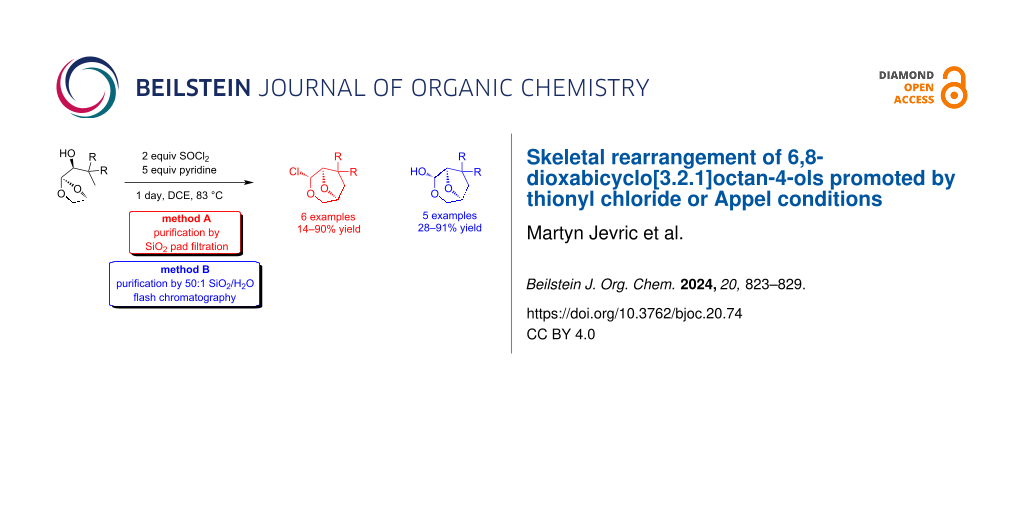
Abstract
A skeletal rearrangement of a series of 6,8-dioxabicyclo[3.2.1]octan-4-ols has been developed using SOCl2 in the presence of pyridine. An oxygen migration from C5 to C4 was observed when the C4 alcohols were treated with SOCl2/pyridine, giving a 2-chloro-3,8-dioxabicyclo[3.2.1]octane ring-system via the chlorosulfite intermediate. Analogous allylic alcohols with endocyclic and exocyclic unsaturations underwent chlorination without rearrangement due to formation of allylic cations. The rearrangement was also demonstrated using Appel conditions, which gave similar results via the alkoxytriphenylphosphonium intermediate. Several reactions of the products were investigated to show the utility of the rearrangement.

Graphical Abstract
Introduction
The 6,8-dioxabicyclo[3.2.1]octane derivative levoglucosenone (1) is produced selectively when cellulose-containing materials, including lignocellulosic biomass, are acidified and pyrolysed [1,2]. Lab scale synthesis of this chiral material can be accomplished in a single step without special glassware [3], while large scale production of the reduction product cyrene (2) allows for its use as a chiral solvent [4]. This product is emerging as a promising platform chemical for the construction of chiral small molecules for pharmaceuticals [5-8], as a building block for catalysts and auxiliaries [9-11], and in materials applications [12-14]. New reactions will increase the number of accessible materials that can be made from this biorenewable starting material, particularly if novel approaches for modifying the connectivity of the bicyclic ring system can be developed.
The 6,8-dioxabicyclo[3.2.1]octane system is known to undergo a number of bond-cleavage reactions and rearrangements when modified at the 4-position [15-17]. Baillargeon and Reddy first reported rearrangements of 6,8-dioxabicyclo[3.2.1]octane derivatives promoted by diethylaminosulfur trifluoride (DAST) [18], and later Karban and co-workers reported a migration of oxygen from the acetal in 3 and 6 to the neighbouring C4-position (Figure 1) [19,20]. A variety of products were reported resulting from fluorination as well as the skeletal rearrangement, with the reaction outcome highly substrate-dependent. A key finding in this work was that the configuration of the alcohol at C4 determined the resultant ring system, as the σ* orbital is not accessible to external nucleophiles due to steric hindrance and the rigid conformation of the bicyclic ring system. When the C4–OH was equatorial, O8 migrated as it was aligned with the σ* orbital giving a 3,8-dioxabicyclo[3.2.1]octane, while O6 migrated when the C4–OH was axial leading to 2,4-dioxabicyclo[2.2.2]octanes. The formation of both anomers from the non-selective addition of fluoride suggested intermediates with oxocarbenium character. This work has recently been extended by Banwell and co-workers to include a set of Diels–Alder adducts of 1, and similar results on the effect of configuration were observed [21].
![[1860-5397-20-74-1]](/bjoc/content/figures/1860-5397-20-74-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Previous work on migration reactions in 6,8-dioxabicyclooctan-4-ols [18].
Figure 1: Previous work on migration reactions in 6,8-dioxabicyclooctan-4-ols [18].
During some recent attempts at the chlorination of the π-stacking chiral auxiliary 10a using SOCl2 [9], we observed the migration of O8 resulting in the formation of anomeric chlorides analogous to the reports of Karban et al. (Scheme 2) [19]. We envisaged that these hexose-derived building blocks with a 2,5-anhydro bond such as 5 could be useful materials for the construction of C-nucleosides such as formycin A (Figure 1) [22-24]. The preparation of C-glycosides usually involves the creation of the glycosidic bond using an organometallic purine or pyrimidine derivative and an electrophilic furanose derivative [23,25]. This process can result in anomeric mixtures, so 5 has potential applications in targeted synthesis, as the configuration of the pseudo-anomeric centre matches the common biological ribosides. This prompted an investigation of the scope of the SOCl2-mediated rearrangement, with the aim of producing useful chiral materials for synthesis.
Results and Discussion
The set of bicyclic systems 10a–f with a C4 alcohol were prepared starting with cyrene (2) by alkylation and then reduction using NaBH4 as per our previously published approach (Scheme 1) [9]. When α-alkylations are performed using 2, the second alkylation step is faster than the first, meaning that only the dialkylated products are formed [16], and the reduction is highly selective with approach of the reductant from the exo-face. This process was used to prepare the known compounds 10a–c, and the novel materials 10d–f [9]. Thus, the reaction of o-dibromoxylene with cyrene gave the alcohol 10d in 71% yield over two-steps through the spirocyclic ketone 9d. Alkylation of 2 with methyl iodide gave an inseparable mixture of ketone 9e and the O-alkylated enol ether by-product, which was then reduced using NaBH4 to give alcohol 10e with 98:2 selectivity. Similarly, the reaction of 4-methoxybenzyl bromide and 2 gave ketone 9f in 35% yield without chromatography, and when reduced resulted in only a single alcohol stereoisomer 10f in 91% yield. The selectivity of the NaBH4 reduction was confirmed for both 10d (see discussion in Supporting Information File 1) and 10e by X-ray crystallography (Figure 2 and Scheme 1, respectively).
![[1860-5397-20-74-i1]](/bjoc/content/inline/1860-5397-20-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structures for 10a–c, preparation of 10d–f, and X-ray structure of 10e.
Scheme 1: Structures for 10a–c, preparation of 10d–f, and X-ray structure of 10e.
The oxygen-migration reaction giving 11a was initially observed using the readily available chiral auxiliary 10a, and following a survey of conditions and isolation protocols, a 90% yield was obtained when 10a was heated in the presence of 2 equivalents of SOCl2 and 5 equivalents of pyridine in DCE. Flash chromatography of the chloroalkyl ether 11a resulted in significant loss due to hydrolysis, although 11a was sufficiently stable for filtration through a pad of silica. Applying these conditions and isolation protocols to all 3,3-disubstituted alcohols 10c–f gave moderate to excellent yields of the rearrangement products 11c–f as single stereoisomers. The reactions of alcohols 10b,d,e also gave some sulfites 13b,d,e, attributed to the reduced steric hindrance in the chlorosulfite intermediate allowing for the second alcohol to approach prior to rearrangement. The isolation of these materials suggested that dialkyl sulfite formation could compete with the rearrangement if the neighbouring groups were small, and attempts to prevent the formation of 13d by slow addition of alcohol 10d to a solution of SOCl2/pyridine in DCE reduced the yield of 11d to 14%. Heating sulfite 13d with tetrabutylammonium chloride led only to hydrolysis back to 10d without rearrangement, indicating that these sulfites were not intermediates in the reaction manifold. Furthermore, the isolation of some starting alcohols in the reactions of 10b and 10d following chromatography was attributed to hydrolysis of the corresponding dialkyl sulfite 13b and 13d on silica, a process that can be acid or base-catalysed [26]. The unsubstituted derivative 11b was difficult to isolate in good yields as multiple products were formed giving complex reaction mixtures. The product 11b was consistently contaminated with a second inseparable product tentatively assigned as 14, which is the expected product of chlorination without skeletal rearrangement (vide infra). Inclusion of the soft-nucleophile allyltrimethylsilane in the reaction of 10b to trap potential oxocarbenium ion intermediates also resulted in a complex mixture.
During the isolation of the chloroalkyl ether products 11a–f, it was apparent that hydrolysis occurred during chromatography, and so an alternate method was developed to generate a single product by promoting the formation of the hemiacetal series 12a–f. Following the rearrangement reaction, chromatography of the chlorides using silica with 2% water added led to the isolation of 12a,c–f in good yield, with the exo-hemiacetals favoured due to steric interactions between the substituents and alcohol, while the attempted preparation of 12b led only to complex mixtures (Scheme 2).
![[1860-5397-20-74-i2]](/bjoc/content/inline/1860-5397-20-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Rearrangement reactions for 10a–f promoted by SOCl2.
Scheme 2: Rearrangement reactions for 10a–f promoted by SOCl2.
The products of the reactions were characterised by 1D and 2D NMR, and X-ray crystallography of members from each class was used to confirm assignments. In the 1H NMR spectra of the chlorides 11a–f, a downfield shift for the anomeric methine was observed to δ ≈6 ppm from δ ≈5.4 ppm in the starting materials 10a–f. There was also a characteristic change in the appearance of the oxymethylene bridge spin system, with the products exhibiting much larger differences in the chemical shifts for the geminal protons relative to those observed in the starting material. For example, in the 1H NMR spectrum of 11e, the H4/H4′ resonances have a difference of 0.94 ppm, while in 10e, the progenitor H7/H7′ methylene resonances are separated by 0.21 ppm. In the 13C NMR spectrum of 11a–f, a ≈10 ppm upfield shift for the chloroacetal was seen to δ ≈92 ppm from the C5 acetal present in the starting materials. Only a single diastereomer was formed for all 3,3-disubstituted rearrangement products due to the hindrance on the endo-face, with the X-ray crystal structures for 11a and 11b allowing for the unambiguous assignment of configuration. The chlorinated product 14 contaminating 11b exhibited a 1H NMR spectrum similar to the starting material 10b, except that the resonance for the H4 methine was shifted from δ 3.60 in 10b to δ 3.90 ppm in 14. The methine had a correlation to a resonance at δC 55.1 ppm in the 2D HSQC spectra consistent with an attached chloride. In the 1H NMR spectra for the hemiacetals 12a,c–f, an upfield shift for the anomeric centre of ≈1.1 ppm was observed relative to the chlorides. There was also evidence of open chain aldehydes present in solution (≈5%) with a doublet at δ 9.6 ppm, with the configuration of the hemiacetal centre confirmed in the solid state by X-ray crystallography on 12a and 12d. The 1H and 13C NMR spectra for 13b,d,e were similar to the starting materials, except that the resonances were doubled due to the diastereotopic ring systems. The different environments were mainly evident in the chemical shifts for H4/H4′ (Δδ 0.06 ppm) and H5/H5′ (Δδ 0.03 ppm), with other resonances only showing broadening due to their remote relationship with the sulfite group. The structures of the dialkyl sulfites were confirmed using X-ray crystallography for 13b, which clearly demonstrated the lack of symmetry across the molecule.
To further examine the scope of the reaction, allylic alcohols 15 and 18 were subjected to the optimised reaction conditions [9,27]. The reaction of 15 with an endocyclic olefin led to a series of separable allylic chlorides 16 and 17a,b, from direct displacement or transposition of the allylic system (Scheme 3). This substrate has previously been examined in the reaction with SOCl2 by Matsumoto et al. in THF and CH2Cl2 and similar results were obtained [28]. When 18 containing an exocyclic alkene was subjected to the reaction conditions, a mixture of benzylic chlorides (20) was formed in low yields, and trace amounts of the allylic chloride 19 was also isolated, the materials differentiated on the basis of the coupling of the acetal H5 with the respective vicinal proton. These results suggested that the formation of the allylic cation occurred readily from alcohols 15 and 18; however, the transition states leading to the rearrangement products were inaccessible and so only chloride addition occurred.
![[1860-5397-20-74-i3]](/bjoc/content/inline/1860-5397-20-74-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of allylic alcohols 15 and 18 with SOCl2.
Scheme 3: Reactions of allylic alcohols 15 and 18 with SOCl2.
The generation of the rearrangement products from the reaction with SOCl2, and the previous work with DAST, suggested that good leaving groups were required to drive the migration reaction. A variety of processes are known for deoxyhalogenation, and it was thought that alternatives to SOCl2 could also be used to promote the oxygen migration. When Appel conditions (PPh3, CCl4) for the deoxychlorination were examined [29], the rearrangement products were observed as the major components in the reaction mixture (Scheme 4). For the reaction of 10a, the starting material was consumed within 1 hour (NMR) to give an intermediate assigned as the alkoxytriphenylphosphonium chloride (26, R = Bn), which then slowly rearranged over 24 hours at 83 °C in DCE, eliminating triphenylphosphine oxide (Figure 2). A single ion was observed in the ESI mass spectrum for the intermediate at m/z 571.1 corresponding to the [M + PPh3 − H]+, and in the 1H NMR, the H4 adjacent to the oxyphosphonium group was observed at δ 4.44 ppm, shifted downfield relative to the starting alcohol 10a along with resonances for the phenyl groups. The isolation of the products using the Appel conditions was more challenging than for the reactions with SOCl2 due to the difficulties separating the products from the byproduct triphenylphosphine oxide, necessitating chromatography which resulted in some hydrolysis. There are a number of catalytic activation strategies for Appel or Mitsunobu reactions such as those described by the Denton group [30], and Rutjes and co-workers [31], and while these may prove useful in future studies, they were not examined in this work.
![[1860-5397-20-74-i4]](/bjoc/content/inline/1860-5397-20-74-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Appel reactions of dioxabicyclo[3.2.1]octan-4-ols 10a,e,f and 15.
Scheme 4: Appel reactions of dioxabicyclo[3.2.1]octan-4-ols 10a,e,f and 15.
To investigate the potential uses for the rearrangement products, a series of reactions on the chloroalkyl ethers 11a and 12a was performed (Scheme 5). The reaction of 11a with allyltrimethylsilane catalysed by aluminium chloride resulted in the displacement of the chloro substituent with the allyl group, affording 21 in good yield. Electrophilic aromatic substitution reactions at the chloroalkyl ether site were possible when promoted by aluminium chloride, with anisole and diphenyl ether giving addition products 22 and 23 containing small amounts of the C2 epimers. Oxidation of the hemiacetal 12a gave a moderate and unoptimised yield of 40% for lactone 24.
![[1860-5397-20-74-i5]](/bjoc/content/inline/1860-5397-20-74-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Some transformations for the skeletal rearrangement products 11a and 12a and X-ray structure for 24.
Scheme 5: Some transformations for the skeletal rearrangement products 11a and 12a and X-ray structure for 24....
The probable mechanism for the transformation with SOCl2 and under Appel conditions is shown in Figure 2. The reaction of alcohol 10 with the electrophiles gives the chlorosulfite 25 or the alkoxytriphenylphosphonium chloride 26, respectively. With heating, SO2 or triphenylphosphine oxide is extruded with a concerted migration of the neighbouring O8 leading to an oxocarbenium ion 27, which is then trapped with chloride giving the observed products. The crystal structure for the precursor alcohol 10d is shown projected along the C4–C5 axis, which demonstrates a 177.3° dihedral angle for the HO–C4–C5–O8 group, aligning O8 antiperiplanar and positioned to migrate during the reaction. The relationship between H4 and O6 is similarly antiperiplanar, with a H4–C4–C5–O6 dihedral angle of 176°, explaining the preference for the different skeletal rearrangements in the two possible configurations at C4 in these rigid ring systems [19,21]. The involvement of the ring-oxygen in nucleophilic displacement reactions in 1,6-anhydroglucose derivatives has also been invoked to explain the observed retention of configuration, showing that substantial interactions between the oxygens of the ring and centres on the larger bridge are possible [32]. The specificity of the rearrangement also eliminates the possibility of an intermediate secondary C4 carbocation, and requires a concerted bond migration. This is a mechanistic difference to the related 1,2-oxygen migration reactions of spiroacetals that involve alkoxy intermediates reported by Suarez and co-workers [33,34]. The presence of oxocarbenium ion 27 is inferred due to the formation of two diastereomers in Karban’s previous work, which suggests a stepwise migration of oxygen from C5 to C4, followed by addition of the halogen nucleophile. Furthermore, if the halogen was involved in the transition state via the σ* orbital (avoiding intermediate 27), the opposite configuration would result at C2 in the products. The single diastereomers isolated in the current work are attributed to the differences in sterics on the faces of the oxocarbenium ion 27, caused by the substitution on the bicyclic ring system.
![[1860-5397-20-74-2]](/bjoc/content/figures/1860-5397-20-74-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Mechanism for the rearrangement of 10, and Newman projection and the X-ray structure of 10d projected along the C4–C5 axis.
Figure 2: Mechanism for the rearrangement of 10, and Newman projection and the X-ray structure of 10d project...
Conclusion
The formation of anomeric chlorides due to bond migrations in the dioxabicyclo[3.2.1]octanol ring system has been described for the first time. The work builds upon the findings of the groups of Karban and Banwell, who described this type of ring transformation using DAST, with two new reagents for promoting the rearrangement reaction. This work adds to the growing set of transformations that are known for levoglucosenone, cyrene and their derivatives, generating a unique set of bicyclic building blocks.
Supporting Information
Supporting information includes detailed experimental details and characterisation data, X-ray crystallography, and copies of 1H and 13C spectra for new compounds.
| Supporting Information File 1: Experimental details, X-ray crystallography and spectra. | ||
| Format: PDF | Size: 11.6 MB | Download |
| Supporting Information File 2: 1H and 13C NMR FIDs, HRMS spectra for all new compounds. | ||
| Format: ZIP | Size: 41.0 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Halpern, Y.; Riffer, R.; Broido, A. J. Org. Chem. 1973, 38, 204–209. doi:10.1021/jo00942a005
Return to citation in text: [1] -
Kudo, S.; Huang, X.; Asano, S.; Hayashi, J.-i. Energy Fuels 2021, 35, 9809–9824. doi:10.1021/acs.energyfuels.1c01062
Return to citation in text: [1] -
Klepp, J.; Dillon, W.; Lin, Y.; Feng, P.; Greatrex, B. W. Org. Synth. 2020, 97, 38–53. doi:10.15227/orgsyn.097.0038
Return to citation in text: [1] -
Zhang, J.; White, G. B.; Ryan, M. D.; Hunt, A. J.; Katz, M. J. ACS Sustainable Chem. Eng. 2016, 4, 7186–7192. doi:10.1021/acssuschemeng.6b02115
Return to citation in text: [1] -
Camp, J. E.; Greatrex, B. W. Front. Chem. (Lausanne, Switz.) 2022, 10, 902239. doi:10.3389/fchem.2022.902239
Return to citation in text: [1] -
Comba, M. B.; Tsai, Y.-h.; Sarotti, A. M.; Mangione, M. I.; Suárez, A. G.; Spanevello, R. A. Eur. J. Org. Chem. 2018, 590–604. doi:10.1002/ejoc.201701227
Return to citation in text: [1] -
Liu, X.; Carr, P.; Gardiner, M. G.; Banwell, M. G.; Elbanna, A. H.; Khalil, Z. G.; Capon, R. J. ACS Omega 2020, 5, 13926–13939. doi:10.1021/acsomega.0c01331
Return to citation in text: [1] -
Sarotti, A. M.; Zanardi, M. M.; Spanevello, R. A. Curr. Org. Synth. 2012, 9, 439–459. doi:10.2174/157017912802651401
Return to citation in text: [1] -
Klepp, J.; Sumby, C. J.; Greatrex, B. W. Synlett 2018, 29, 1441–1446. doi:10.1055/s-0037-1610148
Return to citation in text: [1] [2] [3] [4] [5] -
Sarotti, A. M.; Spanevello, R. A.; Suárez, A. G. Tetrahedron Lett. 2005, 46, 6987–6990. doi:10.1016/j.tetlet.2005.08.073
Return to citation in text: [1] -
Sarotti, A. M.; Spanevello, R. A.; Suárez, A. G. Org. Lett. 2006, 8, 1487–1490. doi:10.1021/ol0603099
Return to citation in text: [1] -
Fadlallah, S.; Mouterde, L. M.; Garnier, G.; Saito, K.; Allais, F. Cellulose-Derived Levoglucosenone, a Great Versatile Chemical Platform for the Production of Renewable Monomers and Polymers. Sustainability & Green Polymer Chemistry Volume 2: Biocatalysis and Biobased Polymers; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2020; pp 77–97. doi:10.1021/bk-2020-1373.ch005
Return to citation in text: [1] -
Banwell, M. G.; Liu, X.; Connal, L. A.; Gardiner, M. G. Macromolecules 2020, 53, 5308–5314. doi:10.1021/acs.macromol.0c01305
Return to citation in text: [1] -
Stanfield, M. K.; Terry, R. S.; Smith, J. A.; Thickett, S. C. Polym. Chem. 2023, 14, 4949–4956. doi:10.1039/d3py01019h
Return to citation in text: [1] -
Ebata, T.; Matsumoto, K.; Yoshikoshi, H.; Koseki, K.; Kawakami, H.; Okano, K.; Matsushita, H. Heterocycles 1993, 36, 1017–1026. doi:10.3987/com-92-9262
Return to citation in text: [1] -
Ledingham, E. T.; Stockton, K. P.; Greatrex, B. W. Aust. J. Chem. 2017, 70, 1146–1150. doi:10.1071/ch17227
Return to citation in text: [1] [2] -
Valeev, F. A.; Gorobets, E. V.; Tsypysheva, I. P.; Singizova, G. S.; Kalimullina, L. K.; Safarov, M. G.; Shitikova, O. V.; Miftakhov, M. S. Chem. Nat. Compd. 2003, 39, 563–568. doi:10.1023/b:conc.0000018110.36123.f2
Return to citation in text: [1] -
Baillargeon, D. J.; Reddy, G. S. Carbohydr. Res. 1986, 154, 275–279. doi:10.1016/s0008-6215(00)90041-7
Return to citation in text: [1] [2] -
Karban, J.; Císařová, I.; Strašák, T.; Šťastná, L. Č.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/c1ob06336g
Return to citation in text: [1] [2] [3] -
Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75
Return to citation in text: [1] -
Pollard, B.; Liu, X.; Connal, L. A.; Banwell, M. G.; Gardiner, M. G. Aust. J. Chem. 2023, 76, 797–811. doi:10.1071/ch23130
Return to citation in text: [1] [2] -
Hori, M.; Itō, E.; Takita, T.; Koyama, G.; Takeuchi, T.; Umezawa, H. J. Antibiot. 1964, 17, 96–99.
Return to citation in text: [1] -
Temburnikar, K.; Seley-Radtke, K. L. Beilstein J. Org. Chem. 2018, 14, 772–785. doi:10.3762/bjoc.14.65
Return to citation in text: [1] [2] -
Štambaský, J.; Hocek, M.; Kočovský, P. Chem. Rev. 2009, 109, 6729–6764. doi:10.1021/cr9002165
Return to citation in text: [1] -
Wu, Q.; Simons, C. Synthesis 2004, 1533–1553. doi:10.1055/s-2004-829106
Return to citation in text: [1] -
Salomon, M. Can. J. Chem. 1977, 55, 2971–2976. doi:10.1139/v77-413
Return to citation in text: [1] -
Comba, M. B.; Mangione, M. I.; Suárez, A. G.; Sarotti, A. M.; Spanevello, R. A. Eur. J. Org. Chem. 2018, 6848–6856. doi:10.1002/ejoc.201801432
Return to citation in text: [1] -
Matsumoto, M.; Ishikawa, H.; Soya, Y.; Ozawa, T. Heterocycles 1994, 38, 2377. doi:10.3987/com-94-6864
Return to citation in text: [1] -
Appel, R. Angew. Chem., Int. Ed. Engl. 1975, 14, 801–811. doi:10.1002/anie.197508011
Return to citation in text: [1] -
Beddoe, R. H.; Andrews, K. G.; Magné, V.; Cuthbertson, J. D.; Saska, J.; Shannon-Little, A. L.; Shanahan, S. E.; Sneddon, H. F.; Denton, R. M. Science 2019, 365, 910–914. doi:10.1126/science.aax3353
Return to citation in text: [1] -
van Kalkeren, H. A.; van Delft, F. L.; Rutjes, F. P. J. T. Pure Appl. Chem. 2013, 85, 817–828. doi:10.1351/pac-con-12-06-13
Return to citation in text: [1] -
Quiquempoix, L.; Wang, Z.; Graton, J.; Latchem, P. G.; Light, M.; Le Questel, J.-Y.; Linclau, B. J. Org. Chem. 2019, 84, 5899–5906. doi:10.1021/acs.joc.9b00310
Return to citation in text: [1] -
Betancor, C.; Dorta, R. L.; Freire, R.; Martín, A.; Prangé, T.; Suárez, E. J. Org. Chem. 1998, 63, 6355–6362. doi:10.1021/jo980834o
Return to citation in text: [1] -
Betancor, C.; Dorta, R. L.; Freire, R.; Prangé, T.; Suárez, E. J. Org. Chem. 2000, 65, 8822–8825. doi:10.1021/jo005593a
Return to citation in text: [1]
| 1. | Halpern, Y.; Riffer, R.; Broido, A. J. Org. Chem. 1973, 38, 204–209. doi:10.1021/jo00942a005 |
| 2. | Kudo, S.; Huang, X.; Asano, S.; Hayashi, J.-i. Energy Fuels 2021, 35, 9809–9824. doi:10.1021/acs.energyfuels.1c01062 |
| 9. | Klepp, J.; Sumby, C. J.; Greatrex, B. W. Synlett 2018, 29, 1441–1446. doi:10.1055/s-0037-1610148 |
| 10. | Sarotti, A. M.; Spanevello, R. A.; Suárez, A. G. Tetrahedron Lett. 2005, 46, 6987–6990. doi:10.1016/j.tetlet.2005.08.073 |
| 11. | Sarotti, A. M.; Spanevello, R. A.; Suárez, A. G. Org. Lett. 2006, 8, 1487–1490. doi:10.1021/ol0603099 |
| 23. | Temburnikar, K.; Seley-Radtke, K. L. Beilstein J. Org. Chem. 2018, 14, 772–785. doi:10.3762/bjoc.14.65 |
| 25. | Wu, Q.; Simons, C. Synthesis 2004, 1533–1553. doi:10.1055/s-2004-829106 |
| 5. | Camp, J. E.; Greatrex, B. W. Front. Chem. (Lausanne, Switz.) 2022, 10, 902239. doi:10.3389/fchem.2022.902239 |
| 6. | Comba, M. B.; Tsai, Y.-h.; Sarotti, A. M.; Mangione, M. I.; Suárez, A. G.; Spanevello, R. A. Eur. J. Org. Chem. 2018, 590–604. doi:10.1002/ejoc.201701227 |
| 7. | Liu, X.; Carr, P.; Gardiner, M. G.; Banwell, M. G.; Elbanna, A. H.; Khalil, Z. G.; Capon, R. J. ACS Omega 2020, 5, 13926–13939. doi:10.1021/acsomega.0c01331 |
| 8. | Sarotti, A. M.; Zanardi, M. M.; Spanevello, R. A. Curr. Org. Synth. 2012, 9, 439–459. doi:10.2174/157017912802651401 |
| 9. | Klepp, J.; Sumby, C. J.; Greatrex, B. W. Synlett 2018, 29, 1441–1446. doi:10.1055/s-0037-1610148 |
| 4. | Zhang, J.; White, G. B.; Ryan, M. D.; Hunt, A. J.; Katz, M. J. ACS Sustainable Chem. Eng. 2016, 4, 7186–7192. doi:10.1021/acssuschemeng.6b02115 |
| 19. | Karban, J.; Císařová, I.; Strašák, T.; Šťastná, L. Č.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/c1ob06336g |
| 3. | Klepp, J.; Dillon, W.; Lin, Y.; Feng, P.; Greatrex, B. W. Org. Synth. 2020, 97, 38–53. doi:10.15227/orgsyn.097.0038 |
| 22. | Hori, M.; Itō, E.; Takita, T.; Koyama, G.; Takeuchi, T.; Umezawa, H. J. Antibiot. 1964, 17, 96–99. |
| 23. | Temburnikar, K.; Seley-Radtke, K. L. Beilstein J. Org. Chem. 2018, 14, 772–785. doi:10.3762/bjoc.14.65 |
| 24. | Štambaský, J.; Hocek, M.; Kočovský, P. Chem. Rev. 2009, 109, 6729–6764. doi:10.1021/cr9002165 |
| 19. | Karban, J.; Císařová, I.; Strašák, T.; Šťastná, L. Č.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/c1ob06336g |
| 20. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 18. | Baillargeon, D. J.; Reddy, G. S. Carbohydr. Res. 1986, 154, 275–279. doi:10.1016/s0008-6215(00)90041-7 |
| 18. | Baillargeon, D. J.; Reddy, G. S. Carbohydr. Res. 1986, 154, 275–279. doi:10.1016/s0008-6215(00)90041-7 |
| 9. | Klepp, J.; Sumby, C. J.; Greatrex, B. W. Synlett 2018, 29, 1441–1446. doi:10.1055/s-0037-1610148 |
| 15. | Ebata, T.; Matsumoto, K.; Yoshikoshi, H.; Koseki, K.; Kawakami, H.; Okano, K.; Matsushita, H. Heterocycles 1993, 36, 1017–1026. doi:10.3987/com-92-9262 |
| 16. | Ledingham, E. T.; Stockton, K. P.; Greatrex, B. W. Aust. J. Chem. 2017, 70, 1146–1150. doi:10.1071/ch17227 |
| 17. | Valeev, F. A.; Gorobets, E. V.; Tsypysheva, I. P.; Singizova, G. S.; Kalimullina, L. K.; Safarov, M. G.; Shitikova, O. V.; Miftakhov, M. S. Chem. Nat. Compd. 2003, 39, 563–568. doi:10.1023/b:conc.0000018110.36123.f2 |
| 12. | Fadlallah, S.; Mouterde, L. M.; Garnier, G.; Saito, K.; Allais, F. Cellulose-Derived Levoglucosenone, a Great Versatile Chemical Platform for the Production of Renewable Monomers and Polymers. Sustainability & Green Polymer Chemistry Volume 2: Biocatalysis and Biobased Polymers; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2020; pp 77–97. doi:10.1021/bk-2020-1373.ch005 |
| 13. | Banwell, M. G.; Liu, X.; Connal, L. A.; Gardiner, M. G. Macromolecules 2020, 53, 5308–5314. doi:10.1021/acs.macromol.0c01305 |
| 14. | Stanfield, M. K.; Terry, R. S.; Smith, J. A.; Thickett, S. C. Polym. Chem. 2023, 14, 4949–4956. doi:10.1039/d3py01019h |
| 21. | Pollard, B.; Liu, X.; Connal, L. A.; Banwell, M. G.; Gardiner, M. G. Aust. J. Chem. 2023, 76, 797–811. doi:10.1071/ch23130 |
| 16. | Ledingham, E. T.; Stockton, K. P.; Greatrex, B. W. Aust. J. Chem. 2017, 70, 1146–1150. doi:10.1071/ch17227 |
| 9. | Klepp, J.; Sumby, C. J.; Greatrex, B. W. Synlett 2018, 29, 1441–1446. doi:10.1055/s-0037-1610148 |
| 32. | Quiquempoix, L.; Wang, Z.; Graton, J.; Latchem, P. G.; Light, M.; Le Questel, J.-Y.; Linclau, B. J. Org. Chem. 2019, 84, 5899–5906. doi:10.1021/acs.joc.9b00310 |
| 33. | Betancor, C.; Dorta, R. L.; Freire, R.; Martín, A.; Prangé, T.; Suárez, E. J. Org. Chem. 1998, 63, 6355–6362. doi:10.1021/jo980834o |
| 34. | Betancor, C.; Dorta, R. L.; Freire, R.; Prangé, T.; Suárez, E. J. Org. Chem. 2000, 65, 8822–8825. doi:10.1021/jo005593a |
| 31. | van Kalkeren, H. A.; van Delft, F. L.; Rutjes, F. P. J. T. Pure Appl. Chem. 2013, 85, 817–828. doi:10.1351/pac-con-12-06-13 |
| 19. | Karban, J.; Císařová, I.; Strašák, T.; Šťastná, L. Č.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/c1ob06336g |
| 21. | Pollard, B.; Liu, X.; Connal, L. A.; Banwell, M. G.; Gardiner, M. G. Aust. J. Chem. 2023, 76, 797–811. doi:10.1071/ch23130 |
| 29. | Appel, R. Angew. Chem., Int. Ed. Engl. 1975, 14, 801–811. doi:10.1002/anie.197508011 |
| 30. | Beddoe, R. H.; Andrews, K. G.; Magné, V.; Cuthbertson, J. D.; Saska, J.; Shannon-Little, A. L.; Shanahan, S. E.; Sneddon, H. F.; Denton, R. M. Science 2019, 365, 910–914. doi:10.1126/science.aax3353 |
| 9. | Klepp, J.; Sumby, C. J.; Greatrex, B. W. Synlett 2018, 29, 1441–1446. doi:10.1055/s-0037-1610148 |
| 27. | Comba, M. B.; Mangione, M. I.; Suárez, A. G.; Sarotti, A. M.; Spanevello, R. A. Eur. J. Org. Chem. 2018, 6848–6856. doi:10.1002/ejoc.201801432 |
| 28. | Matsumoto, M.; Ishikawa, H.; Soya, Y.; Ozawa, T. Heterocycles 1994, 38, 2377. doi:10.3987/com-94-6864 |
© 2024 Jevric et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.