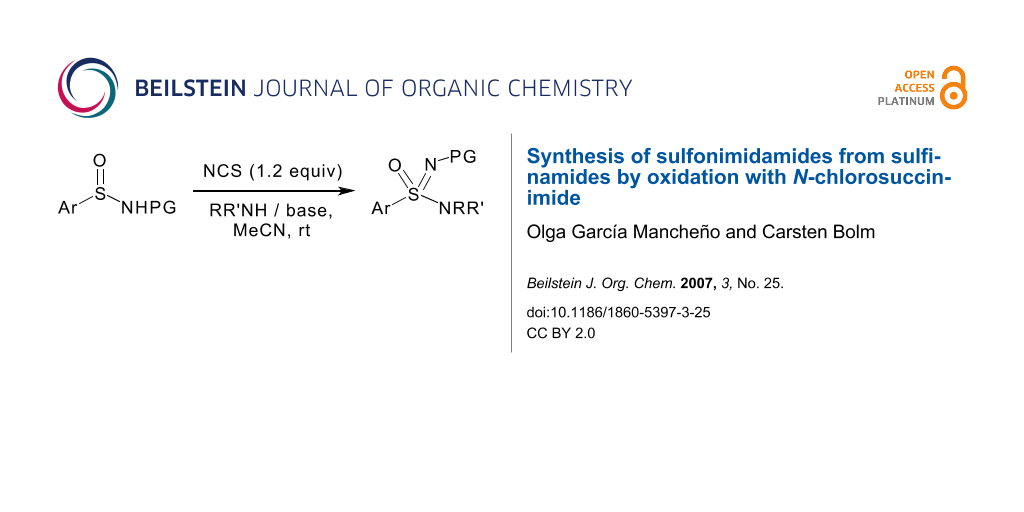
Abstract
Background
The synthesis of sulfonimidamides involves the nucleophilic substitution of a sulfonimidoyl chloride with an amine. However, only four chlorinating systems have been reported for the preparation of the sulfonimidoyl chloride intermediates. Whereas some of them have shown a rather limited substrate spectrum, the most versatile and commonly used tert-butyl hypochlorite is known to be explosive. To establish alternative methods for the synthesis of these molecules is therefore desirable.
Results
The preparation of various p-tolylsulfonimidamides through the reaction of the corresponding N-protected p-tolylsulfinamides and a number of amines in the presence of N-chlorosuccinimide was achieved at room temperature in 50–97% yield.
Conclusion
A convenient alternative procedure for the synthesis of sulfonimidamides from sulfinamides and various amines and sulfonamides using N-chlorosuccinimide as halogenating agent has been developed.

Graphical Abstract
Introduction
Sulfonimidamides 3 are derivatives of sulfonic acid and analogous of sulfonamides, in which one oxygen has been replaced by a nitrogen group. They are known since 1962,[1] and a number of recent investigations focussed on both their reactivity and application in organic synthesis, such as nitrogen sources for metal-catalyzed nitrene transfer reactions, [2-5] and their biological activity, for instance as analogous of oncolytic sulfonylureas [6-8] or mimics of intermediates in protease and amidase reactions. [9] Only a few synthetic approaches for their preparation have been reported, the most direct and common route being the nucleophilic substitution of a sulfonimidoyl chloride 2 with an amine (Scheme 1).
![[1860-5397-3-25-i1]](/bjoc/content/inline/1860-5397-3-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General synthesis of sulfonimidamides 3 from sulfinamides 1.
Scheme 1: General synthesis of sulfonimidamides 3 from sulfinamides 1.
Various chlorinating reagents can be applied for the synthesis of the respective sulfonimidoyl intermediates. Among them, and despite its explosive nature, tert-butyl hypochlorite is the most widely used one. [10-13] Other chlorinating agents present a rather limited substrate scope. For example, chlorine [14-17] is preferred for N-alkyl sulfinamides, reacting very violently with N-aryl derivatives. N-chlorobenzotriazole [15,16,18] is less efficient with bulky amines, and with chloramine-T or -N[3,4,19] only N-tosyl or -nosyl sulfonimidamides can be obtained. In addition, an alternative route to the intermediate N-tosyl or -nosyl sulfonimidoyl chlorides involves the reaction of sulfinyl chlorides with chloramine-T or -N.
In connection with our interest on the application of sulfonimidamides in organic synthesis,[20] we now aimed at exploring an alternative and general procedure for the synthesis of these molecules avoiding the use of potential explosive reagents.
Results and discussion
For the preliminary screening, N-benzoyl sulfinamide 1a was chosen as the model substrate (Scheme 2).
![[1860-5397-3-25-i2]](/bjoc/content/inline/1860-5397-3-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of N-benzoyl sulfonimidamides 3a.
Scheme 2: Synthesis of N-benzoyl sulfonimidamides 3a.
First, the reaction of 1a with different halogenating agents was studied (Table 1).
Table 1: Halogenating agent effect on the synthesis of sulfonimidamides 3a
| Entry | PG | SM | Halogenating agent | Nucleophile | Solvent | Yield of 2 (%)b | Yield of 3 (%)b |
|---|---|---|---|---|---|---|---|
| 1 | Bz | 1a | Chloramine-T | -- | MeCN | 33 | 54 |
| 2 | Bz | 1a | Chloramine-T | -- | MeCN/MS 4Å | 30 | 53 |
| 3 | Bz | 1a | Chloramine-T | -- | CH2Cl2 | 31 | 32 |
| 4 | Bz | 1a | Chloramine-T | -- | Toluene | 68 | -- |
| 5 | Bz | 1a | Chloramine-T | TsNHNa | MeCN | -- | 91 |
| 6 | Bz | 1a | Chloramine-T | TsNHNa | THF | -- | 91 |
| 7 | Bz | 1a | t-BuOCl | TsNHNa | THF | 20 | 73 |
| 8 | Bz | 1a | I2c | TsNHNa | MeCN | -- | <10 |
| 9 | Bz | 1a | Bromamine-T | TsNHNa | MeCN | -- | 71 |
| 10 | Bz | 1a | NBSd | TsNHNa | MeCN | -- | 58 |
| 11 | Bz | 1a | NCS | TsNHNa | THF | -- | 85 |
| 12 | Bz | 1a | NCS | TsNHNa | MeCN | -- | 94 |
| 13 | Bn | 1b | NCS | TsNHNa | MeCN | -- | 56 |
| 14 | Boc | 1c | NCS | TsNHNa | MeCN | -- | 78 |
a Reaction conditions: sulfinamide 1 (1.0 equiv), halogenating agent (1.2 equiv) and TsNHNa (2.0 equiv) in the desired dry solvent (0.1 M) at room temperature for 20 h. b Yield after column chromatography. c Use of 4 equiv of I2. d Okuma et al. mentioned the use of NBS and secondary amines for the preparation of sulfonimidamides from sulfinamides. However, no yield has ever been recorded. [21]
Starting point was the use of chloramine-T as the most common chlorinating reagent for such transformation. As hypothesized, the reaction involved the corresponding sulfonimidoyl chloride. Thus, in the reaction of 1a with chloramine-T in acetonitrile both sulfonimidoyl chloride 2a and sulfinamidamide 3a were isolated in 33 and 54% yield, respectively (Table 1, Entry 1). The use of MS 4Å (1 g/mmol) did not improve this result, leading to a similar mixture of 2a and 3a after 20 h (Entry 2). Other solvents such as dichloromethane gave an unsatisfactory 2a:3a ratio of 1:1 (31 and 32% yield, respectively; Entry 3). Moreover, the reaction in toluene gave exclusively 2a in 68% yield after 24 h (Entry 4). Gratifyingly, using a combination of chloramine-T and TsNHNa, the desired sulfonimidamide 3a was obtained selectively and in high yield (91%) in both acetonitrile and THF as solvents (Table 1, Entries 5 and 6). The reaction with the highly reactive t-BuOCl was surprisingly less efficient, leading to 3a in 73% yield after 20 h, along with unreacted sulfonimidoyl chloride 2a (20%, Table 1, Entry 7). Other halogenating agents, such as I2, bromamine-T, or NBS,[21] were tested as well (Table 1, Entries 8–10), but they exhibited a significantly lower efficiency than the previous chlorinating agents.
Since N-chlorosuccinimide (NCS) had been applied for the oxidation of 4-(methylthio)morpholine towards the synthesis of diazasulfonium salts[22] and the preparation of dialkylamino succinimidosulfonium salts from sulfenamides,[23] this non-explosive and easy to handle oxidizing agent was tested next. To our delight, 1a and NCS reacted well, and in combination with TsNHNa in acetonitrile sulfonimidamide 3a was obtained in excellent yield (94%, Table 1, Entry 12). Noteworthy, the qualitative formation of sulfonimidoyl chloride 2a and its conversion to sulfonimidamide 3a could easily be followed by TLC.
Subsequently, the role of the substituent at the sulfinamide nitrogen was examined. The reactivity of N-benzoyl, -benzyl and -tert-butyl carbamate protected sulfinamides 1a-c, which were prepared according to literature procedures from NH2-free p-tolylsulfinamide using n-BuLi and the corresponding anhydride[24] or by reaction of p-tolylsulfinyl chloride with BnNH2, was compared in the reaction with NCS and TsNHNa in CH3CN at room temperature (Table 1, Entries 12–14).
The best result was obtained with N-benzoyl sulfinamide 1a (94%, Table 1, Entry 12). However, reaction of N-Boc derivative 1c also gave the desired product 3c in good yield (78%, Entry 14). On the other hand, N-benzyl derivative 1b led to 3b in only moderate 56% yield (Entry 13). Therefore, benzoyl was regarded as the N-protecting group of choice for the following studies.
In order to establish the generality of this method, the reaction of 1a with different amines and amides was next investigated (Table 2).
Table 2: Amine scopea
| Entry | RR'NH/Base | Product | R/R' | Yield of 3 (%)b |
|---|---|---|---|---|
| 1 | NsNHNa | 3d | Ns/H | 86 |
| 2 | ThphNHNa | 3e | Thph/H | 94 |
| 3 | BusNHNa | 3f | Bus/H | 50 (28)c |
| 4 | H2NCN/t-BuOK | 3g | CN/H | 85 |
| 5 | PhNH2 | 3h | Ph/H | 94 |
| 6 | Me2NH | 3i | Me/Me | 97 |
| 7 | (TMS)2NH | 3j | H/H | 89 |
a Reaction conditions: sulfinamide 1a (1.0 equiv), NCS (1.2 equiv) and RR'NH/Base (2.0 equiv) in dry acetonitrile (0.1 M) at room temperature. b Yield after column chromatography. c Yield of 2a after column chromatography in brackets.
p-Nitrobenzenesulfonyl and thiophenesulfonylamide sodium salts (NsNHNa and ThphNHNa) were reacted with 1a in the presence of NCS to yield sulfonimidamides 3 in good yields (86 and 94%, Table 2, Entries 1 and 2, respectively). In contrast, when the bulky tert-butylsulfonylamide sodium salt (BusNHNa) was used (Entry 3), the reaction was less efficient. In that case, the desired product 3f was isolated in only moderate yield (50%), together with unreacted sulfonimidoyl chloride 2a (28%), even after prolonged reaction times (24 h). The weakly basic cyanogen amine (pKa ~ 17), which had previously been used in the formation of N-cyano sulfilimines from sulfides using NBS as halogenating agent,[25] was also able to undergo the reaction in the presence of t-BuOK (85%, Table 2, Entry 4). Finally, the more reactive aniline, dimethylamine and hexamethyldisilazane (HMDS) were successfully employed. Even in the absence of an additional base the corresponding products were obtained after short reaction time (2–4 h) in 89–97% yield (Table 2, Entries 5–7).
Ultimately, the cleavage of the N-benzoyl group in 3a was performed (Scheme 3). As expected, the exclusive formation of the most stable regioisomer 4 was observed (73% yield). On the other hand, under the same reaction conditions the attempted deprotection of 3i gave sulfinamide 5 in good yield (87%) as a result of both N-benzoyl cleavage and elimination of the protonated dimethyl amino group.
![[1860-5397-3-25-i3]](/bjoc/content/inline/1860-5397-3-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cleavage of the N-benzoyl group.
Scheme 3: Cleavage of the N-benzoyl group.
In conclusion, we have described a convenient procedure for the synthesis of sulfonimidamides from sulfinamides using a variety of amines and N-chlorosuccinimide as oxidant. The reaction involves sulfonimidoyl chlorides formed in situ, which can be isolated depending on the reaction conditions. The cleavage of the N-benzoyl group has been achieved in the case of N-tosyl derivative 3a. In contrast, the selective deprotection of N-benzoyl sulfonimidamides derived from secondary amine 3i remained unsuccessful due to the concomitant elimination of the substituted amine group under the normal acidic conditions used.
Supporting Information
| Supporting Information File 1: Synthesis of Sulfonimidamides from Sulfinamides by Oxidation with N-Chlorosuccinimide. Experimental Section. Experimental procedures, characterization of new compounds and 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Levchenko, E. S.; Derkach, N. Y.; Kirsanov, A. V. Zh. Obshch. Khim. 1962, 32, 1208–1212.
Return to citation in text: [1] -
Liang, C.; Robert-Peillard, F.; Fruit, C.; Müller, P.; Dodd, R. H.; Dauban, P. Angew. Chem. 2006, 118, 4757–4760. doi:10.1002/ange.200601248
Angew. Chem., Int. Ed. 2006, 45, 4641–4644. doi:10.1002/anie.200601248
Return to citation in text: [1] -
Fruit, C.; Robert-Peillard, F.; Bernardinelli, G.; Müller, P.; Dodd, R. H.; Dauban, P. Tetrahedron: Asymmetry 2005, 16, 3484–3487. doi:10.1016/j.tetasy.2005.07.005
Return to citation in text: [1] [2] -
Di Chenna, P. H.; Robert-Peillard, F.; Dauban, P.; Dodd, R. H. Org. Lett. 2004, 6, 4503–4505. doi:10.1021/ol048167a
Return to citation in text: [1] [2] -
Leca, D.; Toussaint, A.; Mareau, C.; Fensterbank, L.; Lacôte, E.; Malacria, M. Org. Lett. 2004, 6, 3573–3575. doi:10.1021/ol0485520
Return to citation in text: [1] -
Saxena, A.; Agrawal, V. K.; Khadikar, P. V. Oxid. Commun. 2003, 26, 9–13.
Return to citation in text: [1] -
Toth, J. E.; Grindey, G. B.; Ehlhardt, W. J.; Ray, J. E.; Boder, G. B.; Bewley, J. R.; Klingerman, K. K.; Gates, S. B.; Rinzel, S. M.; Schultz, R. M.; Weir, L. C.; Worzalla, J. F. J. Med. Chem. 1997, 40, 1018–1025. doi:10.1021/jm960673l
Return to citation in text: [1] -
Toth, J. E.; Ray, J.; Deeter, J. J. Org. Chem. 1993, 58, 3469–3472. doi:10.1021/jo00064a044
Return to citation in text: [1] -
Cathers, B. E.; Schloss, J. V. Bioorg. Med. Chem. Lett. 1999, 9, 1527–1532. doi:10.1016/S0960-894X(99)00241-3
Return to citation in text: [1] -
Johnson, C. R.; Wambsgans, A. J. Org. Chem. 1979, 44, 2278–2280. doi:10.1021/jo01327a051
Return to citation in text: [1] -
Reggelin, M.; Junker, B. Chem.–Eur. J. 2001, 7, 1232–1239. doi:10.1002/1521-3765(20010316)7:6<1232::AID-CHEM1232>3.0.CO;2-L
Return to citation in text: [1] -
Kluge, R.; Hocke, H.; Schulz, M.; Schilke, F. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 149, 179–206. doi:10.1080/10426509908037031
Return to citation in text: [1] -
Harmata, M. Tetrahedron Lett. 1989, 30, 437–440. doi:10.1016/S0040-4039(00)95221-3
Return to citation in text: [1] -
Jonsson, E. U.; Bacon, C. C.; Johnson, C. R. J. Am. Chem. Soc. 1971, 93, 5306–5308. doi:10.1021/ja00749a084
Return to citation in text: [1] -
Jonsson, E. U.; Bacon, C. C.; Johnson, C. R. J. Am. Chem. Soc. 1971, 93, 5308–5309. doi:10.1021/ja00749a085
Return to citation in text: [1] [2] -
Johnson, C. R.; Jonsson, E. U.; Bacon, C. C. J. Org. Chem. 1979, 44, 2055–2061. doi:10.1021/jo01327a001
Return to citation in text: [1] [2] -
Okuma, K.; Koike, T.; Ohta, H. J. Org. Chem. 1988, 53, 4190–4193. doi:10.1021/jo00253a007
Return to citation in text: [1] -
Pyne, S. G. J. Org. Chem. 1986, 51, 81–87. doi:10.1021/jo00351a017
Return to citation in text: [1] -
Tsushima, S.; Yamada, Y.; Onami, T.; Oshima, K.; Chaney, M. O.; Jones, N. D.; Swartzendruber, J. K. Bull. Chem. Soc. Jpn. 1989, 62, 1167–1178. doi:10.1246/bcsj.62.1167
Return to citation in text: [1] -
Worch, C.; Bolm, C. Synthesis 2007, 1355–1358. doi:10.1055/s-2007-965994
Return to citation in text: [1] -
Okuma, K.; Higuchi, N.; Kaji, S.; Takeuchi, H.; Ohta, H.; Matsuyama, H.; Kamigata, N.; Kobayashi, M. Bull. Chem. Soc. Jpn. 1990, 63, 3223–3229. doi:10.1246/bcsj.63.3223
Return to citation in text: [1] [2] -
Minato, H.; Okuma, K.; Kobayashi, M. J. Org. Chem. 1978, 43, 652–658. doi:10.1021/jo00398a029
Return to citation in text: [1] -
Haacke, M.; Benack, H. Synthesis 1976, 308–310. doi:10.1055/s-1976-24024
Return to citation in text: [1] -
García-Ruano, J. L.; Alonso, R.; Zarzuelo, M. M.; Noheda, P. Tetrahedron: Asymmetry 1995, 6, 1133–1142. doi:10.1016/0957-4166(95)00139-G
Return to citation in text: [1] -
García Mancheño, O.; Bistri, O.; Bolm, C. Org. Lett. 2007, 9, 3809–3811. doi:10.1021/ol7016577
Return to citation in text: [1]
| 1. | Levchenko, E. S.; Derkach, N. Y.; Kirsanov, A. V. Zh. Obshch. Khim. 1962, 32, 1208–1212. |
| 10. | Johnson, C. R.; Wambsgans, A. J. Org. Chem. 1979, 44, 2278–2280. doi:10.1021/jo01327a051 |
| 11. | Reggelin, M.; Junker, B. Chem.–Eur. J. 2001, 7, 1232–1239. doi:10.1002/1521-3765(20010316)7:6<1232::AID-CHEM1232>3.0.CO;2-L |
| 12. | Kluge, R.; Hocke, H.; Schulz, M.; Schilke, F. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 149, 179–206. doi:10.1080/10426509908037031 |
| 13. | Harmata, M. Tetrahedron Lett. 1989, 30, 437–440. doi:10.1016/S0040-4039(00)95221-3 |
| 25. | García Mancheño, O.; Bistri, O.; Bolm, C. Org. Lett. 2007, 9, 3809–3811. doi:10.1021/ol7016577 |
| 9. | Cathers, B. E.; Schloss, J. V. Bioorg. Med. Chem. Lett. 1999, 9, 1527–1532. doi:10.1016/S0960-894X(99)00241-3 |
| 6. | Saxena, A.; Agrawal, V. K.; Khadikar, P. V. Oxid. Commun. 2003, 26, 9–13. |
| 7. | Toth, J. E.; Grindey, G. B.; Ehlhardt, W. J.; Ray, J. E.; Boder, G. B.; Bewley, J. R.; Klingerman, K. K.; Gates, S. B.; Rinzel, S. M.; Schultz, R. M.; Weir, L. C.; Worzalla, J. F. J. Med. Chem. 1997, 40, 1018–1025. doi:10.1021/jm960673l |
| 8. | Toth, J. E.; Ray, J.; Deeter, J. J. Org. Chem. 1993, 58, 3469–3472. doi:10.1021/jo00064a044 |
| 2. |
Liang, C.; Robert-Peillard, F.; Fruit, C.; Müller, P.; Dodd, R. H.; Dauban, P. Angew. Chem. 2006, 118, 4757–4760. doi:10.1002/ange.200601248
Angew. Chem., Int. Ed. 2006, 45, 4641–4644. doi:10.1002/anie.200601248 |
| 3. | Fruit, C.; Robert-Peillard, F.; Bernardinelli, G.; Müller, P.; Dodd, R. H.; Dauban, P. Tetrahedron: Asymmetry 2005, 16, 3484–3487. doi:10.1016/j.tetasy.2005.07.005 |
| 4. | Di Chenna, P. H.; Robert-Peillard, F.; Dauban, P.; Dodd, R. H. Org. Lett. 2004, 6, 4503–4505. doi:10.1021/ol048167a |
| 5. | Leca, D.; Toussaint, A.; Mareau, C.; Fensterbank, L.; Lacôte, E.; Malacria, M. Org. Lett. 2004, 6, 3573–3575. doi:10.1021/ol0485520 |
| 24. | García-Ruano, J. L.; Alonso, R.; Zarzuelo, M. M.; Noheda, P. Tetrahedron: Asymmetry 1995, 6, 1133–1142. doi:10.1016/0957-4166(95)00139-G |
| 21. | Okuma, K.; Higuchi, N.; Kaji, S.; Takeuchi, H.; Ohta, H.; Matsuyama, H.; Kamigata, N.; Kobayashi, M. Bull. Chem. Soc. Jpn. 1990, 63, 3223–3229. doi:10.1246/bcsj.63.3223 |
| 3. | Fruit, C.; Robert-Peillard, F.; Bernardinelli, G.; Müller, P.; Dodd, R. H.; Dauban, P. Tetrahedron: Asymmetry 2005, 16, 3484–3487. doi:10.1016/j.tetasy.2005.07.005 |
| 4. | Di Chenna, P. H.; Robert-Peillard, F.; Dauban, P.; Dodd, R. H. Org. Lett. 2004, 6, 4503–4505. doi:10.1021/ol048167a |
| 19. | Tsushima, S.; Yamada, Y.; Onami, T.; Oshima, K.; Chaney, M. O.; Jones, N. D.; Swartzendruber, J. K. Bull. Chem. Soc. Jpn. 1989, 62, 1167–1178. doi:10.1246/bcsj.62.1167 |
| 22. | Minato, H.; Okuma, K.; Kobayashi, M. J. Org. Chem. 1978, 43, 652–658. doi:10.1021/jo00398a029 |
| 15. | Jonsson, E. U.; Bacon, C. C.; Johnson, C. R. J. Am. Chem. Soc. 1971, 93, 5308–5309. doi:10.1021/ja00749a085 |
| 16. | Johnson, C. R.; Jonsson, E. U.; Bacon, C. C. J. Org. Chem. 1979, 44, 2055–2061. doi:10.1021/jo01327a001 |
| 18. | Pyne, S. G. J. Org. Chem. 1986, 51, 81–87. doi:10.1021/jo00351a017 |
| 14. | Jonsson, E. U.; Bacon, C. C.; Johnson, C. R. J. Am. Chem. Soc. 1971, 93, 5306–5308. doi:10.1021/ja00749a084 |
| 15. | Jonsson, E. U.; Bacon, C. C.; Johnson, C. R. J. Am. Chem. Soc. 1971, 93, 5308–5309. doi:10.1021/ja00749a085 |
| 16. | Johnson, C. R.; Jonsson, E. U.; Bacon, C. C. J. Org. Chem. 1979, 44, 2055–2061. doi:10.1021/jo01327a001 |
| 17. | Okuma, K.; Koike, T.; Ohta, H. J. Org. Chem. 1988, 53, 4190–4193. doi:10.1021/jo00253a007 |
| 21. | Okuma, K.; Higuchi, N.; Kaji, S.; Takeuchi, H.; Ohta, H.; Matsuyama, H.; Kamigata, N.; Kobayashi, M. Bull. Chem. Soc. Jpn. 1990, 63, 3223–3229. doi:10.1246/bcsj.63.3223 |
© 2007 Mancheño and Bolm; licensee Beilstein-Institut
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)