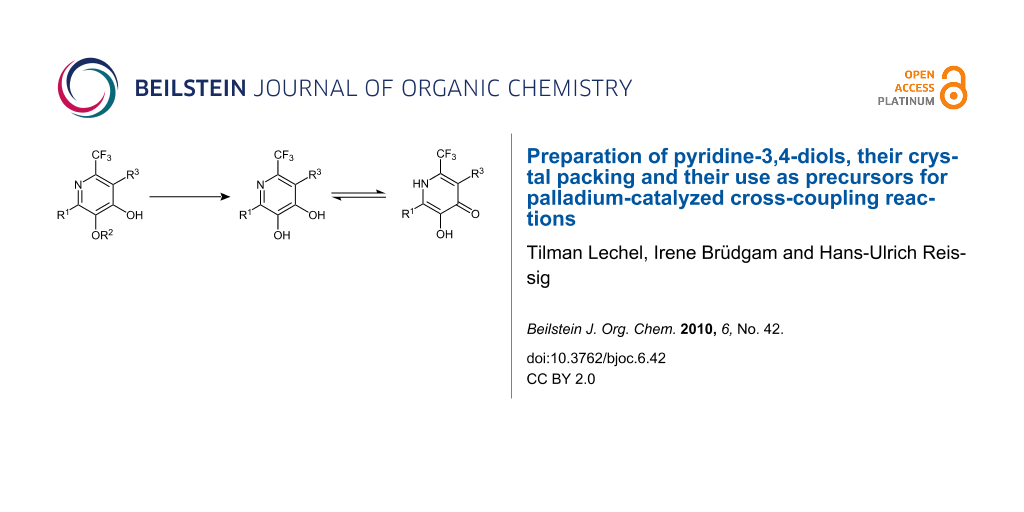
Abstract
A series of trifluoromethyl-substituted 3-alkoxypyridinol derivatives has been deprotected to furnish pyridine-3,4-diol derivatives in good yields. The X-ray crystal structure analysis proved that a 1:1 mixture of pyridine-3,4-diols and their pyridin-4-one tautomers exist in the solid state. Subsequent conversion into bis(perfluoroalkanesulfonate)s were smoothly achieved. The obtained compounds were used as substrates for palladium-catalyzed coupling reactions. Fluorescence measurements of the biscoupled products showed a maximum of emission in the violet region of the spectrum.

Graphical Abstract
Introduction
Pyridine scaffolds have been found in numerous naturally occurring compounds and are also frequently used in functional materials [1-4]. Pyridindiol derivatives are of particular interest as building blocks for the construction of dendritic nanostructures in supramolecular chemistry [5], whereas N-protected pyridine-3,4-diols find applications as potent chelating agents in medicinal chemistry [6]. Furthermore, perfluorinated heteroaromatic compounds are interesting synthetic intermediates for the development of novel pharmaceuticals [7]. Continuing our research on heterocyclic chemistry based on alkoxyallenes [8-17], we focused on the synthesis of trifluoromethyl-substituted pyridine derivatives [18-23]. Herein, we report different methods for the deprotection of a range of 3-alkoxypyridinols 1 to give pyridine-3,4-diols 2 and the corresponding tautomers 2′. This equilibrium between pyridindiols and hydroxypyridinones will be thoroughly investigated in the solid state as well as in solution. Furthermore, subsequent transformations into bistriflate or bisnonaflate derivatives will be described, followed by palladium-catalyzed coupling reactions. The resulting biscoupling products are analysed with regard to their photophysical properties.
Results and Discussion
The preparation of pyridine-3,4-diol derivatives as depicted in Scheme 1 succeeded by using highly substituted trifluoromethyl-substituted 4-hydroxypyridine precursors 1 that have been prepared in two steps from lithiated alkoxyallenes, nitriles and carboxylic acids [21]. It is noteworthy, that the respective protecting group at C-3 of the pyridine core was originally incorporated with the alkoxyallene moiety. The mild cleavage of the benzyl-protected pyridine 1a to diol 2a was achieved by hydrogenolysis in the presence of catalytic amounts of palladium on charcoal. Methyl ethers such as 1b or 1d were cleaved by Lewis-acids. The (2-trimethylsilyl)ethyl-protected pyridine 1c was easily deprotected to diol 2c by a Brønsted acid such as TFA. In most cases, the corresponding pyridindiols 2a–d were obtained in good yields (63%–quant.).
![[1860-5397-6-42-i1]](/bjoc/content/inline/1860-5397-6-42-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Deprotection of 3-alkoxypyridinols 1 to pyridine-3,4-diols 2. aMethod a: Pd/C, H2, MeOH, rt, 1 d; bMethod b: BBr3, CH2Cl2, 0 °C to rt, 1 d; cMethod c: TFA:CH2Cl2 (1:2), rt, 1 h.
Scheme 1: Deprotection of 3-alkoxypyridinols 1 to pyridine-3,4-diols 2. aMethod a: Pd/C, H2, MeOH, rt, 1 d; b...
NMR-measurements in CDCl3 showed that the obtained pyridine-3,4-diols 2 are in equilibrium with their pyridin-4-one tautomers 2′. For instance, in case of 2c the equilibrium is strongly shifted to the pyridin-4-one 2c′ (ratio 2c:2c′ = 30:70). This ratio could be completely shifted to the pyridine-3,4-diol side by a polar protic solvent such as methanol. Surprisingly, the X-ray crystal structure measurement of compound 2c [24] revealed that in the solid state a 1:1 ratio of diol and its pyridinone tautomer 2c′ is preferred. Figure 1 shows that two pyridine-3,4-diol molecules are in one plane with two pyridinone molecules in a perpendicular plane. The two alternating planes are connected by hydrogen bridges.
![[1860-5397-6-42-1]](/bjoc/content/figures/1860-5397-6-42-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of compound 2c/2c′.
Figure 1: X-ray crystal structure of compound 2c/2c′.
As a consequence of our ongoing interest in perfluoroalkyl sulfonate chemistry [25], we converted the two hydroxyl groups into triflates or nonaflates, respectively (Scheme 2). These substituents represent very good leaving groups for subsequent functionalisations such as palladium-catalyzed C–C cross-coupling reactions [26,27]. At first, the pyridindiols 2a and 2c–d were treated with Et3N in dichloromethane and an excess of Tf2O or Nf2O, respectively. This provided bistriflates or bisnonaflates 3a–d as the only products in moderate to very good yields (47–90%). A direct comparison showed that the treatment of 2c with Tf2O led to a higher yield than that with Nf2O. This may be due to lower steric hindrance in the case of the triflating reagent.
![[1860-5397-6-42-i2]](/bjoc/content/inline/1860-5397-6-42-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Conversion of pyridine-3,4-diols 2 into pyridinediyl bistriflates or -nonaflates 3. a) Et3N, Rf2O, CH2Cl2, 0 °C to rt, 1 d.
Scheme 2: Conversion of pyridine-3,4-diols 2 into pyridinediyl bistriflates or -nonaflates 3. a) Et3N, Rf2O, ...
As typical examples of possible palladium-catalyzed cross-couplings we performed several Sonogashira-reactions [28-30]. As described in Scheme 3, the pyridinyl-bistriflates or -nonaflates 3 were coupled with alkynes like phenylacetylene or (triisopropylsilyl)acetylene using Pd(PPh3)4 or alternatively, Pd(OAc)2/PPh3 as catalyst and CuI as co-catalyst in the presence of a 1:2 mixture of iPr2NH and DMF. Only the corresponding biscoupled products 4, which were isolated in moderate yields (30–44%) have been observed. Comparing entries 2 and 3, the coupling of an alkyne with a bisnonaflate gave a slightly lower yield than that with the corresponding bistriflate. A subsequent cleavage of the triisopropylsilyl group with a fluoride source provided product 5 in 58% yield.
![[1860-5397-6-42-i3]](/bjoc/content/inline/1860-5397-6-42-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Sonogashira couplings of pyridinediyl bis(perfluoroalkanesulfonates) 3. a) Pd(PPh3)4 [or Pd(OAc)2/PPh3], CuI, iPr2NH, DMF, 70 °C, 4 h. b) TBAF, THF, rt, 1 h.
Scheme 3: Sonogashira couplings of pyridinediyl bis(perfluoroalkanesulfonates) 3. a) Pd(PPh3)4 [or Pd(OAc)2/P...
The biscoupling reaction led to extended π-systems which might have interesting photophysical properties [31-33]. Hence, absorption and emission of 4b and 4c were studied. The results are depicted in Figure 2 and show absorption maxima in the range of 275–295 nm whereas the emission maxima are located between 385–400 nm.
![[1860-5397-6-42-2]](/bjoc/content/figures/1860-5397-6-42-2.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 2: Absorption and fluorescence spectra of compounds 4b and 4c.
Figure 2: Absorption and fluorescence spectra of compounds 4b and 4c.
Both products are fluorescent, emitting light in the violet region and show similar Stokes shifts. Owing to the additional phenyl substituent at C-2 of pyridine 4c, the π-system is slightly more extended and obviously influences the absorption and emission maxima with a bathochromic shift of 10 nm.
Conclusion
In conclusion, we have successfully demonstrated that 3-alkoxypyridines are ideal precursors for the synthesis of pyridine-3,4-diol derivatives. The coexistence of pyridindiol and pyridinone tautomers in the solid state was discovered by an X-ray structure analysis. It was shown that pyridine-3,4-diols could easily be converted into bis(perfluoroalkanesulfonates) which represent substrates for the construction of extended π-systems using palladium-catalyzed coupling reactions. Moreover, compounds 4b–c show interesting photophysical properties that might be thoroughly investigated in the future. The 3,4-dialkynyl-pyridine derivatives 4 or 5 are also candidates for Bergman cyclizations [34,35] which may establish a route to isoquinoline derivatives.
Experimental
Deprotection of 3-alkoxypyridin-4-ols 1, typical procedures
Cleavage of the benzyloxy group by hydrogenolysis
A mixture of 1a (970 mg, 3.43 mmol) and palladium (365 mg, 10% on charcoal, 0.34 mmol) in methanol (6 mL) was stirred for one day under an atmosphere of hydrogen. Filtration of the reaction mixture through celite with methanol afforded 489 mg (74%) of 2a as a colorless solid, mp 216 °C.
2-Methyl-6-(trifluoromethyl)pyridine-3,4-diol (2a): 1H NMR (CD3OD, 500 MHz): δ = 2.41 (s, 3H, Me), 7.00 (s, 1H, 5-H) ppm. OH-signals could not be detected. 13C NMR (CD3OD, 126 MHz): δ = 17.7 (q, Me), 108.0 (d, C-5), 123.1 (q, 1JCF = 273 Hz, CF3), 139.1 (q, 2JCF = 35.3 Hz, C-6), 139.0, 144.4, 154.1 (3 s, C-2, C-3, C-4) ppm. IR (KBr): ν = 3350–3240 (O-H, N-H), 3110–3040 (=C-H), 3000–2670 (C-H), 1650–1550 (C=O, C=C) cm−1. HRMS (ESI-TOF) calcd. for C12H9F3NO2 [M+H]+: 194.0423, found: 194.0419. C7H6F3NO2 (193.1): calcd. C, 43.53; H, 3.13; N, 7.25; found: C, 43.94; H, 3.10; N, 6.95.
Cleavage of the methoxy group by BBr3
To a solution of 1b (370 mg, 1.14 mmol) in dichloromethane (4 mL) under an argon atmosphere, BBr3 (1 M in CH2Cl2, 3.42 mL, 3.42 mmol) was added dropwise at 0 °C and allowed to warm to room temperature. The reaction was monitored by TLC; upon completion, ice-water was added and the mixture was extracted three times with dichloromethane (5 mL). The combined organic phases were dried over Na2SO4 and concentrated to dryness. Column chromatography on silica gel (hexane/ethyl acetate = 4:1) afforded 222 mg (63%) of 2b as a colorless solid, mp 192 °C.
2-tert-Butyl-5-phenyl-6-(trifluoromethyl)pyridine-3,4-diol (2b): 1H NMR (CD3OD, 500 MHz): δ = 1.45 (s, 9H, t-Bu), 7.24–7.47 (m, 5H, Ph) ppm. OH-signals could not be detected. 13C NMR (CD3OD, 101 MHz): δ = 28.8, 38.6 (q, s, t-Bu), 124.7 (s, C-5), 126.3 (q, 1JCF = 274 Hz, CF3), 129.3, 129.4, 131.5, 133.6 (3 d, s, Ph), 138.6 (q, 2JCF = 32.3 Hz, C-6), 144.3, 151.1, 154.3 (3 s, C-2, C-3, C-4) ppm. IR (KBr): 3450–3260 (O-H, N-H), 3085–3060 (=C-H), 3040–2880 (C-H), 1655–1585 (C=O, C=C) cm−1. HRMS (80 eV, 90 °C) m/z calcd. for C16H16F3NO2: 311.11331; found: 311.11266.
Cleavage of the (2-trimethylsilyl)ethoxy group with TFA
Pyridine derivative 1c (90 mg, 0.268 mmol) was dissolved in a 1:5 mixture of trifluoroacetic acid and dichloromethane (3 mL) and stirred for 1 h at room temperature. After the addition of water and dichloromethane (5 mL) the layers were separated and the aqueous layer was extracted twice with dichloromethane (8 mL). The combined organic layers were dried over Na2SO4 and concentrated to dryness. Column chromatography (silica gel, hexane/ethyl acetate = 2:1) provided 63 mg (quant.) of 2c and 2c′ in a ratio of 30:70 as a colorless solid, mp 102–103 °C.
2-tert-Butyl-6-(trifluoromethyl)pyridine-3,4-diol (2c): 1H NMR (CDCl3, 500 MHz): δ = 1.43 (s, 9H, t-Bu), 7.11 (s, 1H, 5-H) ppm. OH-signals could not be detected. 13C NMR (CDCl3, 126 MHz): δ = 28.2, 37.6 (q, s, t-Bu), 106.5 (d, C-5), 115.6 (q, 1JCF = 294 Hz, CF3), 132.2 (q, 2JCF = 37.0 Hz, C-6), 142.2, 150.7, 154.6 (3 s, C-2, C-3, C-4) ppm.
2-tert-Butyl-3-hydroxy-6-(trifluoromethyl)pyridin-4(1H)-one (2c′): 1H NMR (CDCl3, 500 MHz): δ = 1.51 (s, 9H, t-Bu), 6.82 (s, 1H, 5-H), 8.43 (sbr, 1H, NH) ppm. OH-signal could not be detected. 13C NMR (CDCl3, 126 MHz): δ = 26.7, 34.9 (q, s, t-Bu), 120.1 (q, 1JCF = 273 Hz, CF3), 108.9 (d, C-5), 138.4 (q, 2JCF = 36.4 Hz, C-6), 136.7, 146.5, 170.7 (3 s, C-2, C-3, C-4) ppm. IR (KBr): 3490–3330 (O-H, N-H), 3100–3060 (=C-H), 2960–2870 (C-H), 1710–1580 (C=O, C=C) cm−1. C10H12F3NO2 (235.2): calcd. C, 51.07; H, 5.14; N, 5.96; found: C, 50.87; H, 5.04; N, 5.81.
Conversion into pyridinediyl bis(perfluoroalkanesulfonates), typical procedure
Pyridine-3,4-diol 2c (100 mg, 0.425 mmol) was dissolved in dichloromethane (4 mL) and Et3N (0.24 mL, 1.70 mmol) was added. The solution was cooled to 0 °C and Tf2O (0.29 mL, 1.70 mmol) was added dropwise. After stirring for 1 d at room temperature the reaction mixture was diluted with water (5 mL) and extracted three times with dichloromethane (5 mL). The combined organic phases were dried over Na2SO4 and concentrated to dryness. Column chromatography on silica gel (hexane) afforded 190 mg (90%) of 3b as a colorless oil (volatile under high vacuum).
2-tert-Butyl-6-(trifluoromethyl)pyridine-3,4-diyl bistriflate (3b): 1H NMR (CDCl3, 500 MHz): δ = 1.51 (s, 9H, t-Bu), 7.71 (s, 1H, 5-H) ppm. 13C NMR (CDCl3, 126 MHz): δ = 29.5, 40.0 (q, s, t-Bu), 112.1 (dq, 3JCF = 3.2 Hz, C-5), 118.5, 119.8 (2 q, 1JCF = 321 Hz each, OTf), 120.0 (q, 1JCF = 275 Hz, CF3), 147.2 (q, 2JCF = 36.9 Hz, C-6), 136.0, 149.4, 166.5 (3 s, C-2, C-3, C-4) ppm. z19F NMR (CDCl3, 470 MHz): δ = −68.3 (s, CF3), −71.1, −72.4 (2 s, OTf) ppm. IR (film): ν = 3110–3080 (=C-H), 2980–2880 (C-H), 1600–1575 (C=C) cm−1. C12H10F9NO6S2 (499.3): calcd. C, 28.86; H, 2.02; N, 2.81; found: C, 28.89; H, 1.68; N 2.87.
Sonogashira coupling reaction, typical procedure
A mixture of pyridinediyl bistriflate 3b (245 mg, 0.491 mmol), Pd(PPh3)4 (79 mg, 0.069 mmol), CuI (9.4 mg, 0.049 mmol), (triisopropylsilyl)acetylene (215 mg, 1.18 mmol) in DMF (2.3 mL) and diisopropylamine (1.2 mL) was heated to 60 °C for 4 h under an argon atmosphere. The mixture was allowed to cool to room temperature, diluted with brine (5 mL) and extracted three times with diethyl ether (5 mL). The combined organic phases were dried over Na2SO4 and concentrated to dryness. The residue was purified by column chromatography on silica gel (hexane) followed by HPLC to give 98 mg (35%) of 4a as a colorless oil (volatile under high vacuum).
2-tert-Butyl-6-(trifluoromethyl)-3,4-bis[(triisopropylsilyl)ethynyl] pyridine (4a): 1H NMR (CDCl3, 500 MHz): δ = 1.09–1.17 (m, 42H, TIPS), 1.56 (s, 9H, t-Bu), 7.51 (s, 1H, 5-H) ppm. 13C NMR (CDCl3, 126 MHz): δ = 11.4, 11.6, 18.71, 18.74 (2 d, 2 q, TIPS), 28.7, 40.0 (q, s, t-Bu), 81.6, 90.2, 102.6, 103.7 (4 s, C≡C), 108.2 (s, C-4), 121.3 (q, 1JCF = 274 Hz, CF3), 121.4 (dq, 3JCF = 3.1 Hz, C-5), 143.7 (q, 2JCF = 34.9 Hz, C-6), 137.4 (s, C-3), 170.9 (s, C-2) ppm. 19F NMR (CDCl3, 470 MHz): δ = −68.4 (s, CF3) ppm. IR (film): ν = 2950–2860 (=C-H, C-H), 2145–2065 (C≡C), 1750–1575 (C=C) cm−1. HRMS (ESI-TOF) calcd. for C32H53F3NSi2 [M+H]+: 564.3663; found 564.3690.
Conversion to bisalkyne 5
Pyridine derivative 4a (50 mg, 0.089 mmol) was dissolved in THF (2 mL) and TBAF (0.36 mL, 1 M in THF, 0.356 mmol) was added at room temperature. After stirring for 1 h the reaction mixture was diluted with water (3 mL) and extracted three times with ethyl acetate (3 mL). The combined organic phases were dried over Na2SO4 and concentrated to dryness. Column chromatography on silica gel (hexane/ethyl acetate = 40:1) afforded 13 mg (58%) of 5 as a colorless solid, mp 79–81 °C.
2-tert-Butyl-3,4-diethynyl-6-(trifluoromethyl)pyridine (5): 1H NMR (CDCl3, 500 MHz): δ = 1.54 (s, 9H, t-Bu), 3.57, 3.92 (2 s, 2H, C≡CH), 7.56 (s, 1H, 5-H) ppm. 13C NMR (CDCl3, 126 MHz): δ = 28.7, 39.9 (q, s, t-Bu), 79.5, 79.7, 86.5, 92.5 (2 s, 2 d, C≡CH), 120.1 (dq, 3JCF = 2.8 Hz, C-5), 121.1 (q, 1JCF = 274 Hz, CF3), 121.3 (q, 4JCF = 1.2 Hz, C-4), 137.0 (s, C-3), 144.5 (q, 2JCF = 35.3 Hz, C-6), 171.0 (s, C-2) ppm. 19F NMR (CDCl3, 470 MHz): δ = −68.4 (s, CF3) ppm. IR (KBr): ν = 3305 (≡C-H), 3000–2850 (=C-H, C-H), 2225–2105 (C≡C), 1765–1575 (C=C) cm−1. HRMS (ESI-TOF) calcd. for C14H13F3N [M+H]+: 252.0995; found 252.1009.
Supporting Information
Supporting Information File 1 contains the supplementary data for compounds 2d, 3a, 3c–d and 4b–c.
| Supporting Information File 1: Supplementary data for compounds 2d, 3a, 3c–d and 4b–c. | ||
| Format: PDF | Size: 251.3 KB | Download |
References
-
Müller, T. J. J.; Bunz, U. H. F., Eds. Functional Organic Materials; Wiley-VCH: Weinheim, Germany, 2007.
Return to citation in text: [1] -
McKillop, A.; Boulton, A. J. In Comprehensive Heterocyclic Chemistry; Rees, C. W.; Katritzky, A. R., Eds.; Pergamon: Oxford, U.K., 1984; Vol. 2, pp 67–134.
Return to citation in text: [1] -
Jones, G. In Comprehensive Heterocyclic Chemistry; McKillop, A., Ed.; Pergamon: Oxford, U.K., 1996; Vol. 5, pp 167–243.
Return to citation in text: [1] -
Spitzner, D. Product Class 1: Pyridines. In Six-Membered Hetarenes with One Nitrogen or Phosphorus Atom; Black, D. StC., Ed.; Science of Synthesis, Vol. 15; Thieme: Stuttgart, Germany, 2004; pp 11–284.
Return to citation in text: [1] -
Christinat, N.; Scopelliti, R.; Severin, K. J. Org. Chem. 2007, 72, 2192–2200. doi:10.1021/jo062607p
Return to citation in text: [1] -
Dehkordi, L. S.; Liu, Z. D.; Hider, R. C. Eur. J. Med. Chem. 2008, 43, 1035–1047. doi:10.1016/j.ejmech.2007.07.011
Return to citation in text: [1] -
Negwer, M. Organic-Chemical Drugs and Their Synonyms, 7th ed.; Akademie-Verlag: Berlin, Germany, 1994.
Return to citation in text: [1] -
Reissig, H.-U.; Schade, W.; Okala Amombo, M. G.; Pulz, R.; Hausherr, A. Pure Appl. Chem. 2002, 74, 175–180. doi:10.1351/pac200274010175
Return to citation in text: [1] -
Kaden, S.; Reissig, H.-U. Org. Lett. 2006, 8, 4763–4766. doi:10.1021/ol061538y
Return to citation in text: [1] -
Sörgel, S.; Azap, C.; Reissig, H.-U. Org. Lett. 2006, 8, 4875–4878. doi:10.1021/ol061932w
Return to citation in text: [1] -
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h
Return to citation in text: [1] -
Pfrengle, F.; Lentz, D.; Reissig, H.-U. Angew. Chem. 2009, 121, 3211–3215. doi:10.1002/ange.200805724
Angew. Chem., Int. Ed. 2009, 48, 3165–3169. doi:10.1002/anie.200805724
Return to citation in text: [1] -
Lechel, T.; Lentz, D.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 5432–5435. doi:10.1002/chem.200900386
Return to citation in text: [1] -
Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220
Return to citation in text: [1] -
Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d
Return to citation in text: [1] -
Lechel, T.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2555–2564. doi:10.1002/ejoc.201000056
Return to citation in text: [1] -
Lechel, T.; Reissig, H.-U. Pure Appl. Chem., in press.
Return to citation in text: [1] -
Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322
Return to citation in text: [1] -
Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398
Return to citation in text: [1] -
Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789
Return to citation in text: [1] [2] -
Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2010, 75, 726–732. doi:10.1021/jo9022183
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem., in press.
Return to citation in text: [1] -
CCDC-766558 (for 2c) contains the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566
Return to citation in text: [1] -
Sonogashira, K. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E.-i.; de Meijere, A., Eds.; Wiley: New York, 2002; pp 493–529.
Return to citation in text: [1] -
Marsden, J. A.; Haley, M. M. Cross-Coupling Reactions to sp Carbon Atoms. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere, A.; Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 317–394.
Return to citation in text: [1] -
Negishi, E.-i.; Anastasia, L. Chem. Rev. 2003, 103, 1979–2017. doi:10.1021/cr020377i
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] -
Doucet, H.; Hierso, J.-C. Angew. Chem. 2007, 119, 850–888. doi:10.1002/ange.200602761
Angew. Chem., Int. Ed. 2007, 46, 834–871. doi:10.1002/anie.200602761
Return to citation in text: [1] -
Forrest, S. R.; Thompson, M. E., Eds. Organic Electronics and Optoelectronics. Chem. Rev. 2007, 107, 923–1386. doi:10.1021/cr0501590
Return to citation in text: [1] -
Müllen, K.; Scherf, U., Eds. Organic Light-Emitting Devices: Synthesis, Properties and Applications; Wiley-VCH: Weinheim, Germany, 2006.
Return to citation in text: [1] -
Yamaguchi, Y.; Tanaka, T.; Kobayashi, S.; Wakamiya, T.; Matsubara, Y.; Yoshida, Z.-i. J. Am. Chem. Soc. 2005, 127, 9332–9333. doi:10.1021/ja051588i
Return to citation in text: [1] -
Basak, A.; Mandal, S.; Bag, S. S. Chem. Rev. 2003, 103, 4077–4094. doi:10.1021/cr020069k
Return to citation in text: [1] -
Choy, N.; Blanco, B.; Wen, J.; Krishan, A.; Russell, K. C. Org. Lett. 2000, 2, 3761–3764. doi:10.1021/ol006061j
Return to citation in text: [1]
| 1. | Müller, T. J. J.; Bunz, U. H. F., Eds. Functional Organic Materials; Wiley-VCH: Weinheim, Germany, 2007. |
| 2. | McKillop, A.; Boulton, A. J. In Comprehensive Heterocyclic Chemistry; Rees, C. W.; Katritzky, A. R., Eds.; Pergamon: Oxford, U.K., 1984; Vol. 2, pp 67–134. |
| 3. | Jones, G. In Comprehensive Heterocyclic Chemistry; McKillop, A., Ed.; Pergamon: Oxford, U.K., 1996; Vol. 5, pp 167–243. |
| 4. | Spitzner, D. Product Class 1: Pyridines. In Six-Membered Hetarenes with One Nitrogen or Phosphorus Atom; Black, D. StC., Ed.; Science of Synthesis, Vol. 15; Thieme: Stuttgart, Germany, 2004; pp 11–284. |
| 8. | Reissig, H.-U.; Schade, W.; Okala Amombo, M. G.; Pulz, R.; Hausherr, A. Pure Appl. Chem. 2002, 74, 175–180. doi:10.1351/pac200274010175 |
| 9. | Kaden, S.; Reissig, H.-U. Org. Lett. 2006, 8, 4763–4766. doi:10.1021/ol061538y |
| 10. | Sörgel, S.; Azap, C.; Reissig, H.-U. Org. Lett. 2006, 8, 4875–4878. doi:10.1021/ol061932w |
| 11. | Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h |
| 12. |
Pfrengle, F.; Lentz, D.; Reissig, H.-U. Angew. Chem. 2009, 121, 3211–3215. doi:10.1002/ange.200805724
Angew. Chem., Int. Ed. 2009, 48, 3165–3169. doi:10.1002/anie.200805724 |
| 13. | Lechel, T.; Lentz, D.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 5432–5435. doi:10.1002/chem.200900386 |
| 14. | Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220 |
| 15. | Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d |
| 16. | Lechel, T.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2555–2564. doi:10.1002/ejoc.201000056 |
| 17. | Lechel, T.; Reissig, H.-U. Pure Appl. Chem., in press. |
| 7. | Negwer, M. Organic-Chemical Drugs and Their Synonyms, 7th ed.; Akademie-Verlag: Berlin, Germany, 1994. |
| 6. | Dehkordi, L. S.; Liu, Z. D.; Hider, R. C. Eur. J. Med. Chem. 2008, 43, 1035–1047. doi:10.1016/j.ejmech.2007.07.011 |
| 34. | Basak, A.; Mandal, S.; Bag, S. S. Chem. Rev. 2003, 103, 4077–4094. doi:10.1021/cr020069k |
| 35. | Choy, N.; Blanco, B.; Wen, J.; Krishan, A.; Russell, K. C. Org. Lett. 2000, 2, 3761–3764. doi:10.1021/ol006061j |
| 5. | Christinat, N.; Scopelliti, R.; Severin, K. J. Org. Chem. 2007, 72, 2192–2200. doi:10.1021/jo062607p |
| 25. | Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566 |
| 28. | Negishi, E.-i.; Anastasia, L. Chem. Rev. 2003, 103, 1979–2017. doi:10.1021/cr020377i |
| 29. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 30. |
Doucet, H.; Hierso, J.-C. Angew. Chem. 2007, 119, 850–888. doi:10.1002/ange.200602761
Angew. Chem., Int. Ed. 2007, 46, 834–871. doi:10.1002/anie.200602761 |
| 24. | CCDC-766558 (for 2c) contains the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. |
| 31. | Forrest, S. R.; Thompson, M. E., Eds. Organic Electronics and Optoelectronics. Chem. Rev. 2007, 107, 923–1386. doi:10.1021/cr0501590 |
| 32. | Müllen, K.; Scherf, U., Eds. Organic Light-Emitting Devices: Synthesis, Properties and Applications; Wiley-VCH: Weinheim, Germany, 2006. |
| 33. | Yamaguchi, Y.; Tanaka, T.; Kobayashi, S.; Wakamiya, T.; Matsubara, Y.; Yoshida, Z.-i. J. Am. Chem. Soc. 2005, 127, 9332–9333. doi:10.1021/ja051588i |
| 21. | Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789 |
| 18. | Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322 |
| 19. | Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s |
| 20. | Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398 |
| 21. | Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789 |
| 22. | Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2010, 75, 726–732. doi:10.1021/jo9022183 |
| 23. | Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem., in press. |
| 26. | Sonogashira, K. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E.-i.; de Meijere, A., Eds.; Wiley: New York, 2002; pp 493–529. |
| 27. | Marsden, J. A.; Haley, M. M. Cross-Coupling Reactions to sp Carbon Atoms. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere, A.; Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 317–394. |
© 2010 Lechel et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)