B is a potent κ-opioid receptor agonist and exhibits excellent antinociceptive activity [2]. Lactam C possesses potent antituberculosis activity and excellent selectivity to Mycobacterium tuberculosis strain H37Rv [3]. Araliopsine (D) was isolated from the fruits of Zanthoxylum simulans [4]. Benzofuran E is a potent and relatively selective α2-adrenoceptor antagonist [5].
![[1860-5397-19-74-1]](/bjoc/content/figures/1860-5397-19-74-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of natural and bioactive hydrogenated furo[3,2-c]pyridines.
Figure 1: Examples of natural and bioactive hydrogenated furo[3,2-c]pyridines.
Despite the simplicity of the tetrahydrofuro[3,2-c]pyridine core, only limited approaches for the synthesis of this subclass of heterocycles using furan derivatives as starting compounds have been described [6-8]. The first group of methods is based on the use of 3-substituted furans. For example, the intramolecular Friedel–Crafts alkylation reaction (Scheme 1a) of alcohols [9-11], alkenes [12] or acetylenes [13] affords the desired tetrahydrofuro[3,2-c]pyridines. A related method is based on a Au(I)-catalyzed domino sequence dearomatization/ipso-cyclization/Michael-type Friedel–Crafts alkylation (Scheme 1b) [14-16]. Unfortunately, approaches including the intramolecular alkylation of 3-substituted furans are underinvestigated as these substrates are usually hard to reach and the resulting benzyl carbocation is often prone to undergo rearrangement to a more stable benzhydryl-type cation resulting in the formation of isomeric products.
![[1860-5397-19-74-i1]](/bjoc/content/inline/1860-5397-19-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The described approaches to tetrahydrofuro[3,2-c]pyridines and our work.
Scheme 1: The described approaches to tetrahydrofuro[3,2-c]pyridines and our work.
In an alternative group of methods, more accessible 2-substituted furans are used as starting compounds. For example, a construction of tetrahydrofuro[3,2-c]pyridines based on the Pictet–Spengler reaction [17,18] was described (Scheme 1c). The most studied variation of this cyclization is based on the generation of an acyliminium cation from the corresponding alcohols [19-23] or alkenes [24-29], subsequent attack of furan ring and the formation of annulated tetrahydrofuro[3,2-c]pyridines. Moreover, multistep cascade processes with the simultaneous construction of several cores were described, where the key step is the generation of an acyliminium cation and the Pictet–Spengler cyclization [30-32], including solid-phase synthesis [33-35]. Another route for the construction of tetrahydrofuro[3,2-c]pyridines is based on a Bischler–Napieralski cyclocondensation followed by the C=N bond reduction (Scheme 1d) [36-39]. Despite the simplicity of the latter approach and availability of 2-(furan-2-yl)ethanamine, the instability of intermediate dihydrofuro[3,2-c]pyridines, substantial tarring of reaction mixtures in acidic conditions and a limited number of cyclization examples might constrain its wide application.
Diverse bioactivities and prevalence of hydrogenated furo[3,2-c]pyridines in nature prompted us to develop an approach to substituted tetrahydrofuro[3,2-c]pyridines. From the analysis of the literature data [2,40-43], we suggested that the interaction of furanic amines with various aldehydes in an acidic media should be accompanied by the formation of the corresponding imine, the generation of the iminium cation, and subsequent Pictet–Spengler cyclization to form the key tetrahydrofuro[3,2-c]pyridines. For this method to be effective, the following requirements must be met: 1) the starting compounds must be commercially or readily available; 2) the resulting iminium cation should be stable enough for a smooth cyclization; 3) side processes and tarring of the reaction mixture should be minimized; 4) the possibility for the synthesis of a wide range of desired products should be realized. We have found that all requirements are met for the reaction of 2-(5-methylfuran-2-yl)ethanamine with a set of aromatic aldehydes. Herein, we report the results of our study of this tandem reaction.
Results and Discussion
Initially, we synthesized the key imine 3a by refluxing a solution of the starting 2-(5-methylfuran-2-yl)ethylamine (1a) and commercially available benzaldehyde (2a) in absolute CH3CN for one hour in a quantitative yield. To our delight, when 2 equiv of HCl were subsequently added to the solution of imine 3a at 50 °C, the formation of the desired 2-methyl-4-phenyl-4,5,6,7-tetrahydrofuro[3,2-c]pyridine (4a) was observed in 26% yield (Table 1, entry 1). It should be noted that most part of the product 4a was formed as the hydrochloride salt that was converted to the free base by the treatment with an aqueous solution of NaOH overnight (see Supporting Information File 1 for full experimental data). The yield of the desired product 4a raised significantly when the reaction time was increased (Table 1, entry 2). At the same time, increasing the reaction temperature leads to the formation of the desired product 4a with moderate yield (Table 1, entries 3 and 4). Next, we found that TsOH as a catalyst and several studied solvents (toluene, 1,4-dioxane, AcOH) are inefficient (Table 1, entries 5–10). Finally, we settled on the mixture of acetic and hydrochloric acid previously well-proven for furan chemistry [44-48]. Under these conditions at room temperature for 1 hour, the product yield was 18% (Table 1, entry 11). An increase of the reaction time up to 48 h leads to a significant increase of the yield (Table 1, entries 12 and 13). To reduce the reaction time, we increased the temperature (70 °C) and achieved a comparable result in 3.5 hours (Table 1, entry 14). Moreover, we found that the short-term treatment with aq NaOH solution at rt and at 50 °C overnight were less effective than the treatment with aq NaOH at rt overnight (Table 1, entries 15 and 16). Finally, we found that the optimal reaction time was 5 hours. The yield of the desired product 4a in this case was 67% (Table 1, entry 18). With a shorter time, a slightly reduced reaction yield of 4a was observed, while at longer heating the reaction mixture underwent abundant tarring (Table 1, entries 17 and 19). The treatment of the reaction mixture after the reaction with an aqueous solution of NaHCO3 turned out to be less effective (Table 1, entry 20). It should be noted that 3-(2-oxopropyl)-2-phenylpiperidin-4-one (5a), which is a product of further acid hydrolysis of 4a was observed in most cases in trace amounts; an exception is entry 18 (Table 1), in which the 1,4-diketone 5a was isolated in 10% yield.
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-74-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Time, h | T, °С | Acid, equiv | Yield of 4a, %b |
| 1 | CH3CN | 1.5 | 50 | HCl (2.0) | 26 |
| 2 | CH3CN | 5.5 | 50 | HCl (1.0) | 53 |
| 3 | CH3CN | 4.5 | 82 | HCl (1.5) | 32 |
| 4 | CH3CN | 24 | 82 | HCl (1.5) | 29 |
| 5 | CH3CN | 30 | 70 | TsOH (2.0) | 11 |
| 6 | toluene | 2 | 70 | HCl (1.0) | 58 |
| 7 | toluene | 2 | 110 | HCl (1.0) | 25 |
| 8 | 1,4-dioxane | 2 | 70 | HCl (1.0) | 8 |
| 9 | AcOH | 24 | 50 | TsOH (3.0) | 13 |
| 10 | AcOH | 48 | 118 | TsOH (4.0) | ND |
| 11 | AcOH | 2 | rt | HCl (1.0) | 18 |
| 12 | AcOH | 24 | rt | HCl (2.0) | 31 |
| 13 | AcOH | 48 | rt | HCl (2.0) | 47 |
| 14 | AcOH | 3.5 | 70 | HCl (2.0) | 48 |
| 15c | AcOH | 3.5 | 70 | HCl (2.0) | 41 |
| 16d | AcOH | 3.5 | 70 | HCl (2.0) | 33 |
| 17 | AcOH | 1 | 70 | HCl (2.0) | 52 |
| 18e | AcOH | 5 | 70 | HCl (2.0) | 67 |
| 19 | AcOH | 6.5 | 70 | HCl (2.5) | 38 |
| 20f | AcOH | 5 | 70 | HCl (2.0) | 50 |
aAll reactions were performed at a 1 mmol scale of 1a. The reaction mixture was treated with aq NaOH at rt overnight; bisolated yield; ctreatment of the reaction mixture with aq NaOH at rt for 2 h; dtreatment of the reaction mixture with aq NaOH at 50 °C overnight; e3-(2-oxopropyl)piperidin-4-one (5a) was isolated with 10% yield; ftreatment of the reaction mixture with aq NaHCO3 at rt overnight.
With the optimized reaction conditions in hand, we investigated the scope of the developed tetrahydrofuro[3,2-c]pyridine synthesis. We found that a wide range of substituents at the aromatic cores was tolerant to the reaction conditions, and the yields of the desired products 4 remain moderate to good (Scheme 2). In particular, the use of starting benzaldehydes 2 containing electron-donating groups leads to the desired products 4e–g,i–k in much higher yields than with substrate 4d containing a strong electron-withdrawing group. The product 4n with the nitro group in the para-position of starting benzaldehyde 2n was formed in only trace amounts. A similar result was also observed in the case of isonicotinaldehyde (2m). These results were associated with an insufficient stabilization of the generated iminium cation due to the presence of a strong electron-withdrawing nitro group (for 3n), and due to the fact that the electron deficiency of the pyridine ring is strongly manifested in an acidic media (for 3m). In addition, the disadvantages of the developed method include the impossibility of using aliphatic aldehydes (for example, 2-phenylacetaldehyde (2o)), that is also explained by an insufficient stabilization of the resulting cation and the impossibility of cyclization. We also did not detect the formation of the target product when 1H-pyrrole-2-carbaldehyde (2p) was used. In this case, we observed abundant tarring and decomposition of the reaction mixture, which is probably due to the acidophobic nature of monosubstituted pyrroles. Surprisingly, the reaction of 4-(methylthio)benzaldehyde (2h) with 2-(5-methylfuran-2-yl)ethylamine (1a) under the optimized reaction conditions, in addition to the desired tetrahydrofuro[3,2-c]pyridine (4h), led to the major formation of the corresponding 1,4-diketone 5h.
![[1860-5397-19-74-i2]](/bjoc/content/inline/1860-5397-19-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of tetrahydrofuro[3,2-c]pyridines 4. Conditions: athe reaction was performed at 1 mmol scale of 1a; bthe reaction was performed at 2 mmol scale of 1a; cthe corresponding 3-(2-oxopropyl)piperidin-4-one was isolated with 20% yield; dORTEP diagram with ellipsoid contour probability level of 50%; edetected by GC–MS.
Scheme 2: The synthesis of tetrahydrofuro[3,2-c]pyridines 4. Conditions: athe reaction was performed at 1 mmo...
This unexpected formation of 3-(2-oxopropyl)piperidin-4-one (5h) led us to the idea that the tandem sequence Pictet–Spengler cyclization/furan acid hydrolysis could be a convenient tool for the synthesis of substituted 3-(2-oxopropyl)piperidin-4-ones 5. Using tetrahydrofuro[3,2-c]pyridine 4a as the model compound, we studied the effect of various Brønsted acids, temperature, concentrations, and the nature of the solvent on the efficiency of the reaction. In most cases, we observed the abundant tarring of the reaction mixture and we could not achieve the full conversion of the starting tetrahydrofuro[3,2-c]pyridine 4a in any experiment. The best results were achieved when HCl in 1,4-dioxane was used (Scheme 3). However, upon isolation and purification of the desired product by column chromatography, as well as upon recrystallization from ethyl acetate, 1,4-diketone 5а undergoes Paal–Knorr cyclization with the formation of the starting tetrahydrofuro[3,2-c]pyridine 4а, which ultimately leads to mixtures of compounds 4а and 5а with different ratios. The reversibility of the acid hydrolysis is quite typical for furans and is more pronounced for di- and polysubstituted furans, due to the higher stability of the latter [49-51].
![[1860-5397-19-74-i3]](/bjoc/content/inline/1860-5397-19-74-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The acid-catalyzed reversible transformation of tetrahydrofuro[3,2-c]pyridine 4a and 3-(2-oxopropyl)piperidin-4-one 5a. The product yield was determined by GC–MS using an internal standard.
Scheme 3: The acid-catalyzed reversible transformation of tetrahydrofuro[3,2-c]pyridine 4a and 3-(2-oxopropyl...
3-(2-Oxopropyl)piperidin-4-one 5a could be involved into the Paal–Knorr pyrrole synthesis in a one-pot manner. Thus, without isolation of the intermediate 3-(2-oxopropyl)piperidin-4-one 5a, the reaction mixture after acid hydrolysis was treated with aniline and the desired tetrahydro-1H-pyrrolo[3,2-с]pyridine 6а was isolated in 19% yield only (Scheme 4).
![[1860-5397-19-74-i4]](/bjoc/content/inline/1860-5397-19-74-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of tetrahydropyrrolo[3,2-c]pyridine 6a.
Scheme 4: Synthesis of tetrahydropyrrolo[3,2-c]pyridine 6a.
Finally, we studied some reactions of the obtained tetrahydrofuro[3,2-c]pyridine 4a. We showed that the N-acetyl-protected tetrahydrofuro[3,2-c]pyridine 4q could be obtained by treating the starting tetrahydrofuro[3,2-c]pyridine 4a with acetyl chloride while the N-methylated tetrahydrofuro[3,2-c]pyridine 4r can be formed via subsequent treatment of 4a with NaH and methyl iodide (Scheme 5).
![[1860-5397-19-74-i5]](/bjoc/content/inline/1860-5397-19-74-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reactivity of tetrahydrofuro[3,2-c]pyridine 4a.
Scheme 5: Reactivity of tetrahydrofuro[3,2-c]pyridine 4a.
Conclusion
In conclusion, we have developed a semi-one-pot method for the synthesis of 2,3-annulated furans based on the Pictet–Spengler reaction. This method provides a shortcut to 4-substituted tetrahydrofuro[3,2-c]pyridines in reasonable yields starting from readily available 2-(5-methylfuran-2-yl)ethylamine and aromatic aldehydes. The use of starting benzaldehydes containing electron-donating groups leads to the desired products in much higher yields than with aldehydes containing electron-withdrawing moieties. The availability of the starting materials, mild reaction conditions, and reasonable yields are some of the attractive features of the developed method. In addition, we have also demonstrated some transformations of the products into functionalized derivatives.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H and 13C NMR spectra, HRMS of new compounds, and X-ray crystallography data. | ||
| Format: PDF | Size: 3.1 MB | Download |
| Supporting Information File 2: Crystallographic information file for compound 4a. | ||
| Format: CIF | Size: 224.4 KB | Download |
References
-
Wang, Y.; Huang, W.; Xin, M.; Chen, P.; Gui, L.; Zhao, X.; Zhu, X.; Luo, H.; Cong, X.; Wang, J.; Liu, F. Bioorg. Med. Chem. 2019, 27, 2592–2597. doi:10.1016/j.bmc.2019.03.048
Return to citation in text: [1] -
Naylor, A.; Judd, D. B.; Scopes, D. I. C.; Hayes, A. G.; Birch, P. J. J. Med. Chem. 1994, 37, 2138–2144. doi:10.1021/jm00040a004
Return to citation in text: [1] [2] -
Zhang, W.; Liu, L.-l.; Lun, S.; Wang, S.-S.; Xiao, S.; Gunosewoyo, H.; Yang, F.; Tang, J.; Bishai, W. R.; Yu, L.-F. Eur. J. Med. Chem. 2021, 213, 113202. doi:10.1016/j.ejmech.2021.113202
Return to citation in text: [1] -
Chang, Z. Q.; Liu, F.; Wang, S. L.; Zhao, T. Z.; Wang, M. T. Yaoxue Xuebao 1981, 16, 394–396.
Return to citation in text: [1] -
Van der Mey, M.; Windhorst, A. D.; Klok, R. P.; Herscheid, J. D. M.; Kennis, L. E.; Bischoff, F.; Bakker, M.; Langlois, X.; Heylen, L.; Jurzak, M.; Leysen, J. E. Bioorg. Med. Chem. 2006, 14, 4526–4534. doi:10.1016/j.bmc.2006.02.029
Return to citation in text: [1] -
Nagai, Y.; Uno, H.; Umemoto, S. Chem. Pharm. Bull. 1977, 25, 1911–1922. doi:10.1248/cpb.25.1911
Return to citation in text: [1] -
Imanishi, T.; Yagi, N.; Hanaoka, M. Tetrahedron Lett. 1981, 22, 667–670. doi:10.1016/s0040-4039(01)92518-3
Return to citation in text: [1] -
Imanishi, T.; Yagi, N.; Hanaoka, M. Chem. Pharm. Bull. 1983, 31, 1243–1253. doi:10.1248/cpb.31.1243
Return to citation in text: [1] -
Schneider, C. S.; Pook, K. H. J. Chem. Soc., Perkin Trans. 1 1986, 877–883. doi:10.1039/p19860000877
Return to citation in text: [1] -
Schafroth, M. A.; Rummelt, S. M.; Sarlah, D.; Carreira, E. M. Org. Lett. 2017, 19, 3235–3238. doi:10.1021/acs.orglett.7b01346
Return to citation in text: [1] -
Davies, S. G.; Fletcher, A. M.; Holder, K. E.; Roberts, P. M.; Thomson, J. E.; Zimmer, D. Heterocycles 2019, 99, 919–941. doi:10.3987/com-18-s(f)59
Return to citation in text: [1] -
Fuchs, J. R.; Funk, R. L. Org. Lett. 2001, 3, 3349–3351. doi:10.1021/ol016592n
Return to citation in text: [1] -
Sun, Y.-W.; Tang, X.-Y.; Shi, M. Chem. Commun. 2015, 51, 13937–13940. doi:10.1039/c5cc05808b
Return to citation in text: [1] -
Baba, T.; Oka, J.; Noguchi, K.; Tanaka, K. Eur. J. Org. Chem. 2015, 4374–4382. doi:10.1002/ejoc.201500486
Return to citation in text: [1] -
He, Y.; Li, Z.; Robeyns, K.; Van Meervelt, L.; Van der Eycken, E. V. Angew. Chem., Int. Ed. 2018, 57, 272–276. doi:10.1002/anie.201710592
Return to citation in text: [1] -
He, Y.; Wu, D.; Li, Z.; Robeyns, K.; Van Meervelt, L.; Van der Eycken, E. V. Org. Biomol. Chem. 2019, 17, 6284–6292. doi:10.1039/c9ob01299k
Return to citation in text: [1] -
Calcaterra, A.; Mangiardi, L.; Delle Monache, G.; Quaglio, D.; Balducci, S.; Berardozzi, S.; Iazzetti, A.; Franzini, R.; Botta, B.; Ghirga, F. Molecules 2020, 25, 414. doi:10.3390/molecules25020414
Return to citation in text: [1] -
Gholamzadeh, P. Adv. Heterocycl. Chem. 2019, 127, 153–226. doi:10.1016/bs.aihch.2018.09.002
Return to citation in text: [1] -
Kano, S.; Yuasa, Y.; Yokomatsu, T.; Shibuya, S. Heterocycles 1984, 21, 701. doi:10.3987/s-1984-02-0701
Return to citation in text: [1] -
Tanis, S. P.; Deaton, M. V.; Dixon, L. A.; McMills, M. C.; Raggon, J. W.; Collins, M. A. J. Org. Chem. 1998, 63, 6914–6928. doi:10.1021/jo980718l
Return to citation in text: [1] -
Gao, S.; Tu, Y. Q.; Hu, X.; Wang, S.; Hua, R.; Jiang, Y.; Zhao, Y.; Fan, X.; Zhang, S. Org. Lett. 2006, 8, 2373–2376. doi:10.1021/ol0607185
Return to citation in text: [1] -
Shengule, S. R.; Ryder, G.; Willis, A. C.; Pyne, S. G. Tetrahedron 2012, 68, 10280–10285. doi:10.1016/j.tet.2012.10.014
Return to citation in text: [1] -
Metz, A. E.; Ramalingam, K.; Kozlowski, M. C. Tetrahedron Lett. 2015, 56, 5180–5184. doi:10.1016/j.tetlet.2015.07.058
Return to citation in text: [1] -
Lee, H. I.; Cassidy, M. P.; Rashatasakhon, P.; Padwa, A. Org. Lett. 2003, 5, 5067–5070. doi:10.1021/ol036106r
Return to citation in text: [1] -
Padwa, A.; Lee, H. I.; Rashatasakhon, P.; Rose, M. J. Org. Chem. 2004, 69, 8209–8218. doi:10.1021/jo048647f
Return to citation in text: [1] -
Cassidy, M. P.; Özdemir, A. D.; Padwa, A. Org. Lett. 2005, 7, 1339–1342. doi:10.1021/ol0501323
Return to citation in text: [1] -
Rose, M. D.; Cassidy, M. P.; Rashatasakhon, P.; Padwa, A. J. Org. Chem. 2007, 72, 538–549. doi:10.1021/jo0619783
Return to citation in text: [1] -
Ascic, E.; Jensen, J. F.; Nielsen, T. E. Angew. Chem., Int. Ed. 2011, 50, 5188–5191. doi:10.1002/anie.201100417
Return to citation in text: [1] -
Jebali, K.; Planchat, A.; Amri, H.; Mathé-Allainmat, M.; Lebreton, J. Synthesis 2016, 48, 1502–1517. doi:10.1055/s-0035-1561398
Return to citation in text: [1] -
Zhang, W.; Franzén, J. Adv. Synth. Catal. 2010, 352, 499–518. doi:10.1002/adsc.200900686
Return to citation in text: [1] -
Patil, N. T.; Shinde, V. S.; Sridhar, B. Angew. Chem., Int. Ed. 2013, 52, 2251–2255. doi:10.1002/anie.201208738
Return to citation in text: [1] -
Petersen, R.; Le Quement, S. T.; Nielsen, T. E. Angew. Chem., Int. Ed. 2014, 53, 11778–11782. doi:10.1002/anie.201405747
Return to citation in text: [1] -
Nielsen, T. E.; Meldal, M. J. Org. Chem. 2004, 69, 3765–3773. doi:10.1021/jo049918p
Return to citation in text: [1] -
Diness, F.; Beyer, J.; Meldal, M. Chem. – Eur. J. 2006, 12, 8056–8066. doi:10.1002/chem.200600138
Return to citation in text: [1] -
Komnatnyy, V. V.; Givskov, M.; Nielsen, T. E. Chem. – Eur. J. 2012, 18, 16793–16800. doi:10.1002/chem.201202745
Return to citation in text: [1] -
Herz, W.; Tocker, S. J. Am. Chem. Soc. 1955, 77, 3554–3556. doi:10.1021/ja01618a038
Return to citation in text: [1] -
Sano, T.; Toda, J.; Shoda, M.; Yamamoto, R.; Ando, H.; Isobe, K.; Hosoi, S.; Tsuda, Y. Chem. Pharm. Bull. 1992, 40, 3145–3156. doi:10.1248/cpb.40.3145
Return to citation in text: [1] -
Tamayo, N. A.; Bo, Y.; Gore, V.; Ma, V.; Nishimura, N.; Tang, P.; Deng, H.; Klionsky, L.; Lehto, S. G.; Wang, W.; Youngblood, B.; Chen, J.; Correll, T. L.; Bartberger, M. D.; Gavva, N. R.; Norman, M. H. J. Med. Chem. 2012, 55, 1593–1611. doi:10.1021/jm2013634
Return to citation in text: [1] -
Kreituss, I.; Chen, K.-Y.; Eitel, S. H.; Adam, J.-M.; Wuitschik, G.; Fettes, A.; Bode, J. W. Angew. Chem., Int. Ed. 2016, 55, 1553–1556. doi:10.1002/anie.201509256
Return to citation in text: [1] -
Wang, Y.; Huang, W.; Xin, M.; Chen, P.; Gui, L.; Zhao, X.; Tang, F.; Wang, J.; Liu, F. Bioorg. Med. Chem. 2017, 25, 75–83. doi:10.1016/j.bmc.2016.10.011
Return to citation in text: [1] -
Lepovitz, L. T.; Martin, S. F. Tetrahedron 2019, 75, 130637. doi:10.1016/j.tet.2019.130637
Return to citation in text: [1] -
Tan, Q.; Yang, Z.; Jiang, D.; Cheng, Y.; Yang, J.; Xi, S.; Zhang, M. Angew. Chem., Int. Ed. 2019, 58, 6420–6424. doi:10.1002/anie.201902155
Return to citation in text: [1] -
García-Muñoz, M. J.; Foubelo, F.; Yus, M. J. Org. Chem. 2016, 81, 10214–10226. doi:10.1021/acs.joc.6b01047
Return to citation in text: [1] -
Butin, A. V.; Uchuskin, M. G.; Pilipenko, A. S.; Tsiunchik, F. A.; Cheshkov, D. A.; Trushkov, I. V. Eur. J. Org. Chem. 2010, 920–926. doi:10.1002/ejoc.200901241
Return to citation in text: [1] -
Butin, A. V.; Nevolina, T. A.; Shcherbinin, V. A.; Uchuskin, M. G.; Serdyuk, O. V.; Trushkov, I. V. Synthesis 2010, 2969–2978. doi:10.1055/s-0030-1258165
Return to citation in text: [1] -
Uchuskin, M. G.; Molodtsova, N. V.; Lysenko, S. A.; Strel'nikov, V. N.; Trushkov, I. V.; Butin, A. V. Eur. J. Org. Chem. 2014, 2508–2515. doi:10.1002/ejoc.201301762
Return to citation in text: [1] -
Merkushev, A. A.; Strel'nikov, V. N.; Uchuskin, M. G.; Trushkov, I. V. Tetrahedron 2017, 73, 6523–6529. doi:10.1016/j.tet.2017.09.043
Return to citation in text: [1] -
Zelina, E. Y.; Nevolina, T. A.; Sorotskaja, L. N.; Skvortsov, D. A.; Trushkov, I. V.; Uchuskin, M. G. J. Org. Chem. 2018, 83, 11747–11757. doi:10.1021/acs.joc.8b01669
Return to citation in text: [1] -
Lutz, R. E.; Welstead, W. J. J. Am. Chem. Soc. 1963, 85, 755–761. doi:10.1021/ja00889a023
Return to citation in text: [1] -
Popp, F. D.; Schleigh, W. R.; Katz, L. E. J. Chem. Soc. C 1968, 2253–2257. doi:10.1039/j39680002253
Return to citation in text: [1] -
Grochowski, M. R.; Yang, W.; Sen, A. Chem. – Eur. J. 2012, 18, 12363–12371. doi:10.1002/chem.201201522
Return to citation in text: [1]
| 49. | Lutz, R. E.; Welstead, W. J. J. Am. Chem. Soc. 1963, 85, 755–761. doi:10.1021/ja00889a023 |
| 50. | Popp, F. D.; Schleigh, W. R.; Katz, L. E. J. Chem. Soc. C 1968, 2253–2257. doi:10.1039/j39680002253 |
| 51. | Grochowski, M. R.; Yang, W.; Sen, A. Chem. – Eur. J. 2012, 18, 12363–12371. doi:10.1002/chem.201201522 |
| 2. | Naylor, A.; Judd, D. B.; Scopes, D. I. C.; Hayes, A. G.; Birch, P. J. J. Med. Chem. 1994, 37, 2138–2144. doi:10.1021/jm00040a004 |
| 40. | Wang, Y.; Huang, W.; Xin, M.; Chen, P.; Gui, L.; Zhao, X.; Tang, F.; Wang, J.; Liu, F. Bioorg. Med. Chem. 2017, 25, 75–83. doi:10.1016/j.bmc.2016.10.011 |
| 41. | Lepovitz, L. T.; Martin, S. F. Tetrahedron 2019, 75, 130637. doi:10.1016/j.tet.2019.130637 |
| 42. | Tan, Q.; Yang, Z.; Jiang, D.; Cheng, Y.; Yang, J.; Xi, S.; Zhang, M. Angew. Chem., Int. Ed. 2019, 58, 6420–6424. doi:10.1002/anie.201902155 |
| 43. | García-Muñoz, M. J.; Foubelo, F.; Yus, M. J. Org. Chem. 2016, 81, 10214–10226. doi:10.1021/acs.joc.6b01047 |
| 44. | Butin, A. V.; Uchuskin, M. G.; Pilipenko, A. S.; Tsiunchik, F. A.; Cheshkov, D. A.; Trushkov, I. V. Eur. J. Org. Chem. 2010, 920–926. doi:10.1002/ejoc.200901241 |
| 45. | Butin, A. V.; Nevolina, T. A.; Shcherbinin, V. A.; Uchuskin, M. G.; Serdyuk, O. V.; Trushkov, I. V. Synthesis 2010, 2969–2978. doi:10.1055/s-0030-1258165 |
| 46. | Uchuskin, M. G.; Molodtsova, N. V.; Lysenko, S. A.; Strel'nikov, V. N.; Trushkov, I. V.; Butin, A. V. Eur. J. Org. Chem. 2014, 2508–2515. doi:10.1002/ejoc.201301762 |
| 47. | Merkushev, A. A.; Strel'nikov, V. N.; Uchuskin, M. G.; Trushkov, I. V. Tetrahedron 2017, 73, 6523–6529. doi:10.1016/j.tet.2017.09.043 |
| 48. | Zelina, E. Y.; Nevolina, T. A.; Sorotskaja, L. N.; Skvortsov, D. A.; Trushkov, I. V.; Uchuskin, M. G. J. Org. Chem. 2018, 83, 11747–11757. doi:10.1021/acs.joc.8b01669 |
| 1. | Wang, Y.; Huang, W.; Xin, M.; Chen, P.; Gui, L.; Zhao, X.; Zhu, X.; Luo, H.; Cong, X.; Wang, J.; Liu, F. Bioorg. Med. Chem. 2019, 27, 2592–2597. doi:10.1016/j.bmc.2019.03.048 |
| 5. | Van der Mey, M.; Windhorst, A. D.; Klok, R. P.; Herscheid, J. D. M.; Kennis, L. E.; Bischoff, F.; Bakker, M.; Langlois, X.; Heylen, L.; Jurzak, M.; Leysen, J. E. Bioorg. Med. Chem. 2006, 14, 4526–4534. doi:10.1016/j.bmc.2006.02.029 |
| 33. | Nielsen, T. E.; Meldal, M. J. Org. Chem. 2004, 69, 3765–3773. doi:10.1021/jo049918p |
| 34. | Diness, F.; Beyer, J.; Meldal, M. Chem. – Eur. J. 2006, 12, 8056–8066. doi:10.1002/chem.200600138 |
| 35. | Komnatnyy, V. V.; Givskov, M.; Nielsen, T. E. Chem. – Eur. J. 2012, 18, 16793–16800. doi:10.1002/chem.201202745 |
| 4. | Chang, Z. Q.; Liu, F.; Wang, S. L.; Zhao, T. Z.; Wang, M. T. Yaoxue Xuebao 1981, 16, 394–396. |
| 36. | Herz, W.; Tocker, S. J. Am. Chem. Soc. 1955, 77, 3554–3556. doi:10.1021/ja01618a038 |
| 37. | Sano, T.; Toda, J.; Shoda, M.; Yamamoto, R.; Ando, H.; Isobe, K.; Hosoi, S.; Tsuda, Y. Chem. Pharm. Bull. 1992, 40, 3145–3156. doi:10.1248/cpb.40.3145 |
| 38. | Tamayo, N. A.; Bo, Y.; Gore, V.; Ma, V.; Nishimura, N.; Tang, P.; Deng, H.; Klionsky, L.; Lehto, S. G.; Wang, W.; Youngblood, B.; Chen, J.; Correll, T. L.; Bartberger, M. D.; Gavva, N. R.; Norman, M. H. J. Med. Chem. 2012, 55, 1593–1611. doi:10.1021/jm2013634 |
| 39. | Kreituss, I.; Chen, K.-Y.; Eitel, S. H.; Adam, J.-M.; Wuitschik, G.; Fettes, A.; Bode, J. W. Angew. Chem., Int. Ed. 2016, 55, 1553–1556. doi:10.1002/anie.201509256 |
| 3. | Zhang, W.; Liu, L.-l.; Lun, S.; Wang, S.-S.; Xiao, S.; Gunosewoyo, H.; Yang, F.; Tang, J.; Bishai, W. R.; Yu, L.-F. Eur. J. Med. Chem. 2021, 213, 113202. doi:10.1016/j.ejmech.2021.113202 |
| 24. | Lee, H. I.; Cassidy, M. P.; Rashatasakhon, P.; Padwa, A. Org. Lett. 2003, 5, 5067–5070. doi:10.1021/ol036106r |
| 25. | Padwa, A.; Lee, H. I.; Rashatasakhon, P.; Rose, M. J. Org. Chem. 2004, 69, 8209–8218. doi:10.1021/jo048647f |
| 26. | Cassidy, M. P.; Özdemir, A. D.; Padwa, A. Org. Lett. 2005, 7, 1339–1342. doi:10.1021/ol0501323 |
| 27. | Rose, M. D.; Cassidy, M. P.; Rashatasakhon, P.; Padwa, A. J. Org. Chem. 2007, 72, 538–549. doi:10.1021/jo0619783 |
| 28. | Ascic, E.; Jensen, J. F.; Nielsen, T. E. Angew. Chem., Int. Ed. 2011, 50, 5188–5191. doi:10.1002/anie.201100417 |
| 29. | Jebali, K.; Planchat, A.; Amri, H.; Mathé-Allainmat, M.; Lebreton, J. Synthesis 2016, 48, 1502–1517. doi:10.1055/s-0035-1561398 |
| 2. | Naylor, A.; Judd, D. B.; Scopes, D. I. C.; Hayes, A. G.; Birch, P. J. J. Med. Chem. 1994, 37, 2138–2144. doi:10.1021/jm00040a004 |
| 30. | Zhang, W.; Franzén, J. Adv. Synth. Catal. 2010, 352, 499–518. doi:10.1002/adsc.200900686 |
| 31. | Patil, N. T.; Shinde, V. S.; Sridhar, B. Angew. Chem., Int. Ed. 2013, 52, 2251–2255. doi:10.1002/anie.201208738 |
| 32. | Petersen, R.; Le Quement, S. T.; Nielsen, T. E. Angew. Chem., Int. Ed. 2014, 53, 11778–11782. doi:10.1002/anie.201405747 |
| 13. | Sun, Y.-W.; Tang, X.-Y.; Shi, M. Chem. Commun. 2015, 51, 13937–13940. doi:10.1039/c5cc05808b |
| 17. | Calcaterra, A.; Mangiardi, L.; Delle Monache, G.; Quaglio, D.; Balducci, S.; Berardozzi, S.; Iazzetti, A.; Franzini, R.; Botta, B.; Ghirga, F. Molecules 2020, 25, 414. doi:10.3390/molecules25020414 |
| 18. | Gholamzadeh, P. Adv. Heterocycl. Chem. 2019, 127, 153–226. doi:10.1016/bs.aihch.2018.09.002 |
| 12. | Fuchs, J. R.; Funk, R. L. Org. Lett. 2001, 3, 3349–3351. doi:10.1021/ol016592n |
| 19. | Kano, S.; Yuasa, Y.; Yokomatsu, T.; Shibuya, S. Heterocycles 1984, 21, 701. doi:10.3987/s-1984-02-0701 |
| 20. | Tanis, S. P.; Deaton, M. V.; Dixon, L. A.; McMills, M. C.; Raggon, J. W.; Collins, M. A. J. Org. Chem. 1998, 63, 6914–6928. doi:10.1021/jo980718l |
| 21. | Gao, S.; Tu, Y. Q.; Hu, X.; Wang, S.; Hua, R.; Jiang, Y.; Zhao, Y.; Fan, X.; Zhang, S. Org. Lett. 2006, 8, 2373–2376. doi:10.1021/ol0607185 |
| 22. | Shengule, S. R.; Ryder, G.; Willis, A. C.; Pyne, S. G. Tetrahedron 2012, 68, 10280–10285. doi:10.1016/j.tet.2012.10.014 |
| 23. | Metz, A. E.; Ramalingam, K.; Kozlowski, M. C. Tetrahedron Lett. 2015, 56, 5180–5184. doi:10.1016/j.tetlet.2015.07.058 |
| 9. | Schneider, C. S.; Pook, K. H. J. Chem. Soc., Perkin Trans. 1 1986, 877–883. doi:10.1039/p19860000877 |
| 10. | Schafroth, M. A.; Rummelt, S. M.; Sarlah, D.; Carreira, E. M. Org. Lett. 2017, 19, 3235–3238. doi:10.1021/acs.orglett.7b01346 |
| 11. | Davies, S. G.; Fletcher, A. M.; Holder, K. E.; Roberts, P. M.; Thomson, J. E.; Zimmer, D. Heterocycles 2019, 99, 919–941. doi:10.3987/com-18-s(f)59 |
| 6. | Nagai, Y.; Uno, H.; Umemoto, S. Chem. Pharm. Bull. 1977, 25, 1911–1922. doi:10.1248/cpb.25.1911 |
| 7. | Imanishi, T.; Yagi, N.; Hanaoka, M. Tetrahedron Lett. 1981, 22, 667–670. doi:10.1016/s0040-4039(01)92518-3 |
| 8. | Imanishi, T.; Yagi, N.; Hanaoka, M. Chem. Pharm. Bull. 1983, 31, 1243–1253. doi:10.1248/cpb.31.1243 |
| 14. | Baba, T.; Oka, J.; Noguchi, K.; Tanaka, K. Eur. J. Org. Chem. 2015, 4374–4382. doi:10.1002/ejoc.201500486 |
| 15. | He, Y.; Li, Z.; Robeyns, K.; Van Meervelt, L.; Van der Eycken, E. V. Angew. Chem., Int. Ed. 2018, 57, 272–276. doi:10.1002/anie.201710592 |
| 16. | He, Y.; Wu, D.; Li, Z.; Robeyns, K.; Van Meervelt, L.; Van der Eycken, E. V. Org. Biomol. Chem. 2019, 17, 6284–6292. doi:10.1039/c9ob01299k |
© 2023 Mendogralo and Uchuskin; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.