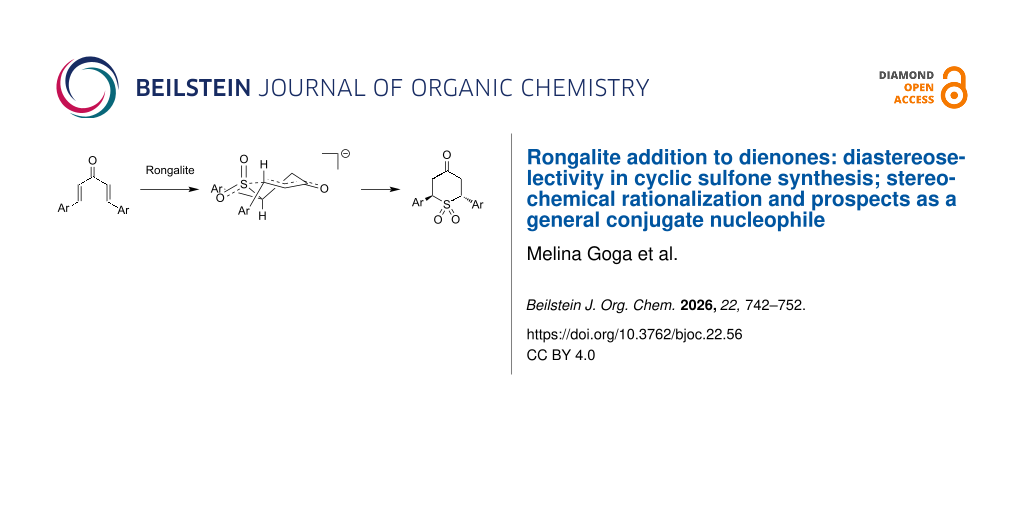
Abstract
A detailed experimental and computational study of the diastereoselectivity of cyclic sulfone synthesis by reaction of Rongalite with doubly electrophilic dienones is presented. Computational methods, including density functional theory, conformational search methods, and internal reaction coordinate methods, have been used to rationalize the diastereoselectivity of the reaction by transition-state modeling, and showcases the conformational subtleties of sulfur-containing rings. Extension of the original reaction to double intermolecular additions is also demonstrated along with a comparative study of Rongalite with the reactivity of other readily available sulfinate nucleophiles using competition and exchange experiments.

Graphical Abstract
Introduction
Sulfonyl groups are widespread in licensed drugs [1]. New methods for the incorporation of sulfonyl groups into organic molecules are desirable, particularly in the light of the general need for more sustainable chemical synthesis. Recently, there has been significant progress in the use of SO2 equivalents and bulk inorganic high oxidation state sulfur compounds for the direct incorporation of sulfones [2,3]. Despite these advances, use of a low-valent sulfur nucleophile for initial installation of the sulfur atom followed by oxidation remains a widespread method, even though it is far from ideal environmentally and also in terms of functional group tolerance [4].
RongaliteTM is a low-cost commodity chemical that has been used as a bleaching agent in the dyeing industry for more than a century [5,6]. Over the last 10 years particularly, it has begun to join the toolbox of reagents in organic synthesis, and shows diverse reactivity. It has been employed in reductions, radical processes and as a reagent for C1 transfer [7-14]. Importantly for our work, it can also act as an equivalent of the unstable hyposulfite (SO22−) ion (Figure 1) [15,16].
![[1860-5397-22-56-1]](/bjoc/content/figures/1860-5397-22-56-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Rongalite as an equivalent of the unstable hyposulfite ion.
Figure 1: Rongalite as an equivalent of the unstable hyposulfite ion.
The reagent was first used for the preparation of symmetrical sulfones as early as 1971 [17], but interest in the reagent has increased dramatically over the last two decades [5,6]. Modified reagents in which the Rongalite hydroxy group has been protected have allowed for consecutive and different reactions at sulfur, giving access to a range of unsymmetrical sulfones [18,19]. Although sulfur nucleophiles often participate in conjugate additions, few reports documented this behavior for Rongalite (Scheme 1). This may have been due to its known reactivity as a conjugate reductant [17,20]. We recently published a preliminary report on the reaction of Rongalite with dienones leading to cyclic sulfones (Scheme 1) [21]. We now describe our efforts to resolve a stereochemical ambiguity, the modelling of transition states to explain the diastereoselectivity of the one-pot reaction, a full exploration of its scope and limitations and further mechanistic and competition experiments that place the nucleophilicity of Rongalite in context by comparison with other readily available sulfinates.
![[1860-5397-22-56-i1]](/bjoc/content/inline/1860-5397-22-56-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Progression of Rongalite conjugate additions.
Scheme 1: Progression of Rongalite conjugate additions.
Results and Discussion
Confirmation of diastereoselectivity
In our initial report, we supposed that the major product was the trans-isomer, following the analysis of Detty and co-workers [22], who prepared sulfones 1a and 1b (Figure 2) by a conventional sulfide oxidation. In that early work, the stereochemical analysis was dependent on conformational analysis and comparison of coupling constants in the 1H NMR spectra. Experiments discussed in our inital report confirmed that the reaction proceeded under kinetic control [21].
![[1860-5397-22-56-2]](/bjoc/content/figures/1860-5397-22-56-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Sulfone diastereomers 1a and 1b.
Figure 2: Sulfone diastereomers 1a and 1b.
Closer examination of the 1995 paper [22] revealed a slight ambiguity – the 1H NMR signals that were discussed and used to assign relative stereochemistry in the main text were found to be swapped over in the experimental section. We had no reason to think that this was anything but a typographical error, but we nevertheless sought confirmation of the structures of the minor and major diastereomers in our own hands. Noting that preparation of sulfides by double conjugate addition of Na2S or NaSH has been widely studied, and that the stereochemical assignments presented therein are in some cases supported by X-ray crystallographic studies, we prepared trans-sulfide 2 using an established protocol [23], and then oxidized it to trans-sulfone 1a (Scheme 2). This material was identical to the major product from Rongalite double conjugate addition.
![[1860-5397-22-56-i2]](/bjoc/content/inline/1860-5397-22-56-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Verification of relative stereochemistry by i) sulfide oxidation and ii) derivatization.
Scheme 2: Verification of relative stereochemistry by i) sulfide oxidation and ii) derivatization.
Additional confirmation of the structures of major and minor diastereomers 1a and 1b was provided by derivatization of both molecules in ways that confirmed their relative stereochemistry by taking advantage of the different symmetry elements present in each case. Thus, both reductive amination and hydride reduction of C2-symmetric 1a led to the isolation of the only possible (chiral) diastereomer of the products 3 and 4 in each case – reaction of the nucleophile with either face of the iminium ion or ketone gives the same diastereomer, as illustrated. By contrast, hydride reduction of 1b gave an approx. 9:1 mixture of products 5 and 6, as judged by the crude 1H NMR spectrum. The major product was isolated and assigned as 5 (from axial attack of a small hydride reagent) [24]. From analysis of the NMR spectra of 5, it was clear that the molecule retained symmetry following reduction, reflected in the reduced number of signals in both 1H and 13C spectra compared to alcohol 4.
Steric limits and advanced computation
Substrate 7 (Scheme 3), with four ortho-methyl groups, showed no sign of product formation under the standard reaction conditions. In order to determine whether this was due to an unreactive conformational preference of the substrate or to difficulty in forming the product, we used density functional theory (DFT) and conformational search techniques based on tight-binding methods to find the energetically minimized structure of 7 (see Supporting Information File 1 for details on all computations). The lowest energy structure is depicted in Figure 3. It shows a structure somewhat rotated out of planarity (dihedral angles between aromatic C–C and vinylic CH are 34°). This presumably still allows for some conjugative stabilization from the aromatic rings to the enone π-systems and reduces eclipsing interactions between the ortho-methyl groups and the vinylic H (indicated). There is nothing to suggest from the calculations that an inherent reactivity problem prevents the initial addition of Rongalite to the enone, especially given that the singly electrophilic 2,6-dimethylbenzalacetone is known to participate in reactions with conjugate nucleophiles [25]. It is therefore more likely that steric effects in the transition state for the putative cyclization to give 8 are prohibitive. Indeed, we were unable to locate a transition state for this process computationally.
![[1860-5397-22-56-i3]](/bjoc/content/inline/1860-5397-22-56-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of sterically hindered substrates.
Scheme 3: Reactions of sterically hindered substrates.
![[1860-5397-22-56-3]](/bjoc/content/figures/1860-5397-22-56-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Minimized structure of dienone 7 (calculated using the PBE0 functional [26] and the def2-SVP basis set [27] (see Supporting Information File 1 for full computational details)).
Figure 3: Minimized structure of dienone 7 (calculated using the PBE0 functional [26] and the def2-SVP basis set [27]...
In order to test the limits of the reaction with related substitution patterns in this process, we prepared unsymmetrical substrate 9 from benzalacetone and 2,6-dimethylbenzaldehyde. Reaction of 9 with Rongalite under our standard conditions did indeed give the expected product 10a. In this case only the major (trans) isomer was isolated, but the minor diastereomer did appear to be present in the crude reaction mixture (≈9:1 ratio by 1H NMR). Evidence for the sterically congested nature of the sulfone 10a was provided by the NMR spectra, which show evidence of restricted rotation (Scheme 3). In the 1H NMR spectrum the signals for the two ortho-methyl groups are inequivalent, and indeed differ in chemical shift by a remarkable 1.0 ppm. Similarly, in the 13C NMR spectrum, the ortho-disubsituted ring produces 6 aromatic and 2 aliphatic signals, rather than the 4 aromatic and 1 aliphatic signals that would be expected given the symmetrical aromatic substitution pattern. These data indicate that the ortho-methyl groups cannot freely rotate past the nearby sulfone oxygen atoms. Calculations suggested a rotation barrier of 18 kcal/mol, consistent with it being insurmountable at room temperature, vs only 4 kcal/mol for the unsubstituted phenyl ring (see Supporting Information File 1).
The combined results of substrates 7 and 9 (Scheme 3) suggested that a maximum in permitted steric congestion might be demonstrated by reaction of dibenzalacetone substrate 11 [28] with Rongalite. Pleasingly, this reaction was successful, leading to adduct 12a. Notably, in this case, no trace of the minor isomer was observed in the crude 1H NMR spectrum (Supporting Information File 1). We thus used computation to model the cyclization to give products trans-12a and cis-12b, as this substrate appeared to represent a limiting case for both reaction diastereoselectivity and steric tolerance. Given that the reaction takes place in a high concentration of ≈80% acetic acid (pH ≈3), it seemed plausible that activation of the dienone carbonyl group would occur by hydrogen-bonding, rather than complete proton transfer to the carbonyl oxygen lone pairs [29]. We therefore included explicit solvation with acetic acid in our computational work, by which we were able to locate transition states for reactions to give trans-12a and the unobserved cis-12b (Figure 4). The activation energy difference to reach TS-12a, leading to 12a and TS-12b, leading to 12b was found to be a significant 4.7 kcal/mol in favor of TS-12a (see Supporting Information File 1 for a graphical representation of this and TS images directly generated from the computational data). The analogous calculations for 1a and 1b showed a much smaller difference in activation barrier, 1.0 kcal/mol, which is consistent with the lower selectivity observed for that substrate, within the expected accuracy of the calculations (see also Supporting Information File 1) [30]. Similar transition states have been invoked for other cyclizations leading to 6-membered rings [31-33], but this is the first time that they have been computationally modeled for a sulfur nucleophile in a reaction of this type. What follows is an examination of the transition state structures leading to the trans and cis-isomers 12a and 12b; although the transition states leading to 1a and 1b may differ slightly in energy, they are very similar in appearance to those for 12a and 12b. Examination of the calculated transition state structure TS-12a shows a flattened half-chair conformer, which has the aryl rings in pseudo-equatorial positions. TS-12b adopts a different shape, closer to a flattened chair which again allows both aryl groups to be equatorial. There has been much debate in the literature regarding the conformational preferences of sulfur-containing 6-membered rings – spectroscopy of structurally related 5,6-dihydro-2H-thiopyrans suggests a preference for a half-chair in the unsubstituted ring but there is some evidence for boat-like major conformers in substituted systems [23,34-36]. There are clearly some subtle interactions that contribute to the observed energy differences between the differently shaped two transition states, but undoubtedly their aggregation leads to a significant overall energy difference. Although one of the aryl rings is close to eclipsing one of the S–O bonds in TS-trans, there is a significant distortion in the chair shape in TS-cis: the two highlighted CH bonds are not parallel – they are converging and this 1,3-diaxial interaction may be a contributing factor in increasing the activation barrier relative to TS-trans. Further technical discussion of the calculations is provided in Supporting Information File 1.
![[1860-5397-22-56-4]](/bjoc/content/figures/1860-5397-22-56-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Modeling of cyclization transition states (the solvating acetic acid molecules were omitted for clarity, and the curvature of the 6-membered rings is exaggerated for the same reason. Calculated using the PBE0 functional [26] and the def2-SVP basis set [27] (see Supporting Information File 1 for full computational details)).
Figure 4: Modeling of cyclization transition states (the solvating acetic acid molecules were omitted for cla...
Current limitations
Shown in Figure 5 are some substrates that were not successful, which serve to inform the scope and current limitations of this process. Substrates 13, 14, and 15 [33,37,38] are all very electron-poor and resulted in unidentifiable mixtures. This is most likely due to decomposition processes initiated by reduction. Rongalite has been reported to react in this manner with other electron-poor aromatics [39]. Substrate 16 [40], in contrast, is very electron-rich. This produced sluggish reactivity, incomplete conversion and unknown products. Typical signals for the trans-product could be seen in the crude 1H NMR spectrum, but we were not able to isolate this compound in sufficient purity for characterization. We were intrigued to study the cyclooctanone-derived substrate 17 [41], as it would give an interesting bicyclic system 18. However, we did not obtain any of the desired product, with low conversion and a mixture of products, none of which were sulfone 18. The absence of cyclized product is likely due to the necessity of forming a bridgehead enol intermediate which violates Bredt’s rule [42]. A larger ring substrate may have circumvented this problem, but the necessary double aldol reactions to prepare such substrates, using cyclononanone or cyclodecanone are not known. The difficulties in forming medium-ring ketone enolates have been well described [41].
![[1860-5397-22-56-5]](/bjoc/content/figures/1860-5397-22-56-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Rongalite reactivity vs other sulfinates
We finally decided to place the reactivity of Rongalite in context by comparison with other commonly available sulfinates, namely, sodium methanesulfinate and sodium p-toluenesulfinate.
In the course of our previous studies, we had established that Rongalite did not react productively with mesityl oxide (19) to give 20, in contrast to the successful reaction with phorone (21) to give 22 [43] (Scheme 4). As a control reaction, we conducted the addition of commercially available sodium p-toluenesulfinate to mesityl oxide under analogous conditions, which produced known sulfone 23 [44] in high yield. This demonstrates the general viability of sulfinate addition to produce C-tertiary sulfones, but also implies steric limits to these processes in double intermolecular conjugate addition with Rongalite as the nucleophile.
![[1860-5397-22-56-i4]](/bjoc/content/inline/1860-5397-22-56-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Initial reactivity comparison with p-toluenesulfinate.
Scheme 4: Initial reactivity comparison with p-toluenesulfinate.
Our approach was to use competition experiments to indicate the relative reactivity of Rongalite to related aryl- and alkylsulfinates, so that future predictions will be possible about its reactivity with other electrophiles. In anticipation of the likely products from such test reactions, we prepared the mono- and diadducts 24–27 arising from addition of methanesulfinate and p-toluenesulfinate to dibenzalacetone (Scheme 5). Diadducts 25 and 27 were isolated as inseparable 3:2 and 4:3 mixtures of diastereomers, respectively. From these experiments, we observed the following: 1) double conjugate addition of methanesulfinate occurs readily, such that selective preparation of monoadduct 24 requires an excess of dibenzalacetone to prevent double addition; 2) the second addition of p-toluenesulfinate is significantly slower than the first – however this could be explained by the crystallinity of the monoadduct 26, which precipitates much more readily from the reaction mixture than does 24. A longer reaction time and an excess of sodium p-toluenesulfinate is needed to convert 26 to 27 in a high yield; 3) both reactions are cleaner and higher yielding in the case of p-toluenesulfinate addition.
![[1860-5397-22-56-i5]](/bjoc/content/inline/1860-5397-22-56-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of anticipated products in competition experiments with Rongalite and other sulfinates.
Scheme 5: Preparation of anticipated products in competition experiments with Rongalite and other sulfinates.
Competition experiments were then performed by treatment of dibenzalacetone with 1 equivalent of methanesulfinate or p-toluenesulfinate and 1 equivalent of Rongalite (50 °C, AcOH/H2O, o/n) led to a mixture of products. These results are tabulated in Table 1. Mixtures of cyclic and acyclic products were obtained in both cases. Our initial interpretation was that the ratios reflected the relative rates of reaction of Rongalite and the other sulfinates. However, we also considered the possibility that initial sulfinate addition could be reversible. To test this hypothesis, we treated sulfone 26 with Rongalite under standard conditions (Scheme 6) and found approximately a 1:1 mixture of 26 and the cyclic sulfones 1a/1b in the same diastereomeric ratio (≈7:1) as observed earlier. This supports the idea that sulfinate addition is reversible. Adducts 1a and 1b most likely form from dibenzalacetone generated in situ. Therefore, prior elimination of p-toluenesulfinate to give dibenzalacetone, followed by Rongalite addition, would account for the formation of the cyclic products here. This reaction occurs to a significant extent on the timescale of the competition experiments at 50 °C, the results of which cannot therefore be fully reflective of the relative nucleophilicities of Rongalite and the other sulfinates. In contrast, performing the competition experiment at room temperature gave a different product profile in which a good yield of 26 (62%) precipitated from the reaction mixture. No other products were observed in either the precipitate or the extracted filtrate from that reaction. The high selectivity for formation of 26 at room temperature suggests a profile that is more reflective of kinetic control, such that we can argue that 26 forms between 1 and 2 orders of magnitude more rapidly than cyclic products 1a and 1b, such that they are not detectable in the 1H NMR spectra.
![[Graphic 1]](/bjoc/content/inline/1860-5397-22-56-i8.svg?max-width=637&scale=1.0)
![[1860-5397-22-56-i6]](/bjoc/content/inline/1860-5397-22-56-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Further anecdotal evidence that p-toluenesulfinate is more reactive than Rongalite is provided by the fact that monoadduct 26 forms readily at room temperature in high yield. This is not the case for the formation of the cyclic Rongalite adducts, which require higher temperatures and a longer reaction time. However, this does not conclusively indicate beyond all doubt the lower nucleophilicity of Rongalite – it is still possible that loss of formaldehyde from an intermediate such as 28 (inset in scheme to Table 1) is rate-determining. We can, however, conclude that conjugate addition of p-toluenesulfinate is significantly faster than the slowest step of the double conjugate addition to give 1a and 1b. However, the simplest explanation for these results is that Rongalite is significantly less nucleophilic than p-toluenesulfinate. This could be related to the inductive withdrawal of electron density by the hydroxy oxygen, which is absent in the other sulfinates.
We finally elected to examine double intermolecular conjugate addition with some commercially available enones (Scheme 7). These reactions were sluggish, but yielded the expected adducts 30, 32, and 34 as 1:1 diastereomeric mixtures (30 was obtained in a 1:1 mixture according to the crude 1H NMR spectrum, but an analytical sample obtained by crystallization consisted of a 3:1 mixture). In contrast, a control reaction of sodium p-toluenesulfinate with benzalacetone to give known sulfone 35 [45], occurred readily at room temperature in high yield. It appears that there is scope for further development of Rongalite as a nucleophile in double intermolecular conjugate additions, but full realization of this goal is likely to require the intervention of catalysis. Suitable modes of activation might include well-chosen Lewis acids [46], hydrogen-bond donors [47] or, given the high solubility of Rongalite in water, phase-transfer catalysis [48]. Our earlier results indicate that tolerance of steric effects is greater in reactions with doubly electrophilic substrates that render the second conjugate addition intramolecular, justifying our choice of dienone substrates for this study.
![[1860-5397-22-56-i7]](/bjoc/content/inline/1860-5397-22-56-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Double intermolecular additions.
Scheme 7: Double intermolecular additions.
Conclusion
Methodical investigation of substrate scope allied with detailed computational modelling have improved the understanding of diastereoselective sulfone formation from the reaction of Rongalite and readily available dienones. We have also established that Rongalite, despite apparent lower inherent reactivity than other sulfinates, can be used to prepare sterically encumbered systems and even participate in double intermolecular conjugate additions with β-substituted enones. Our work will be of interest to other workers involved in the synthesis of sulfur heterocycles and also those looking to use Rongalite as a source of nucleophilic sulfur in other transformations. We are currently examining extensions of this method and other sulfinate reaction development and catalysis.
Supporting Information
| Supporting Information File 1: Experimental procedures, copies of 1H and 13C NMR spectra and further computational details. | ||
| Format: PDF | Size: 2.4 MB | Download |
Data Availability Statement
Data generated and analyzed during this study is available from the corresponding author upon reasonable request.
References
-
Ilardi, E. A.; Vitaku, E.; Njardarson, J. T. J. Med. Chem. 2014, 57, 2832–2842. doi:10.1021/jm401375q
Return to citation in text: [1] -
Emmett, E. J.; Willis, M. C. Asian J. Org. Chem. 2015, 4, 602–611. doi:10.1002/ajoc.201500103
Return to citation in text: [1] -
Qian, B.-C.; Zhu, C.-Z.; Shen, G.-B. ACS Omega 2022, 7, 39531–39561. doi:10.1021/acsomega.2c04205
Return to citation in text: [1] -
Matavos-Aramyan, S.; Soukhakian, S.; Jazebizadeh, M. H. Phosphorus, Sulfur Silicon Relat. Elem. 2020, 195, 181–193. doi:10.1080/10426507.2019.1672691
Return to citation in text: [1] -
Kotha, S.; Khedkar, P. Chem. Rev. 2012, 112, 1650–1680. doi:10.1021/cr100175t
Return to citation in text: [1] [2] -
Kotha, S.; Khedkar, P.; Dommaraju, Y. Tetrahedron Lett. 2019, 60, 631–648. doi:10.1016/j.tetlet.2019.01.031
Return to citation in text: [1] [2] -
Yu, F.; Mao, R.; Yu, M.; Gu, X.; Wang, Y. J. Org. Chem. 2019, 84, 9946–9956. doi:10.1021/acs.joc.9b01113
Return to citation in text: [1] -
Golla, S.; Kokatla, H. P. J. Org. Chem. 2022, 87, 9915–9925. doi:10.1021/acs.joc.2c00936
Return to citation in text: [1] -
Laha, J. K.; Gupta, P. J. Org. Chem. 2022, 87, 4204–4214. doi:10.1021/acs.joc.1c03031
Return to citation in text: [1] -
Zhang, G.; Sang, Z.; Wu, W.; Fan, Q.; Ding, C. ChemistrySelect 2022, 7, e202202833. doi:10.1002/slct.202202833
Return to citation in text: [1] -
Wang, M.; Tang, B.-C.; Xiang, J.-C.; Chen, X.-L.; Ma, J.-T.; Wu, Y.-D.; Wu, A.-X. Org. Lett. 2019, 21, 8934–8937. doi:10.1021/acs.orglett.9b03212
Return to citation in text: [1] -
Chen, X.-L.; Wang, H.-Y.; Wu, C.-Y.; Tang, B.-C.; Hu, Y.-L.; Ma, J.-T.; Zhuang, S.-Y.; Yu, Z.-C.; Wu, Y.-D.; Wu, A.-X. Org. Lett. 2022, 24, 7659–7664. doi:10.1021/acs.orglett.2c03194
Return to citation in text: [1] -
Golla, S.; Jalagam, S.; Poshala, S.; Kokatla, H. P. Org. Biomol. Chem. 2022, 20, 4926–4932. doi:10.1039/d2ob00665k
Return to citation in text: [1] -
Chen, X.-L.; Wu, C.-Y.; Ma, J.-T.; Zhuang, S.-Y.; Yu, Z.-C.; Wu, Y.-D.; Wu, A.-X. Org. Lett. 2022, 24, 223–227. doi:10.1021/acs.orglett.1c03877
Return to citation in text: [1] -
Shavnya, A.; Coffey, S. B.; Hesp, K. D.; Ross, S. C.; Tsai, A. S. Org. Lett. 2016, 18, 5848–5851. doi:10.1021/acs.orglett.6b02894
Return to citation in text: [1] -
Alvarez, E. M.; Plutschack, M. B.; Berger, F.; Ritter, T. Org. Lett. 2020, 22, 4593–4596. doi:10.1021/acs.orglett.0c00982
Return to citation in text: [1] -
Kerber, R.; Starnick, J. Chem. Ber. 1971, 104, 2035–2043. doi:10.1002/cber.19711040703
Return to citation in text: [1] [2] -
Kim, D.-K.; Um, H.-S.; Park, H.; Kim, S.; Choi, J.; Lee, C. Chem. Sci. 2020, 11, 13071–13078. doi:10.1039/d0sc02947e
Return to citation in text: [1] -
Shavnya, A.; Hesp, K. D.; Tsai, A. S. Adv. Synth. Catal. 2018, 360, 1768–1774. doi:10.1002/adsc.201800071
Return to citation in text: [1] -
Chen, X.-L.; Tang, B.-C.; He, C.; Ma, J.-T.; Zhuang, S.-Y.; Wu, Y.-D.; Wu, A.-X. Chem. Commun. 2020, 56, 13653–13656. doi:10.1039/d0cc05800a
Return to citation in text: [1] -
Goga, M.; Zong, H.; Prana, J.; Michel, R.; Muro, A.; Rubin, E.; Brenya, J.; Bebbington, M. W. P. Synth. Commun. 2023, 53, 1351–1359. doi:10.1080/00397911.2023.2222316
Return to citation in text: [1] [2] -
Rule, N. G.; Detty, M. R.; Kaeding, J. E.; Sinicropi, J. A. J. Org. Chem. 1995, 60, 1665–1673. doi:10.1021/jo00111a027
Return to citation in text: [1] [2] -
Gendron, T.; Kessedjian, H.; Davioud‐Charvet, E.; Lanfranchi, D. A. Eur. J. Org. Chem. 2015, 1790–1796. doi:10.1002/ejoc.201403516
Return to citation in text: [1] [2] -
Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, Part A: Structure and Mechanisms, 4th ed.; Kluwer Academic Publishers: New York, NY, USA, 2000. doi:10.1007/b114222
Return to citation in text: [1] -
Zhang, W.; Benmohamed, R.; Arvanites, A. C.; Morimoto, R. I.; Ferrante, R. J.; Kirsch, D. R.; Silverman, R. B. Bioorg. Med. Chem. 2012, 20, 1029–1045. doi:10.1016/j.bmc.2011.11.039
Return to citation in text: [1] -
Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158–6170. doi:10.1063/1.478522
Return to citation in text: [1] [2] -
Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a
Return to citation in text: [1] [2] -
Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Bioorg. Med. Chem. 2009, 17, 2623–2631. doi:10.1016/j.bmc.2008.10.044
Return to citation in text: [1] -
Bordwell pKa Table. https://organicchemistrydata.org/hansreich/resources/pka/ (accessed Feb 10, 2026).
Return to citation in text: [1] -
Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Angew. Chem., Int. Ed. 2022, 61, e202205735. doi:10.1002/anie.202205735
Return to citation in text: [1] -
Tan, J.; Xu, X.; Zhang, L.; Li, Y.; Liu, Q. Angew. Chem., Int. Ed. 2009, 48, 2868–2872. doi:10.1002/anie.200805703
Return to citation in text: [1] -
Bi, X.; Dong, D.; Liu, Q.; Pan, W.; Zhao, L.; Li, B. J. Am. Chem. Soc. 2005, 127, 4578–4579. doi:10.1021/ja043023c
Return to citation in text: [1] -
Cao, H.; Lin, V.; Wan, P.; Liu, W.; Zeng, J.; Cui, H. Heterocycles 2021, 102, 93–104. doi:10.3987/com-20-14364
Return to citation in text: [1] [2] -
Leal, L. A.; Lister, D. G.; Alonso, J. L.; Tecklenburg, M. M. J.; Villarreal, J. R.; Laane, J. J. Chem. Soc., Faraday Trans. 1994, 90, 2849–2855. doi:10.1039/ft9949002849
Return to citation in text: [1] -
Rondeau, D.; Raoult, E.; Tallec, A.; Sinbandhit, S.; Toupet, L.; Imberty, A.; Pradère, J. P. J. Chem. Soc., Perkin Trans. 2 1996, 2623–2629. doi:10.1039/p29960002623
Return to citation in text: [1] -
Baxter, C. A. R.; Whiting, D. A. J. Chem. Soc. C 1968, 1174–1178. doi:10.1039/j39680001174
Return to citation in text: [1] -
Cao, B.; Wang, Y.; Ding, K.; Neamati, N.; Long, Y.-Q. Org. Biomol. Chem. 2012, 10, 1239–1245. doi:10.1039/c1ob06773g
Return to citation in text: [1] -
Wang, R.-Y.; Li, C.-W.; Cho, S.-T.; Chang, C.-H.; Chen, J.-J.; Shih, T.-L. Arch. Pharm. (Weinheim, Ger.) 2022, 355, 2100448. doi:10.1002/ardp.202100448
Return to citation in text: [1] -
Jarvis, W. F.; Hoey, M. D.; Finocchio, A. L.; Dittmer, D. C. J. Org. Chem. 1988, 53, 5750–5756. doi:10.1021/jo00259a026
Return to citation in text: [1] -
Xue, T.; Li, Y.; Li, X.; Huang, B.; Song, Q.; Nie, J.; Zhu, X. J. Photochem. Photobiol., A 2021, 418, 113395. doi:10.1016/j.jphotochem.2021.113395
Return to citation in text: [1] -
Farrell, P. G.; Read, B. A. Can. J. Chem. 1968, 46, 3685–3690. doi:10.1139/v68-609
Return to citation in text: [1] [2] -
Buchanan, G. L. Chem. Soc. Rev. 1974, 3, 41–63. doi:10.1039/cs9740300041
Return to citation in text: [1] -
Borsdorf, R.; Remane, H. J. Prakt. Chem. 1980, 322, 152–154. doi:10.1002/prac.19803220119
Return to citation in text: [1] -
Sato, T.; Homma, I. Bull. Chem. Soc. Jpn. 1971, 44, 1885–1891. doi:10.1246/bcsj.44.1885
Return to citation in text: [1] -
Lu, G.-p.; Cai, C.; Chen, F.; Ye, R.-l.; Zhou, B.-j. ACS Sustainable Chem. Eng. 2016, 4, 1804–1809. doi:10.1021/acssuschemeng.5b01784
Return to citation in text: [1] -
Walsh, P. J.; Kozlowski, M. C. Fundamentals of Asymmetric Catalysis; University Science Books: Melville, NJ, USA, 2009.
Return to citation in text: [1] -
Steppeler, F.; Iwan, D.; Wojaczyńska, E.; Wojaczyński, J. Molecules 2020, 25, 401. doi:10.3390/molecules25020401
Return to citation in text: [1] -
Lee, H.-J.; Maruoka, K. Nat. Rev. Chem. 2024, 8, 851–869. doi:10.1038/s41570-024-00642-x
Return to citation in text: [1]
| 39. | Jarvis, W. F.; Hoey, M. D.; Finocchio, A. L.; Dittmer, D. C. J. Org. Chem. 1988, 53, 5750–5756. doi:10.1021/jo00259a026 |
| 40. | Xue, T.; Li, Y.; Li, X.; Huang, B.; Song, Q.; Nie, J.; Zhu, X. J. Photochem. Photobiol., A 2021, 418, 113395. doi:10.1016/j.jphotochem.2021.113395 |
| 41. | Farrell, P. G.; Read, B. A. Can. J. Chem. 1968, 46, 3685–3690. doi:10.1139/v68-609 |
| 1. | Ilardi, E. A.; Vitaku, E.; Njardarson, J. T. J. Med. Chem. 2014, 57, 2832–2842. doi:10.1021/jm401375q |
| 7. | Yu, F.; Mao, R.; Yu, M.; Gu, X.; Wang, Y. J. Org. Chem. 2019, 84, 9946–9956. doi:10.1021/acs.joc.9b01113 |
| 8. | Golla, S.; Kokatla, H. P. J. Org. Chem. 2022, 87, 9915–9925. doi:10.1021/acs.joc.2c00936 |
| 9. | Laha, J. K.; Gupta, P. J. Org. Chem. 2022, 87, 4204–4214. doi:10.1021/acs.joc.1c03031 |
| 10. | Zhang, G.; Sang, Z.; Wu, W.; Fan, Q.; Ding, C. ChemistrySelect 2022, 7, e202202833. doi:10.1002/slct.202202833 |
| 11. | Wang, M.; Tang, B.-C.; Xiang, J.-C.; Chen, X.-L.; Ma, J.-T.; Wu, Y.-D.; Wu, A.-X. Org. Lett. 2019, 21, 8934–8937. doi:10.1021/acs.orglett.9b03212 |
| 12. | Chen, X.-L.; Wang, H.-Y.; Wu, C.-Y.; Tang, B.-C.; Hu, Y.-L.; Ma, J.-T.; Zhuang, S.-Y.; Yu, Z.-C.; Wu, Y.-D.; Wu, A.-X. Org. Lett. 2022, 24, 7659–7664. doi:10.1021/acs.orglett.2c03194 |
| 13. | Golla, S.; Jalagam, S.; Poshala, S.; Kokatla, H. P. Org. Biomol. Chem. 2022, 20, 4926–4932. doi:10.1039/d2ob00665k |
| 14. | Chen, X.-L.; Wu, C.-Y.; Ma, J.-T.; Zhuang, S.-Y.; Yu, Z.-C.; Wu, Y.-D.; Wu, A.-X. Org. Lett. 2022, 24, 223–227. doi:10.1021/acs.orglett.1c03877 |
| 23. | Gendron, T.; Kessedjian, H.; Davioud‐Charvet, E.; Lanfranchi, D. A. Eur. J. Org. Chem. 2015, 1790–1796. doi:10.1002/ejoc.201403516 |
| 47. | Steppeler, F.; Iwan, D.; Wojaczyńska, E.; Wojaczyński, J. Molecules 2020, 25, 401. doi:10.3390/molecules25020401 |
| 5. | Kotha, S.; Khedkar, P. Chem. Rev. 2012, 112, 1650–1680. doi:10.1021/cr100175t |
| 6. | Kotha, S.; Khedkar, P.; Dommaraju, Y. Tetrahedron Lett. 2019, 60, 631–648. doi:10.1016/j.tetlet.2019.01.031 |
| 24. | Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, Part A: Structure and Mechanisms, 4th ed.; Kluwer Academic Publishers: New York, NY, USA, 2000. doi:10.1007/b114222 |
| 48. | Lee, H.-J.; Maruoka, K. Nat. Rev. Chem. 2024, 8, 851–869. doi:10.1038/s41570-024-00642-x |
| 4. | Matavos-Aramyan, S.; Soukhakian, S.; Jazebizadeh, M. H. Phosphorus, Sulfur Silicon Relat. Elem. 2020, 195, 181–193. doi:10.1080/10426507.2019.1672691 |
| 21. | Goga, M.; Zong, H.; Prana, J.; Michel, R.; Muro, A.; Rubin, E.; Brenya, J.; Bebbington, M. W. P. Synth. Commun. 2023, 53, 1351–1359. doi:10.1080/00397911.2023.2222316 |
| 45. | Lu, G.-p.; Cai, C.; Chen, F.; Ye, R.-l.; Zhou, B.-j. ACS Sustainable Chem. Eng. 2016, 4, 1804–1809. doi:10.1021/acssuschemeng.5b01784 |
| 2. | Emmett, E. J.; Willis, M. C. Asian J. Org. Chem. 2015, 4, 602–611. doi:10.1002/ajoc.201500103 |
| 3. | Qian, B.-C.; Zhu, C.-Z.; Shen, G.-B. ACS Omega 2022, 7, 39531–39561. doi:10.1021/acsomega.2c04205 |
| 22. | Rule, N. G.; Detty, M. R.; Kaeding, J. E.; Sinicropi, J. A. J. Org. Chem. 1995, 60, 1665–1673. doi:10.1021/jo00111a027 |
| 46. | Walsh, P. J.; Kozlowski, M. C. Fundamentals of Asymmetric Catalysis; University Science Books: Melville, NJ, USA, 2009. |
| 18. | Kim, D.-K.; Um, H.-S.; Park, H.; Kim, S.; Choi, J.; Lee, C. Chem. Sci. 2020, 11, 13071–13078. doi:10.1039/d0sc02947e |
| 19. | Shavnya, A.; Hesp, K. D.; Tsai, A. S. Adv. Synth. Catal. 2018, 360, 1768–1774. doi:10.1002/adsc.201800071 |
| 21. | Goga, M.; Zong, H.; Prana, J.; Michel, R.; Muro, A.; Rubin, E.; Brenya, J.; Bebbington, M. W. P. Synth. Commun. 2023, 53, 1351–1359. doi:10.1080/00397911.2023.2222316 |
| 43. | Borsdorf, R.; Remane, H. J. Prakt. Chem. 1980, 322, 152–154. doi:10.1002/prac.19803220119 |
| 5. | Kotha, S.; Khedkar, P. Chem. Rev. 2012, 112, 1650–1680. doi:10.1021/cr100175t |
| 6. | Kotha, S.; Khedkar, P.; Dommaraju, Y. Tetrahedron Lett. 2019, 60, 631–648. doi:10.1016/j.tetlet.2019.01.031 |
| 22. | Rule, N. G.; Detty, M. R.; Kaeding, J. E.; Sinicropi, J. A. J. Org. Chem. 1995, 60, 1665–1673. doi:10.1021/jo00111a027 |
| 44. | Sato, T.; Homma, I. Bull. Chem. Soc. Jpn. 1971, 44, 1885–1891. doi:10.1246/bcsj.44.1885 |
| 17. | Kerber, R.; Starnick, J. Chem. Ber. 1971, 104, 2035–2043. doi:10.1002/cber.19711040703 |
| 15. | Shavnya, A.; Coffey, S. B.; Hesp, K. D.; Ross, S. C.; Tsai, A. S. Org. Lett. 2016, 18, 5848–5851. doi:10.1021/acs.orglett.6b02894 |
| 16. | Alvarez, E. M.; Plutschack, M. B.; Berger, F.; Ritter, T. Org. Lett. 2020, 22, 4593–4596. doi:10.1021/acs.orglett.0c00982 |
| 17. | Kerber, R.; Starnick, J. Chem. Ber. 1971, 104, 2035–2043. doi:10.1002/cber.19711040703 |
| 20. | Chen, X.-L.; Tang, B.-C.; He, C.; Ma, J.-T.; Zhuang, S.-Y.; Wu, Y.-D.; Wu, A.-X. Chem. Commun. 2020, 56, 13653–13656. doi:10.1039/d0cc05800a |
| 41. | Farrell, P. G.; Read, B. A. Can. J. Chem. 1968, 46, 3685–3690. doi:10.1139/v68-609 |
| 27. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 25. | Zhang, W.; Benmohamed, R.; Arvanites, A. C.; Morimoto, R. I.; Ferrante, R. J.; Kirsch, D. R.; Silverman, R. B. Bioorg. Med. Chem. 2012, 20, 1029–1045. doi:10.1016/j.bmc.2011.11.039 |
| 26. | Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158–6170. doi:10.1063/1.478522 |
| 27. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 33. | Cao, H.; Lin, V.; Wan, P.; Liu, W.; Zeng, J.; Cui, H. Heterocycles 2021, 102, 93–104. doi:10.3987/com-20-14364 |
| 37. | Cao, B.; Wang, Y.; Ding, K.; Neamati, N.; Long, Y.-Q. Org. Biomol. Chem. 2012, 10, 1239–1245. doi:10.1039/c1ob06773g |
| 38. | Wang, R.-Y.; Li, C.-W.; Cho, S.-T.; Chang, C.-H.; Chen, J.-J.; Shih, T.-L. Arch. Pharm. (Weinheim, Ger.) 2022, 355, 2100448. doi:10.1002/ardp.202100448 |
| 23. | Gendron, T.; Kessedjian, H.; Davioud‐Charvet, E.; Lanfranchi, D. A. Eur. J. Org. Chem. 2015, 1790–1796. doi:10.1002/ejoc.201403516 |
| 34. | Leal, L. A.; Lister, D. G.; Alonso, J. L.; Tecklenburg, M. M. J.; Villarreal, J. R.; Laane, J. J. Chem. Soc., Faraday Trans. 1994, 90, 2849–2855. doi:10.1039/ft9949002849 |
| 35. | Rondeau, D.; Raoult, E.; Tallec, A.; Sinbandhit, S.; Toupet, L.; Imberty, A.; Pradère, J. P. J. Chem. Soc., Perkin Trans. 2 1996, 2623–2629. doi:10.1039/p29960002623 |
| 36. | Baxter, C. A. R.; Whiting, D. A. J. Chem. Soc. C 1968, 1174–1178. doi:10.1039/j39680001174 |
| 26. | Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158–6170. doi:10.1063/1.478522 |
| 30. | Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Angew. Chem., Int. Ed. 2022, 61, e202205735. doi:10.1002/anie.202205735 |
| 31. | Tan, J.; Xu, X.; Zhang, L.; Li, Y.; Liu, Q. Angew. Chem., Int. Ed. 2009, 48, 2868–2872. doi:10.1002/anie.200805703 |
| 32. | Bi, X.; Dong, D.; Liu, Q.; Pan, W.; Zhao, L.; Li, B. J. Am. Chem. Soc. 2005, 127, 4578–4579. doi:10.1021/ja043023c |
| 33. | Cao, H.; Lin, V.; Wan, P.; Liu, W.; Zeng, J.; Cui, H. Heterocycles 2021, 102, 93–104. doi:10.3987/com-20-14364 |
| 28. | Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Bioorg. Med. Chem. 2009, 17, 2623–2631. doi:10.1016/j.bmc.2008.10.044 |
| 29. | Bordwell pKa Table. https://organicchemistrydata.org/hansreich/resources/pka/ (accessed Feb 10, 2026). |
© 2026 Goga et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.