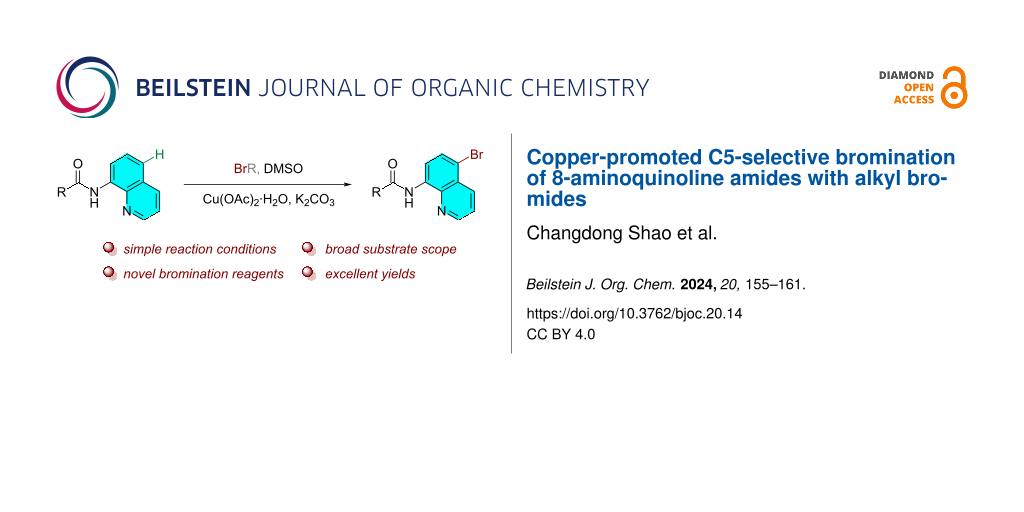
Abstract
An efficient and practical method for the synthesis of C5-brominated 8-aminoquinoline amides via a copper-promoted selective bromination of 8-aminoquinoline amides with alkyl bromides was developed. The reaction proceeds smoothly in dimethyl sulfoxide (DMSO) under air, employing activated and unactivated alkyl bromides as the halogenation reagents without additional external oxidants. This method features outstanding site selectivity, broad substrate scope, and excellent yields.

Graphical Abstract
Introduction
Over the past decades, the 8-aminoquinoline motif that could be found in several natural products [1] has attracted significant attention for its widespread usage in pharmaceuticals [2], agrochemicals [3], and functional materials [4].
In particular, the aminoquinoline scaffold has emerged as an important auxiliary group for the proximal C–H activation with the efforts of Daugulis [5] and others [6]. Results from medical research indicated that the introduction of halogen atoms into quinoline motifs has positive effects on their bioactivities, such as antimalarial, antitumor, and so on [7]. Therefore, it is of great importance to develop syntheses for 8-aminoquinoline derivatives with diverse substituent groups, especially those leading to halogenated derivatives.
Recent years have witnessed much progress in halogenation reactions of the quinoline ring in the C2–C7 positions [8,9]. Among them, the synthesis of important C5-halogenated products gained particular attention since Stahl et al. reported the first chlorination example using LiCl as the halogen source [10]. Following this pioneering work, elegant strategies for the C5–H bromination of the quinoline ring employing simple inorganic and organic bromine-containing compounds such as Br2, CuBr, CuBr2, LiBr, NaBr, KBr, HBr, NH4Br, and tetrabutylammonium bromide (TBAB) as halogen sources have been realized (Scheme 1, reaction 1) [11-20]. Another category of extensively used bromination reagents are brominated imides, such as N-bromosuccinimide (NBS), N-bromosaccharin (NBSA), tribromoisocyanuric acid (TBCA), 1,3-dibromo-5,5-dimethylhydantoin (DBDMH), and so on (Scheme 1, reaction 2) [21-24]. However, reports on the bromination of C5–H of 8-aminoquinolines employing acyl bromides, alkyl bromides, and aryl bromides as bromination reagents are limited. Wan and Li, respectively, demonstrated a few examples of a one-pot N-acylation and C5–H bromination of 8‑aminoquinolines using acyl bromines acting as both acyl and halide donors [25,26]. The groups of Lei and Fang independently realized the selective C5-bromination of 8-aminoquinoline amides using carbon tetrabromide and dibromomethane under photo- and electrocatalysis conditions [27,28]. In 2017, Xia and co-workers reported a novel, mild, metal-free, and regioselective bromination of amides, wherein the organic dye eosin Y acted as the bromine source in combination with Selectfluor® (Scheme 1, reaction 3) [29].
![[1860-5397-20-14-i1]](/bjoc/content/inline/1860-5397-20-14-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Methods for the C5-selective bromination of 8-aminoquinoline amides.
Scheme 1: Methods for the C5-selective bromination of 8-aminoquinoline amides.
Despite significant progress in this area, most of these methodologies still suffer from the use of external oxidants, complex reaction equipment, and expensive and/or toxic halogen sources, which limit the practicality for large-scale use. Therefore, novel bromination reagents and simple bromination approaches are still required to be established. Very recently, we reported a copper-catalyzed C5-bromination and difluoromethylation reaction of 8-aminoquinolines using ethyl bromodifluoroacetate as bifunctional reagent [30] and the feasibility of using activated alkyl bromides as bromination reagents has been preliminarily demonstrated. To further expand the scope of bromination reagents and enhance the utility of the reaction method, herein, we wish to report a simple and efficient copper-promoted C5-selective bromination reaction of 8-aminoquinoline amides using activated and unactivated alkyl bromides as the bromine source (Scheme 1, reaction 4).
Results and Discussion
At the beginning of this investigation, N-(quinolin-8-yl)benzamide (1a) and ethyl bromoacetate (2a) were selected as model substrates to screen the reaction parameters (Table 1). The treatment of 1a with 2a (4.0 equiv) in the presence of FeCl3 (20 mol %) and K3PO4 (1.0 equiv) in DMSO at 100 °C for 12 h gave the brominated product 3aa in 65% yield (Table 1, entry 1). The bromination was found to selectively take place at the C5-position of the quinoline ring of 1a in this reaction. Other competitive site-selective C–H bromination products and multiple brominated products were not observed. Subsequently, the bromination reaction was examined with various catalysts such as CoCl2·6H2O, Ni(OAc)2·4H2O, MnSO4·H2O, CuCl, CuBr, CuCl2, CuBr2, and Cu(OAc)2·H2O (Table 1, entries 2–9). To our delight, copper salts were effective, giving the desired product 3aa in excellent yields of 88–95% (Table 1, entries 7–9). Cuprous salts, cobalt chloride, and nickel acetate were partially efficient for the reaction, providing product 3aa in 85% yield (Table 1, entries 3–6). The catalytic efficiency of MnSO4·H2O was consistent with FeCl3, affording 3aa in 64% yield (Table 1, entry 2). The reaction was further examined with a series of bases such as Li2CO3, Na2CO3, K2CO3, Cs2CO3, K2HPO4, KHCO3, KH2PO4, and KOAc, and the results demonstrated that the reaction proceeds under alkaline conditions (Table 1, entries 10–17). Among the bases screened, K2CO3 was the most effective one, yielding the desired product 3aa quantitatively (Table 1, entry 12). Other bases, such as Li2CO3, Na2CO3, Cs2CO3, and K2HPO4, were also very efficient, giving the product in excellent yields (Table 1, entries 10, 11, 13, and 14). The other bases were found to be less effective (Table 1, entries 15–17). Notably, the bromination reaction was still efficient with a lower catalyst loading (10 mol %) and lower base loading (0.5 equiv), respectively (Table 1, entries 18 and 19). However, the amount of alkyl bromide and temperature affected the reaction significantly (Table 1, entries 20 and 21). Finally, control experiments demonstrated that copper promoted the transformation and a base was the indispensable factor for the reaction (Table 1, entries 22–24). Therefore, a facile and highly efficient C5-bromination protocol has been established.
Table 1: Optimization of the reaction conditions for the copper-promoted C5-brominationa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-14-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst | Base | Yield (%)b |
| 1 | FeCl3 | K3PO4 | 65 |
| 2 | MnSO4·H2O | K3PO4 | 64 |
| 3 | CoCl2·6H2O | K3PO4 | 85 |
| 4 | Ni(OAc)2·4H2O | K3PO4 | 85 |
| 5 | CuCl | K3PO4 | 85 |
| 6 | CuBr | K3PO4 | 85 |
| 7 | CuCl2 | K3PO4 | 92 |
| 8 | CuBr2 | K3PO4 | 88 |
| 9 | Cu(OAc)2·H2O | K3PO4 | 95 |
| 10 | Cu(OAc)2·H2O | Li2CO3 | 93 |
| 11 | Cu(OAc)2·H2O | Na2CO3 | 97 |
| 12 | Cu(OAc)2·H2O | K2CO3 | 100 (99) |
| 13 | Cu(OAc)2·H2O | Cs2CO3 | 96 |
| 14 | Cu(OAc)2·H2O | K2HPO4 | 96 |
| 15 | Cu(OAc)2·H2O | KHCO3 | 84 |
| 16 | Cu(OAc)2·H2O | KH2PO4 | 70 |
| 17 | Cu(OAc)2·H2O | KOAc | 56 |
| 18c | Cu(OAc)2·H2O | K2CO3 | 93 |
| 19d | Cu(OAc)2·H2O | K2CO3 | 94 |
| 20e | Cu(OAc)2·H2O | K2CO3 | 76 |
| 21f | Cu(OAc)2·H2O | K2CO3 | 31 |
| 22 | – | K3PO4 | 52 |
| 23 | Cu(OAc)2·H2O | – | – |
| 24 | – | – | – |
aReaction conditions: 1a (0.2 mmol), 2a (0.8 mmol), catalyst (20 mol %), base (0.2 mmol), DMSO (1.0 mL), stirred under air in a 35 mL sealed tube. b1H NMR yield with dibromomethane as the internal standard, isolated yield in parentheses. cCatalyst loading was 10 mol %. d0.1 mmol K2CO3 was used. e0.6 mmol 2a was used. fStirred at 80 °C.
Having identified the optimal reaction conditions for the bromination of N-(quinolin-8-yl)benzamide (1a) with ethyl bromoacetate (2a) (Table 1, entry 12), we next examined the substrate scope and limitations of our method with an array of 8-aminoquinoline amides, and the results are illustrated in Scheme 2. A wide variety of 8-aminoquinoline amides with different substitutions efficiently participated in this mild and versatile bromination. Benzamides with both electron-donating groups (3ba–ca) and electron-withdrawing groups (3da–fa) were well tolerated, affording the desired products in excellent yields (92‒99%). These results indicated that the electronic effect of substituents affected the reaction only slightly. Products with substituents with derivatization feasibilities such as halogen (3ga–ia), acetyloxy (3ja), and ester groups (3ka) also were obtained in high yields (92‒99%), which demonstrated the practical value of this methodology. Moreover, the reaction showed a good tolerance to sterically hindered substrates like trimethylbenzamide, affording the corresponding brominated product in 70% yield (3la). Also the substrate derived from trimethylgallic acid bearing multiple methoxy groups afforded the desired product 3ma in excellent yield (99%). Noteworthy to mention, substrates containing naphthalene, furan, and thiophene rings also efficiently underwent reaction leading to the quinoline C5-brominated products in excellent yields (3na–pa). In contrast, side products, substituted on the naphthalene, furan, or thiophene ring were not detected. This protocol was also compatible with linear, branched, and cyclic aliphatic acid-derived substrates (3qa–va). Surprisingly, small ring-containing substrates displaying significant ring strain were stable under the reaction conditions and afforded the bromination products 3ua and 3va in 93 and 98% yield. Finally, some other acyl motifs were investigated (3wa–za) and the results showed that the protocol could be successfully applied to sulfonamides, albeit giving the target product 3za in only 53% yield.
![[1860-5397-20-14-i2]](/bjoc/content/inline/1860-5397-20-14-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of the 8-aminoquinoline amides. Reaction conditions: 1 (0.2 mmol), 2a (0.8 mmol), Cu(OAc)2·H2O (20 mol %), K2CO3 (0.2 mmol), DMSO (1.0 mL), stirred under air at 100 ºC for 12 h. Isolated yield.
Scheme 2: Substrate scope of the 8-aminoquinoline amides. Reaction conditions: 1 (0.2 mmol), 2a (0.8 mmol), C...
The scope of the bromination reaction was further extended with various alkyl bromides. As shown in Scheme 3, a series of activated alkyl bromides containing ester, difluoromethylene, benzyl motifs (2b–f), and unactivated alkyl bromides (2g–i) were evaluated in this reaction. Activated alkyl bromides such as 2d, 2e, and 2f performed well, affording the brominated product 3aa in excellent yields (90–99%). However, transformations of amide 1a to the brominated product 3aa employing unactivated alkyl bromides (2g–i) as reaction partners proceeded with low efficiency (0–53%). Notably, the reactivity of primary alkyl bromides is higher than that of secondary alkyl bromides, while the reactivity of tertiary alkyl bromides is the lowest (2a–c, 2g–i). Finally, dibromomethane (2j) proceeded well in the reaction, furnishing 3aa in 65% yield.
![[1860-5397-20-14-i3]](/bjoc/content/inline/1860-5397-20-14-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of the bromoalkanes. Reaction conditions: 1a (0.2 mmol), 2 (0.8 mmol), Cu(OAc)2·H2O (20 mol %), K2CO3 (0.2 mmol), DMSO (1.0 mL), stirred under air at 100 ºC for 12 h. Isolated yield.
Scheme 3: Substrate scope of the bromoalkanes. Reaction conditions: 1a (0.2 mmol), 2 (0.8 mmol), Cu(OAc)2·H2O...
As showcased in Scheme 4, this methodology is also applicable to substrates containing substituents on the quinoline ring. For example, the 6-OMe-substituted quinoline derivative N-(6-methoxyquinolin-8-yl)benzamide (4) underwent bromination at the C5 position to give 5 in nearly quantitative yield (Scheme 4, reaction 1). Subsequently, we attempted to apply this method to chlorination and iodination reactions using ethyl chlorodifluoroacetate (6) and 1-iodobutane (8) as the respective halogenation reagents. However, these attempts ended in failure (Scheme 4, reactions 2 and 3). Furthermore, to demonstrate the synthetic usefulness of the protocol for industrial production, a gram-scale preparation was carried out using 1a, that afforded the desired product in 96% yield (Scheme 4, reaction 4).
![[1860-5397-20-14-i4]](/bjoc/content/inline/1860-5397-20-14-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Further substrate scope investigations and gram-scale application.
Scheme 4: Further substrate scope investigations and gram-scale application.
To gain more insight into the reaction mechanism, several control experiments were carried out (Scheme 5). On one hand, the failure of substrates 10–15 to participate in the reaction indicated that both the acylamino and quinoline N motifs played a significant role. On the other hand, the stoichiometric amount of free radical inhibitors, including TEMPO and BHT, could not comprehensively suppress the reaction. Based on these experimental results and previous works [30-32], a probable mechanism is proposed. As shown in Scheme 5, ethyl bromoacetate (2a) undergoes attack by the dipolar aprotic solvent DMSO to afford the intermediate A. This intermediate then reacts with the bromine anion to give intermediate B. Dimethylsulfonium bromide or dimethyl thioether/molecular bromine intermediate C is then generated, followed by the combination of the bromine anion with intermediate B. Finally, selective C5 bromination is accomplished via aromatic electrophilic substitution of 1a with intermediate C promoted by the copper catalyst to afford the desired product 3aa.
![[1860-5397-20-14-i5]](/bjoc/content/inline/1860-5397-20-14-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Control experiments and proposed mechanism.
Scheme 5: Control experiments and proposed mechanism.
Conclusion
In summary, we have developed an efficient and practical method for the synthesis of C5-brominated 8-aminoquinoline amides in good to excellent yields via a copper-promoted selective bromination using alkyl bromides as the bromide source. This methodology is scalable, proceeds well with various aromatic and aliphatic amide substrates, and proceeds well with activated and unactivated alkyl bromides. Further studies on mechanistic details and the persistent exploration of halogen sources are ongoing in our laboratory.
Experimental
A 35 mL sealed tube equipped with a stirring bar was charged with 8-amidequinolines (1, 0.2 mmol, 1.0 equiv), BrR (2, 0.8 mmol, 4.0 equiv), Cu(OAc)2·H2O (8.0 mg, 0.04 mmol, 20 mol %), K2CO3 (27.6 mg, 0.2 mmol, 1.0 equiv), and DMSO (1.0 mL). The tube was sealed with a Teflon cap under air, then the mixture was stirred at 100 ºC for 12 h. After completion, the reaction mixture was diluted with ethyl acetate (20 mL) and washed with saturated sodium bicarbonate and brine successively. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified on preparative thin-layer chromatography (PTLC) to afford the desired product 3.
Supporting Information
| Supporting Information File 1: General information, experimental procedures for all the products, characterization data, and NMR spectra. | ||
| Format: PDF | Size: 3.2 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Michael, J. P. Nat. Prod. Rep. 2008, 25, 166–187. doi:10.1039/b612168n
Return to citation in text: [1] -
Solomon, V. R.; Lee, H. Curr. Med. Chem. 2011, 18, 1488–1508. doi:10.2174/092986711795328382
Return to citation in text: [1] -
Bond, J. A.; Walker, T. W. Weed Technol. 2012, 26, 183–188. doi:10.1614/wt-d-11-00136.1
Return to citation in text: [1] -
Mohamad, N. S.; Zakaria, N. H.; Daud, N.; Tan, L. L.; Ta, G. C.; Heng, L. Y.; Hassan, N. I. Sensors 2021, 21, 311. doi:10.3390/s21010311
Return to citation in text: [1] -
Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154–13155. doi:10.1021/ja054549f
Return to citation in text: [1] -
Rouquet, G.; Chatani, N. Angew. Chem., Int. Ed. 2013, 52, 11726–11743. doi:10.1002/anie.201301451
Return to citation in text: [1] -
Kuang, W.-B.; Huang, R.-Z.; Fang, Y.-L.; Liang, G.-B.; Yang, C.-H.; Ma, X.-L.; Zhang, Y. RSC Adv. 2018, 8, 24376–24385. doi:10.1039/c8ra04640a
Return to citation in text: [1] -
Xu, Z.; Yang, X.; Yin, S.-F.; Qiu, R. Top. Curr. Chem. 2020, 378, 42. doi:10.1007/s41061-020-00303-9
Return to citation in text: [1] -
Zhu, L.; Cao, X.; Li, Y.; Liu, T.; Wang, X.; Qiu, R.; Yin, S. Chin. J. Org. Chem. 2017, 37, 1613. doi:10.6023/cjoc201703020
Return to citation in text: [1] -
Suess, A. M.; Ertem, M. Z.; Cramer, C. J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 9797–9804. doi:10.1021/ja4026424
Return to citation in text: [1] -
Long, Y.; Pan, L.; Zhou, X. Molecules 2019, 24, 535. doi:10.3390/molecules24030535
Return to citation in text: [1] -
He, X.; Xu, Y.-z.; Kong, L.-x.; Wu, H.-h.; Ji, D.-z.; Wang, Z.-b.; Xu, Y.-g.; Zhu, Q.-h. Org. Chem. Front. 2017, 4, 1046–1050. doi:10.1039/c6qo00644b
Return to citation in text: [1] -
Zhu, L.; Qiu, R.; Cao, X.; Xiao, S.; Xu, X.; Au, C.-T.; Yin, S.-F. Org. Lett. 2015, 17, 5528–5531. doi:10.1021/acs.orglett.5b02511
Return to citation in text: [1] -
Rao, N. S.; Reddy, G. M.; Sridhar, B.; Sarma, M. H. Eur. J. Org. Chem. 2017, 438–442. doi:10.1002/ejoc.201601151
Return to citation in text: [1] -
Chen, X.-X.; Wang, J.-X.; Ren, J.-T.; Xie, H.; Zhao, Y.; Li, Y.-M.; Sun, M. Synlett 2017, 28, 1840–1844. doi:10.1055/s-0036-1588420
Return to citation in text: [1] -
Jiao, J.-Y.; Mao, Y.-J.; Feng, A.-W.; Li, X.-F.; Li, M.-T.; Zhang, X.-H. Tetrahedron 2017, 73, 1482–1488. doi:10.1016/j.tet.2017.01.060
Return to citation in text: [1] -
Qiao, H.; Sun, S.; Yang, F.; Zhu, Y.; Kang, J.; Wu, Y.; Wu, Y. Adv. Synth. Catal. 2017, 359, 1976–1980. doi:10.1002/adsc.201601053
Return to citation in text: [1] -
Qiao, L.; Cao, X.; Chai, K.; Shen, J.; Xu, J.; Zhang, P. Tetrahedron Lett. 2018, 59, 2243–2247. doi:10.1016/j.tetlet.2018.04.036
Return to citation in text: [1] -
Yang, X.; Yang, Q.-L.; Wang, X.-Y.; Xu, H.-H.; Mei, T.-S.; Huang, Y.; Fang, P. J. Org. Chem. 2020, 85, 3497–3507. doi:10.1021/acs.joc.9b03223
Return to citation in text: [1] -
Zhao, H.-Y.; Yang, X.-Y.; Lei, H.; Xin, M.; Zhang, S.-Q. Synth. Commun. 2019, 49, 1406–1415. doi:10.1080/00397911.2019.1598558
Return to citation in text: [1] -
Li, Y.; Zhu, L.; Cao, X.; Au, C.-T.; Qiu, R.; Yin, S.-F. Adv. Synth. Catal. 2017, 359, 2864–2873. doi:10.1002/adsc.201700391
Return to citation in text: [1] -
Dutta, H. S.; Khan, B.; Khan, A. A.; Raziullah; Ahmad, A.; Kant, R.; Koley, D. ChemistrySelect 2017, 2, 6488–6492. doi:10.1002/slct.201701649
Return to citation in text: [1] -
Motati, D. R.; Uredi, D.; Watkins, E. B. Chem. Sci. 2018, 9, 1782–1788. doi:10.1039/c7sc04107a
Return to citation in text: [1] -
He, X.; Jiang, Y.; Zhou, H.; Zhu, Q. ChemistrySelect 2021, 6, 5191–5194. doi:10.1002/slct.202100616
Return to citation in text: [1] -
Du, Y.; Liu, Y.; Wan, J.-P. J. Org. Chem. 2018, 83, 3403–3408. doi:10.1021/acs.joc.8b00068
Return to citation in text: [1] -
Li, D.; Jia, Z.; Jiang, Y.; Jia, J.; Zhao, X.; Li, Z.; Xu, Z. ChemistrySelect 2019, 4, 13964–13967. doi:10.1002/slct.201904286
Return to citation in text: [1] -
Ma, B.; Lu, F.; Yang, H.; Gu, X.; Li, Z.; Li, R.; Pei, H.; Luo, D.; Zhang, H.; Lei, A. Asian J. Org. Chem. 2019, 8, 1136–1140. doi:10.1002/ajoc.201900293
Return to citation in text: [1] -
Lin, X.; Zeng, C.; Liu, C.; Fang, Z.; Guo, K. Org. Biomol. Chem. 2021, 19, 1352–1357. doi:10.1039/d0ob02055a
Return to citation in text: [1] -
Huang, B.; Zhao, Y.; Yang, C.; Gao, Y.; Xia, W. Org. Lett. 2017, 19, 3799–3802. doi:10.1021/acs.orglett.7b01427
Return to citation in text: [1] -
Shao, C.; Xu, T.; Chen, C.; Yang, Q.; Tang, C.; Chen, P.; Lu, M.; Hu, Z.; Hu, H.; Zhang, T. RSC Adv. 2023, 13, 6993–6999. doi:10.1039/d3ra00088e
Return to citation in text: [1] [2] -
Li, J.-Q.; Tan, H.-L.; Ma, D.-D.; Zhu, X.-X.; Cui, H.-L. J. Org. Chem. 2021, 86, 10118–10128. doi:10.1021/acs.joc.1c00844
Return to citation in text: [1] -
Liang, Y.-F.; Wu, K.; Song, S.; Li, X.; Huang, X.; Jiao, N. Org. Lett. 2015, 17, 876–879. doi:10.1021/ol5037387
Return to citation in text: [1]
| 5. | Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154–13155. doi:10.1021/ja054549f |
| 30. | Shao, C.; Xu, T.; Chen, C.; Yang, Q.; Tang, C.; Chen, P.; Lu, M.; Hu, Z.; Hu, H.; Zhang, T. RSC Adv. 2023, 13, 6993–6999. doi:10.1039/d3ra00088e |
| 4. | Mohamad, N. S.; Zakaria, N. H.; Daud, N.; Tan, L. L.; Ta, G. C.; Heng, L. Y.; Hassan, N. I. Sensors 2021, 21, 311. doi:10.3390/s21010311 |
| 30. | Shao, C.; Xu, T.; Chen, C.; Yang, Q.; Tang, C.; Chen, P.; Lu, M.; Hu, Z.; Hu, H.; Zhang, T. RSC Adv. 2023, 13, 6993–6999. doi:10.1039/d3ra00088e |
| 31. | Li, J.-Q.; Tan, H.-L.; Ma, D.-D.; Zhu, X.-X.; Cui, H.-L. J. Org. Chem. 2021, 86, 10118–10128. doi:10.1021/acs.joc.1c00844 |
| 32. | Liang, Y.-F.; Wu, K.; Song, S.; Li, X.; Huang, X.; Jiao, N. Org. Lett. 2015, 17, 876–879. doi:10.1021/ol5037387 |
| 3. | Bond, J. A.; Walker, T. W. Weed Technol. 2012, 26, 183–188. doi:10.1614/wt-d-11-00136.1 |
| 27. | Ma, B.; Lu, F.; Yang, H.; Gu, X.; Li, Z.; Li, R.; Pei, H.; Luo, D.; Zhang, H.; Lei, A. Asian J. Org. Chem. 2019, 8, 1136–1140. doi:10.1002/ajoc.201900293 |
| 28. | Lin, X.; Zeng, C.; Liu, C.; Fang, Z.; Guo, K. Org. Biomol. Chem. 2021, 19, 1352–1357. doi:10.1039/d0ob02055a |
| 2. | Solomon, V. R.; Lee, H. Curr. Med. Chem. 2011, 18, 1488–1508. doi:10.2174/092986711795328382 |
| 29. | Huang, B.; Zhao, Y.; Yang, C.; Gao, Y.; Xia, W. Org. Lett. 2017, 19, 3799–3802. doi:10.1021/acs.orglett.7b01427 |
| 10. | Suess, A. M.; Ertem, M. Z.; Cramer, C. J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 9797–9804. doi:10.1021/ja4026424 |
| 21. | Li, Y.; Zhu, L.; Cao, X.; Au, C.-T.; Qiu, R.; Yin, S.-F. Adv. Synth. Catal. 2017, 359, 2864–2873. doi:10.1002/adsc.201700391 |
| 22. | Dutta, H. S.; Khan, B.; Khan, A. A.; Raziullah; Ahmad, A.; Kant, R.; Koley, D. ChemistrySelect 2017, 2, 6488–6492. doi:10.1002/slct.201701649 |
| 23. | Motati, D. R.; Uredi, D.; Watkins, E. B. Chem. Sci. 2018, 9, 1782–1788. doi:10.1039/c7sc04107a |
| 24. | He, X.; Jiang, Y.; Zhou, H.; Zhu, Q. ChemistrySelect 2021, 6, 5191–5194. doi:10.1002/slct.202100616 |
| 8. | Xu, Z.; Yang, X.; Yin, S.-F.; Qiu, R. Top. Curr. Chem. 2020, 378, 42. doi:10.1007/s41061-020-00303-9 |
| 9. | Zhu, L.; Cao, X.; Li, Y.; Liu, T.; Wang, X.; Qiu, R.; Yin, S. Chin. J. Org. Chem. 2017, 37, 1613. doi:10.6023/cjoc201703020 |
| 25. | Du, Y.; Liu, Y.; Wan, J.-P. J. Org. Chem. 2018, 83, 3403–3408. doi:10.1021/acs.joc.8b00068 |
| 26. | Li, D.; Jia, Z.; Jiang, Y.; Jia, J.; Zhao, X.; Li, Z.; Xu, Z. ChemistrySelect 2019, 4, 13964–13967. doi:10.1002/slct.201904286 |
| 7. | Kuang, W.-B.; Huang, R.-Z.; Fang, Y.-L.; Liang, G.-B.; Yang, C.-H.; Ma, X.-L.; Zhang, Y. RSC Adv. 2018, 8, 24376–24385. doi:10.1039/c8ra04640a |
| 6. | Rouquet, G.; Chatani, N. Angew. Chem., Int. Ed. 2013, 52, 11726–11743. doi:10.1002/anie.201301451 |
| 11. | Long, Y.; Pan, L.; Zhou, X. Molecules 2019, 24, 535. doi:10.3390/molecules24030535 |
| 12. | He, X.; Xu, Y.-z.; Kong, L.-x.; Wu, H.-h.; Ji, D.-z.; Wang, Z.-b.; Xu, Y.-g.; Zhu, Q.-h. Org. Chem. Front. 2017, 4, 1046–1050. doi:10.1039/c6qo00644b |
| 13. | Zhu, L.; Qiu, R.; Cao, X.; Xiao, S.; Xu, X.; Au, C.-T.; Yin, S.-F. Org. Lett. 2015, 17, 5528–5531. doi:10.1021/acs.orglett.5b02511 |
| 14. | Rao, N. S.; Reddy, G. M.; Sridhar, B.; Sarma, M. H. Eur. J. Org. Chem. 2017, 438–442. doi:10.1002/ejoc.201601151 |
| 15. | Chen, X.-X.; Wang, J.-X.; Ren, J.-T.; Xie, H.; Zhao, Y.; Li, Y.-M.; Sun, M. Synlett 2017, 28, 1840–1844. doi:10.1055/s-0036-1588420 |
| 16. | Jiao, J.-Y.; Mao, Y.-J.; Feng, A.-W.; Li, X.-F.; Li, M.-T.; Zhang, X.-H. Tetrahedron 2017, 73, 1482–1488. doi:10.1016/j.tet.2017.01.060 |
| 17. | Qiao, H.; Sun, S.; Yang, F.; Zhu, Y.; Kang, J.; Wu, Y.; Wu, Y. Adv. Synth. Catal. 2017, 359, 1976–1980. doi:10.1002/adsc.201601053 |
| 18. | Qiao, L.; Cao, X.; Chai, K.; Shen, J.; Xu, J.; Zhang, P. Tetrahedron Lett. 2018, 59, 2243–2247. doi:10.1016/j.tetlet.2018.04.036 |
| 19. | Yang, X.; Yang, Q.-L.; Wang, X.-Y.; Xu, H.-H.; Mei, T.-S.; Huang, Y.; Fang, P. J. Org. Chem. 2020, 85, 3497–3507. doi:10.1021/acs.joc.9b03223 |
| 20. | Zhao, H.-Y.; Yang, X.-Y.; Lei, H.; Xin, M.; Zhang, S.-Q. Synth. Commun. 2019, 49, 1406–1415. doi:10.1080/00397911.2019.1598558 |
© 2024 Shao et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.