Abstract
We report on the block copolymerization of two structurally different norbornene monomers (±)-endo,exo-bicyclo[2.2.1]-hept-5-ene-2,3-dicarboxylic acid dimethylester (7), and (±)-endo,exo-bicyclo[2.2.1]-hept-5-ene-2,3-dicarboxylic acid bis(1-oxyl-2,2,6,6-tetramethyl-piperidin-4-yl) ester (9) using ruthenium based Grubbs’ type initiators [(PCy3)2Cl2Ru(benzylidene)] G1 (PCy3 = tricyclohexylphosphine), [(H2IMes)(PCy3)Cl2Ru(benzylidene)] G2 (H2IMes = 1,3-bis(mesityl)-2-imidazolidinylidene), [(H2IMes)(py)2Cl2Ru(benzylidene)] G3 (py = pyridine or 3-bromopyridine) and Umicore type initiators [(PCy3)2Cl2Ru(3-phenylinden-1-ylidene)] U1 (PCy3 = tricyclohexylphosphine), [(H2IMes)(PCy3)Cl2Ru(3-phenylinden-1-ylidene)] U2 (H2IMes = 1,3-bis(mesityl)-2-imidazolidinylidene), [(H2IMes)(py)Cl2Ru(3-phenylinden-1-ylidene)] U3 (py = pyridine or 3-bromopyridine) via ring opening polymerization (ROMP). The crossover reaction and the polymerization kinetics were investigated using matrix assisted laser desorption ionization mass spectroscopy (MALDI-TOF) and nuclear magnetic resonance (NMR), respectively. MALDI showed that there was a complete crossover reaction after the addition of 25 equivalents of the second monomer. NMR investigation showed that U3 gave a faster rate of polymerization in comparison to U1. The synthesis of block copolymers with molecular weights up to Mn = 31 000 g/mol with low polydispersities (Mw/Mn = 1.2) is reported.
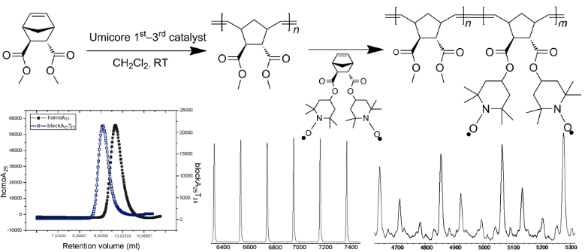
Graphical Abstract
Introduction
Block copolymers are macromolecules composed of linear or non-linear arrangements of chemically different polymeric chains. If the different blocks are incompatible, a rich variety of well defined self-assembled structures both in bulk and selective solvents arises [1]. The synthetic approach to block copolymers has been widely discussed [1] and achieved extensively via living polymerization methods. Thus, besides acyclic diene metathesis polymerization (ADMET) [2], ring opening metathesis polymerization (ROMP) [3-5] is another type of olefin metathesis polymerization that can be used for the synthesis of block copolymers.
Early examples of catalysts for ROMP were based on molybdenum alkylidene catalysts, however, the true breakthrough of the method was hampered by the restricted functional group tolerance of Schrock initiators due to their sensitivity towards protic solvents and air [6]. With the advent of the Grubbs’ catalyst G1 (see Scheme 1) and related complexes as initiators, polymerization reactions can now not only be performed in protic media but also without rigorous exclusion of molecular oxygen. However, these advantages are hampered by the considerable lower activity of catalysts such as G1 when compared with Schrock’s molybdenum catalysts [7-9]. Often, the polydispersity indices of the resulting polymers obtained with initiator G1 are large with values ranging between 1.3 and 1.5 arising from an unfavorable rate of initiation (ki) relative to propagation (kp) as well as considerable secondary metathesis (backbiting). Grubbs’ second generation catalyst G2 displays an activity comparable to the Schrock type initiators. It exhibits a higher functional group tolerance than G1, but initiation by catalyst G2 is often slow as a result of the slow dissociation of the phosphine group, sometimes limiting its application in polymer synthesis. Alternatively, Grubbs’ third generation catalyst G3 introduced by Grubbs et al. [10] has an ultrafast initiating ruthenium benzylidene. The rate of reaction of G3 with ethyl vinyl ether thus is six orders of magnitude higher than for G2 [10], leading to a faster initiation and often lower polydispersities of the resulting polymers.
![[1860-5397-6-59-i1]](/bjoc/content/inline/1860-5397-6-59-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Grubbs G1–G3 and Umicore U1–U3 catalyst.
Scheme 1: Grubbs G1–G3 and Umicore U1–U3 catalyst.
Recently, structural variations of G1–G3 catalysts generated a new series of catalysts U1–U3 bearing indenyl-carbenes instead of benzylidene-carbenes. These new catalysts are now commercially available and are well known as the Umicore catalysts (NEOLYST™). However, their synthetic profile with respect to the synthesis of block copolymers is largely unexplored. As we recently have reported extensively on the use of ROMP methods in blockcopolymer synthesis [11-13], either via direct copolymerization or coupled to postmodification methods via azide/alkyne-“click”-chemistry [14-17], the crossover reaction of more complex monomers remains the crucial factor in achieving defined BCP’s with low polydispersities. In a recent example, the crossover reaction of various monomers with the Grubbs’ type catalysts G1–G3 was studied in detail via MALDI mass spectrometry [11], revealing a more detailed picture of the crossover reaction (Scheme 2). Thus mass spectrometry could often demonstrate insufficient crossover reactions between monomers of different reactivity such as monomer A 7 and monomer T 9, despite a low polydispersity when the crossover reaction was monitored by conventional GPC methods [11]. A semi-quantification method of the respective spectra allowed a good correlation between the rate-constants of initiation and propagation of the different monomers.
![[1860-5397-6-59-i2]](/bjoc/content/inline/1860-5397-6-59-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic pathway to BCP-AnTm using Grubbs’ (1–3) and Umicore (4–6) type catalysts.
Scheme 2: Synthetic pathway to BCP-AnTm using Grubbs’ (1–3) and Umicore (4–6) type catalysts.
The current publication describes the synthesis of block copolymers AnTm composed of monomers 7 and 9, initiated via the catalysts U1–U3, as well as mass spectrometric investigations of the crossover reactions via MALDI methods. The incorporation of the free radical 9 into block copolymer is an important contribution in the generation of polymers for reversible charge storage materials, as monomer 9 can accept or donate electrons reversibly.
Results and Discussion
The polymerization of monomer 7 was investigated using catalysts U1–U3 (see Table 1). Basically, the catalyst U3 showed good polymerization behavior, furnishing the homopolymers (entries 2, 3) with excellent control of chain length and low polydispersities (Mw/Mn = 1.2). The catalysts U1 and U2 gave poor results (see entries 4 and 5) presumably due to slow initiation and fast polymerization, which is in accord with the structurally similar catalysts G1 and G2 (see Scheme 1). Similarly, the polymerization of monomer T 9 was investigated, which gave good results with the catalysts G2, G3, and U3 (entries 6, 7, and 8). The other catalysts G1, U1, and U2 did not yield good polymerization results (data not shown) and were therefore not considered further for the synthesis of the respective block copolymers.
Table 1: Overview of polymerization result of monomer A 7 and monomer T 9 with the catalysts U1–U3 and G1–G3.
| entry | polymer | 1st monomer | 2nd monomer | catalyst | molecular weight (g/mol) | PDI | |
|---|---|---|---|---|---|---|---|
| GPC | calculated | ||||||
| 1 | Homo-A15a | Mon-A | — | G3 | 3700 | 3100 | 1.1 |
| 2 | Homo-A15a | Mon-A | — | U3 | 2700 | 3150 | 1.2 |
| 3 | Homo-A25a | Mon-A | — | U3 | 4410 | 5250 | 1.2 |
| 4 | Homo-A50b | Mon-A | — | U1 | 8500 | 10500 | 1.2 |
| 5 | Homo-A50b | Mon-A | — | U2 | 418000 | 10500 | 1.4 |
| 6 | Homo-T20 | Mon-T | — | G2 | 12900 | 9800 | 1.7 |
| 7 | Homo-T100b | Mon-T | — | G3 | 48800 | 49000 | 1.3 |
| 8 | Homo-T50b | Mon-T | — | U3 | 24100 | 24500 | 1.3 |
| 9 | BCP-A15T1a | Mon-A | Mon-T | U3 | 3200 | 3640 | 1.1 |
| 10 | BCP-A15T2a | Mon-A | Mon-T | U3 | 4020 | 4130 | 1.2 |
| 11 | BCP-A15T4a | Mon-A | Mon-T | U3 | 4400 | 5110 | 1.1 |
| 12 | BCP-A25T1a | Mon-A | Mon-T | U3 | 5100 | 5740 | 1.1 |
| 13 | BCP-A25T2a | Mon-A | Mon-T | U3 | 5500 | 6230 | 1.1 |
| 14 | BCP-A25T4a | Mon-A | Mon-T | U3 | 6100 | 7770 | 1.1 |
| 15 | BCP-A15T1a | Mon-A | Mon-T | G3 | 4600 | 3640 | 1.1 |
| 16 | BCP-A15T2a | Mon-A | Mon-T | G3 | 4900 | 4140 | 1.1 |
| 17 | BCP-A15T5a | Mon-A | Mon-T | U3 | 5680 | 7700 | 1.1 |
| 18 | BCP-A25T25 | Mon-A | Mon-T | U3 | 17000 | 17500 | 1.1 |
| 19 | BCP-A50T50 | Mon-A | Mon-T | U3 | 31300 | 35000 | 1.2 |
| 20 | BCP-A10T10 | Mon-A | Mon-T | U3 | 7300 | 7000 | 1.1 |
| 21 | BCP-A20T20 | Mon-A | Mon-T | U3 | 13400 | 14000 | 1.1 |
aThe polymer was synthesized for MALDI analysis.
bThe experiment was performed for kinetic measurements by taking samples every 5 minutes.
The 1H NMR spectrum (Figure 1) of the respective homopolymer (A50) clearly shows the expected resonances together with the resonances of the indenyl-moieties of the initiator-structure. The spectrum further revealed that the unsaturated polymer exhibited no stereoregularity (cis/trans ~ 50/50 see the peaks at 5.2 and 5.4 ppm in Figure 1) which is in accordance with results reported in the literature [18]. Figure 2 shows the relevant region of the 13C NMR spectrum, with the approximately equal peak intensities indicating an equal m:r ratio. Thus the polymerization yields the respective polymer, although in poor yields which is underlined when monitoring the kinetics of the polymerization of monomer A 7 using catalysts U1 and U3 (see Figure 3, Figure 4 and Figure 5). As expected in accordance with the known polymerization reactions of the respective Grubbs’ type catalysts, catalyst U3 polymerizes significantly faster (the polymerization reaction is complete after ~20 seconds) whereas the polymerization initiated with catalyst U1 takes significantly longer and never yields significant amounts of the homopolymer (yield < 10 %).
![[1860-5397-6-59-1]](/bjoc/content/figures/1860-5397-6-59-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectrum of the homopolymer A50 synthesized with the catalyst U3.
Figure 1: 1H NMR spectrum of the homopolymer A50 synthesized with the catalyst U3.
![[1860-5397-6-59-2]](/bjoc/content/figures/1860-5397-6-59-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 13C NMR spectrum of the homopolymer A50 synthesized with catalyst U3.
Figure 2: 13C NMR spectrum of the homopolymer A50 synthesized with catalyst U3.
![[1860-5397-6-59-3]](/bjoc/content/figures/1860-5397-6-59-3.svg?scale=1.8&max-width=1024&background=FFFFFF)
Figure 3: Kinetic progress monitored via 1H NMR spectroscopy of the polymerization of monomer A 7 with catalyst U1; [7]/[U1] = 20/1.
Figure 3: Kinetic progress monitored via 1H NMR spectroscopy of the polymerization of monomer A 7 with cataly...
![[1860-5397-6-59-4]](/bjoc/content/figures/1860-5397-6-59-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Kinetic progress monitored via 1H NMR spectroscopy of the polymerization of 7 with catalyst U3; [7]/[U3] = 20/1.
Figure 4: Kinetic progress monitored via 1H NMR spectroscopy of the polymerization of 7 with catalyst U3; [7]...
As the polymerization kinetics of 7 using catalyst U3 could not be monitored effectively with GPC because it was too fast (50 units were polymerized in less than 1 minute), the kinetics were monitored by NMR. NMR measurements were conducted every 8 seconds and the result showed that the polymerization was complete within ~20 seconds as shown in Figure 4 and Figure 5. The rate of polymerization (kp) was calculated by integrating the peaks (6.25 and 6.07 ppm) corresponding to the alkene protons of monomer A 7 as they disappeared. Plotting ln([M]0/[M]t) vs. time (t) gave a straight line as shown in Figure 5 and indicated linear chain growth. The slope of the straight line was divided by the initial initiator concentration [I]0 assuming a first order kinetics which gave kp as 18.5 l·mol−1·s−1.
![[1860-5397-6-59-5]](/bjoc/content/figures/1860-5397-6-59-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ln([M]0/[M]t) vs. time (t) plot obtained from 1H NMR spectra for homopolymer-A50 using catalyst U3; [7]/[U3] = 20/1.
Figure 5: ln([M]0/[M]t) vs. time (t) plot obtained from 1H NMR spectra for homopolymer-A50 using catalyst U3;...
As monomer T 9 is a free stable radical, the progress of its polymerization with catalyst U3 could not be monitored by 1H NMR spectroscopy. Therefore, the conventional method of following the Mn vs. time (t) profile was carried out as shown in Figure 6. Chain growth with a maximum polydispersity of Mw/Mn ~1.3 was observed, clearly proving the high precision of this type of polymerization reaction.
![[1860-5397-6-59-6]](/bjoc/content/figures/1860-5397-6-59-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Mn vs. time (t) kinetic plot and Mw/Mn (PDI) of the polymerization of monomer T 9 with catalyst U3; [9]/[U3] = 50/1.
Figure 6: Mn vs. time (t) kinetic plot and Mw/Mn (PDI) of the polymerization of monomer T 9 with catalyst U3;...
As the polymerization of both, monomer A 7 and monomer T 9 proceeded well with catalyst U3, the synthesis of the BCP was achieved by use of this initiating system to yield the respective BCP-A10T10, A20T20, A25T25 and A50T50 with the expected molecular weight and with low polydispersity (see Table 1, entries 18–21). The GPC traces of A25 block and the A25T25 block copolymer are shown in Figure 7, indicating the expected shift in the retention time after addition of monomer T 9 after all of the monomer A 7 had been consumed.
![[1860-5397-6-59-7]](/bjoc/content/figures/1860-5397-6-59-7.svg?scale=1.9&max-width=1024&background=FFFFFF)
Figure 7: GPC trace of the block copolymer A25T25 synthesized with catalyst U3.
Figure 7: GPC trace of the block copolymer A25T25 synthesized with catalyst U3.
In order to achieve a deeper insight into the exact nature of the crossover reaction when changing from monomer A 7 to monomer T 9 with catalyst U3, the respective reaction was monitored according to our previous methods using MALDI mass spectrometry [11]. Thus homopolymer A25 was initiated with catalyst U3 and subsequently reacted with 1, 2, 5 and 25 equivalents of monomer T after all of the monomer A 7 had been consumed. The respective samples were then quenched with ethyl vinyl ether, and subsequently analyzed by MALDI-TOF mass spectrometry and GPC. The GPC results are shown in Table 1, entries 9–14 and 18–21, indicating that with increasing amount of added monomer T an equal increase of Mn can be observed. However, in order to check for the detailed composition of the reaction mixture, MALDI spectra were measured. As shown in Figure 8, homopolymer A25 can be desorbed well in MALDI, featuring the respective AnNa+-ions as a pure series. Thus the homopolymer An can serve as molecular probe for the subsequent desorption of the individual AnT1, 2, 5-species.
![[1860-5397-6-59-8]](/bjoc/content/figures/1860-5397-6-59-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: MALDI-TOF mass spectra of homopolymer A25 4 synthesized with catalyst U3: (a) full spectrum, (b) expansion.
Figure 8: MALDI-TOF mass spectra of homopolymer A25 4 synthesized with catalyst U3: (a) full spectrum, (b) ex...
The MALDI spectrum of the crossover reaction of A25 with exactly one equivalent of monomer T 9 using U3 as initiator is shown in Figure 9. Thus, together with the still present An-series (visible as AnNa+-series), the respective crossover species AnT1, and AnT2 can be seen as the respective Na+-ions. These results demonstrate that a large amount of An-species did not participate in the crossover reaction, since due to the fast polymerization of monomer T 9, it was rapidly consumed, leading to AnT2-species and its respective higher homologues.
![[1860-5397-6-59-9]](/bjoc/content/figures/1860-5397-6-59-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: MALDI-TOF mass spectrum of BCP-A25T1 synthesized with catalyst U3.
Figure 9: MALDI-TOF mass spectrum of BCP-A25T1 synthesized with catalyst U3.
The respective MALDI spectrum of the crossover reaction of the homopolymer A25 with exactly two equivalents of monomer T 9 is shown in Figure 10. Again, a significant amount of homopolymer An (visible as AnNa+-series) is present in the reaction mixture, the respective crossover-species AnT1, and AnT2 can be seen as the respective Na+-ions. Additionally, the respective series AnT3 is visible, indicative of the further chain growth process after the crossover reaction. Again, despite the excess of Tn-species a large amount of An-species did not participate in the crossover reaction due to the fast polymerization of monomer T 9. MALDI spectra of a further series of block copolymers A25T5 and A25T25 was carried out in order to check for the presence of residual homopolymer A25 in the polymer mixture (see Figure 11). We could not detect any residual homopolymer in either of these final samples. As it is known from our previous investigations, that the homopolymer An in MALDI is desorbed preferentially by a factor of 13 with respect to the A25Tn-species, this now indicates a complete cross-over reaction and thus the successful preparation of block copolymers AnTm via this methodology. Basically, this synthetic approach now allows the synthesis of AT type BCP’s with high precision and chain length control up to molecular weights of ~31000 g/mol.
![[1860-5397-6-59-10]](/bjoc/content/figures/1860-5397-6-59-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: MALDI-TOF mass spectrum of BCP-A25T2 13 synthesized with catalyst U3.
Figure 10: MALDI-TOF mass spectrum of BCP-A25T2 13 synthesized with catalyst U3.
![[1860-5397-6-59-11]](/bjoc/content/figures/1860-5397-6-59-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: MALDI-TOF mass spectrum of BCP-A25T25 synthesized with catalyst U3.
Figure 11: MALDI-TOF mass spectrum of BCP-A25T25 synthesized with catalyst U3.
Conclusion
The synthesis of new block copolymers containing free radical centers within one block via ROMP has been described. MALDI analyses especially provide a detailed picture of the crossover reaction. Basically, the NEOLYST™ catalysts are comparable to the well known Grubbs’ catalysts, indicating a similar profile of initiation and propagation. However, the catalyst U3 is especially a highly potent catalyst for ROMP and displays a broad profile of tolerance against functional groups within the monomer, enabling the successful synthesis of block copolymers containing free-radical species in high densities.
Experimental
General Remarks
Solvents/Reagents/Materials: Catalysts G1, G2 and G3 were obtained from Sigma-Aldrich. Catalysts U1, U2 and U3 were obtained as gifts from the Umicore chemical company. All reagents used for the synthesis of norbornene monomers 7 and 9 were obtained from Sigma-Aldrich Chemical Co. (Germany) and used as received without further purification unless otherwise indicated. Bicyclopentadiene (100%), fumaric acid (99+%), thionyl chloride (99+%, Fluka), pyridine (99.8%), methanol and 4-hydroxy-2,2,6,6-tetramethyl-piperidin-1-oxyl (TEMPOL) were obtained from Sigma-Aldrich and used without further purification. Dichloromethane (CH2Cl2) was freshly distilled over CaH2 and degassed with argon prior to use. Other solvents such as hexane and ethyl acetate were used after distillation.
Instrumentation: 1H NMR spectra were recorded on a Varian Gemini 400 MHz FT-NMR spectrometer, and MestRec (4.9.9.9) was used for data interpretation. The polymerization kinetics of the polymerization reactions with both catalysts U1 and U3 were measured at 25 °C on a 200 MHz FT-NMR spectrometer using CDCl3 as a solvent. GPC analysis was performed on a Viscotek VE2001 system with THF as the eluant at a flow rate of 1 ml/min and an injection volume of 100 µL. Polystyrene standards were used for conventional external calibration using a Viscotek VE3580 refractive index detector. Positive ion MALDI-TOF (matrix-assisted laser desorption ionization time-of-flight) measurements were performed on a Bruker Autoflex-III instrument equipped with a smart ion beam laser. Measurements were carried out in linear and reflector mode. Samples were prepared from THF solution by mixing matrix (20 mg/ml), polymer (20 mg/ml), and salt (20 mg/ml solution) in a ratio of 100:10:1. Dithranol (1,8-dihydroxy-9(10H)-anthracetone, Aldrich 97%) was used as the matrix. Sodium trifluoroacetate (Aldrich, 98%), silver trifluoroacetate (Aldrich, 99.99%) or lithium trifluoroacetate (Aldrich, 99.8%) were added for ion formation, with sodium trifluoroacetate as the optimal salt for obtaining the highest S/N ratio.
Monomer synthesis
5-Norbornene-endo,exo-2,3-dicarboxylic acid dimethylester, monomer A 7 was synthesised according to reference [11], 5-norbornene-endo, exo-2,3-dicarboxylic acid bis(1-oxyl-2,2,6,6-tetramethyl-piperidin-4-yl) ester, monomer T 9, was prepared according to references [19,20].
Synthesis of homopolymers A15 and T20
Monomer A 7 (50.0 mg, 0.23 mmol) dissolved in 1 ml of CH2Cl2 was introduced into a heated and argon flushed glass tube equipped with a magnetic stirring bar. A solution of catalyst U3 (11.8 mg, 0.015 mmol) dissolved in 1 ml of CH2Cl2 was then added. After 5 min of stirring at room temperature, the total consumption of monomer A 7 was confirmed by thin layer chromatography (TLC). The reaction was then quenched with 5 drops of cold ethyl vinyl ether, and the resulting polymer purified by column chromatography (SiO2). The homo-polymerization of monomer T 9 was carried out in the same manner with catalyst G2. Homopolymers (An) with different chain lengths (n = 15, 50, 25) with the catalysts G3, U1, U2 and U3 as initiators were also synthesized using the same procedure by adopting the required polymerization times.
Block copolymer syntheses
The synthesis of block copolymers (An-b-Tn) was carried out analogously to methods developed previously in our laboratory [14,16]. For example the synthesis of BCP-A50T50 was performed by sequential addition of monomers. Monomer A 7 (15 mg, 0.071 mmol) dissolved in 1 ml of CH2Cl2 was introduced into a heated and argon flushed glass tube equipped with a magnetic stirring bar. To this solution, catalyst G3 (1.26 mg, 0.0014 mmol) dissolved in 1 ml of CH2Cl2 was then added. The mixture was allowed to stir at room temperature for 1 h until all of the monomer A 7 was consumed as confirmed by GPC and TLC. Subsequently, monomer T 9 (35 mg, 0.071 mmol) dissolved in 1 ml of CH2Cl2 was then added to the above reaction mixture and stirred for 2 h at room temperature until all of monomer T 9 was consumed, as confirmed by GPC and TLC. The polymerization was quenched by the addition of cold ethyl vinyl ether. The polymer was isolated by column chromatography (SiO2) (eluent: DCM).
Kinetic experiments
A pyrene stock solution was prepared from 70 mg of pyrene dissolved in 2 ml of CDCl3. Monomer A 7 (20.83 mg, 0.099 mmol) dissolved in CDCl3 (0.2 ml) was first introduced into the NMR tube and then the pyrene stock solution (0.2 ml) was added. Before adding the initiator solution, the ratio of the monomer to the internal standard was determined by NMR. On the basis of this value, the monomer concentration at t = 0 was determined. A solution of the catalyst U3 (1.48 mg, 0.0019 mmol), dissolved in CDCl3 (0.2 ml) (in case of catalyst U1 (1.83 mg, 0.0019 mmol)) dissolved in CDCl3 (0.2 ml) was added via a syringe to yield the desired monomer to initiator ratio. After shaking, the tube was inserted into the NMR spectrometer, and the decrease in the monomer with respect to time was monitored by integrating the resonance peaks at 6.27 and 6.07 ppm. For determination of the monomer concentration at t = 0 and the monomer consumption, the corresponding signals at 6.27 and 6.07 ppm from monomer A 7 compared with the one at 8.20 ppm from the internal standard pyrene were integrated. The time between the addition of the initiator solution and the first measurement was added to the first measuring point.
References
-
Hadjichristidis, N.; Pispas, S.; Floudas, G. A. Block Copolymers-synthetic strategies, physical properties and application; John Wiley and Sons: New York, 2002.
Return to citation in text: [1] [2] -
Baughman, T. W.; Wagener, K. B. Adv. Polym. Sci. 2005, 176, 1–42.
Return to citation in text: [1] -
Buchmeiser, M. R. Chem. Rev. 2000, 100, 1565–1604. doi:10.1021/cr990248a
Return to citation in text: [1] -
Frenzel, U.; Nuyken, O. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 2895–2916. doi:10.1002/pola.10324
Return to citation in text: [1] -
Schrock, R. R. Acc. Chem. Res. 1990, 23, 158–165. doi:10.1021/ar00173a007
Return to citation in text: [1] -
Slugovc, C. Macromol. Rapid Commun. 2004, 25, 1283–1297. doi:10.1002/marc.200400150
Return to citation in text: [1] -
Novak, B. M.; Grubbs, R. H. J. Am. Chem. Soc. 1988, 110, 960–961. doi:10.1021/ja00211a043
Return to citation in text: [1] -
Bazan, G. C.; Schrock, R. R.; Choj, H. N.; Gibson, V. C. Macromolecules 1991, 24, 4495–4502. doi:10.1021/ma00016a003
Return to citation in text: [1] -
Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H. Nature 2008, 456, 933–937. doi:10.1038/nature07594
Letter.
Return to citation in text: [1] -
Love, J. A.; Morgan, J. P.; Trnka, T. M.; Grubbs, R. H. Angew. Chem., Int. Ed. 2002, 41, 4035–4037. doi:10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I
Return to citation in text: [1] [2] -
Binder, W. H.; Pulamagatta, B.; Kir, O.; Kurzhals, S.; Barqawi, H.; Tanner, S. Macromolecules 2009, 42, 9457–9466. doi:10.1021/ma902115j
Return to citation in text: [1] [2] [3] [4] [5] -
Binder, W. H.; Kurzhals, S.; Pulamagatta, B.; Decker, U.; Manohar Pawar, G.; Wang, D.; Kühnel, C.; Buchmeiser, M. R. Macromolecules 2008, 41, 8405–8412. doi:10.1021/ma801465r
Return to citation in text: [1] -
Kluger, C.; Binder, W. H. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 485–499. doi:10.1002/pola.21867
Return to citation in text: [1] -
Binder, W. H.; Kluger, C.; Josipovic, M.; Straif, C. J.; Friedbacher, G. Macromolecules 2006, 39, 8092–8101. doi:10.1021/ma061256d
Return to citation in text: [1] [2] -
Binder, W. H.; Kluger, C.; Straif, C. J.; Friedbacher, G. Macromolecules 2005, 38, 9405–9410. doi:10.1021/ma0518252
Return to citation in text: [1] -
Binder, W. H.; Kluger, C. Macromolecules 2004, 37, 9321–9330. doi:10.1021/ma0480087
Return to citation in text: [1] [2] -
Binder, W. H.; Lomoschitz, M.; Sachsenhofer, R.; Friedbacher, G. J. Nanomater. 2009. doi:10.1155/2009/613813.
Article ID 613813.
Return to citation in text: [1] -
Lee, W. L.; Register, A. R. Macromolecules 2005, 38, 1216–1222. doi:10.1021/ma048013a
Return to citation in text: [1] -
Katsumata, T.; Qu, J.; Shiotsuki, M.; Satoh, M.; Wada, J.; Igarashi, J.; Mizoguchi, K.; Masuda, T. Macromolecules 2008, 41, 1175–1183. doi:10.1021/ma7020425
Return to citation in text: [1] -
Katsumata, T.; Satoh, M.; Wada, J.; Shiotsuki, M.; Sanda, F.; Masuda, T. Macromol. Rapid Commun. 2006, 27, 1206–1211. doi:10.1002/marc.200600286
Return to citation in text: [1]
| 14. | Binder, W. H.; Kluger, C.; Josipovic, M.; Straif, C. J.; Friedbacher, G. Macromolecules 2006, 39, 8092–8101. doi:10.1021/ma061256d |
| 16. | Binder, W. H.; Kluger, C. Macromolecules 2004, 37, 9321–9330. doi:10.1021/ma0480087 |
| 1. | Hadjichristidis, N.; Pispas, S.; Floudas, G. A. Block Copolymers-synthetic strategies, physical properties and application; John Wiley and Sons: New York, 2002. |
| 6. | Slugovc, C. Macromol. Rapid Commun. 2004, 25, 1283–1297. doi:10.1002/marc.200400150 |
| 11. | Binder, W. H.; Pulamagatta, B.; Kir, O.; Kurzhals, S.; Barqawi, H.; Tanner, S. Macromolecules 2009, 42, 9457–9466. doi:10.1021/ma902115j |
| 3. | Buchmeiser, M. R. Chem. Rev. 2000, 100, 1565–1604. doi:10.1021/cr990248a |
| 4. | Frenzel, U.; Nuyken, O. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 2895–2916. doi:10.1002/pola.10324 |
| 5. | Schrock, R. R. Acc. Chem. Res. 1990, 23, 158–165. doi:10.1021/ar00173a007 |
| 19. | Katsumata, T.; Qu, J.; Shiotsuki, M.; Satoh, M.; Wada, J.; Igarashi, J.; Mizoguchi, K.; Masuda, T. Macromolecules 2008, 41, 1175–1183. doi:10.1021/ma7020425 |
| 20. | Katsumata, T.; Satoh, M.; Wada, J.; Shiotsuki, M.; Sanda, F.; Masuda, T. Macromol. Rapid Commun. 2006, 27, 1206–1211. doi:10.1002/marc.200600286 |
| 18. | Lee, W. L.; Register, A. R. Macromolecules 2005, 38, 1216–1222. doi:10.1021/ma048013a |
| 1. | Hadjichristidis, N.; Pispas, S.; Floudas, G. A. Block Copolymers-synthetic strategies, physical properties and application; John Wiley and Sons: New York, 2002. |
| 11. | Binder, W. H.; Pulamagatta, B.; Kir, O.; Kurzhals, S.; Barqawi, H.; Tanner, S. Macromolecules 2009, 42, 9457–9466. doi:10.1021/ma902115j |
| 11. | Binder, W. H.; Pulamagatta, B.; Kir, O.; Kurzhals, S.; Barqawi, H.; Tanner, S. Macromolecules 2009, 42, 9457–9466. doi:10.1021/ma902115j |
| 12. | Binder, W. H.; Kurzhals, S.; Pulamagatta, B.; Decker, U.; Manohar Pawar, G.; Wang, D.; Kühnel, C.; Buchmeiser, M. R. Macromolecules 2008, 41, 8405–8412. doi:10.1021/ma801465r |
| 13. | Kluger, C.; Binder, W. H. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 485–499. doi:10.1002/pola.21867 |
| 11. | Binder, W. H.; Pulamagatta, B.; Kir, O.; Kurzhals, S.; Barqawi, H.; Tanner, S. Macromolecules 2009, 42, 9457–9466. doi:10.1021/ma902115j |
| 10. | Love, J. A.; Morgan, J. P.; Trnka, T. M.; Grubbs, R. H. Angew. Chem., Int. Ed. 2002, 41, 4035–4037. doi:10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I |
| 11. | Binder, W. H.; Pulamagatta, B.; Kir, O.; Kurzhals, S.; Barqawi, H.; Tanner, S. Macromolecules 2009, 42, 9457–9466. doi:10.1021/ma902115j |
| 10. | Love, J. A.; Morgan, J. P.; Trnka, T. M.; Grubbs, R. H. Angew. Chem., Int. Ed. 2002, 41, 4035–4037. doi:10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I |
| 7. | Novak, B. M.; Grubbs, R. H. J. Am. Chem. Soc. 1988, 110, 960–961. doi:10.1021/ja00211a043 |
| 8. | Bazan, G. C.; Schrock, R. R.; Choj, H. N.; Gibson, V. C. Macromolecules 1991, 24, 4495–4502. doi:10.1021/ma00016a003 |
| 9. |
Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H. Nature 2008, 456, 933–937. doi:10.1038/nature07594
Letter. |
| 14. | Binder, W. H.; Kluger, C.; Josipovic, M.; Straif, C. J.; Friedbacher, G. Macromolecules 2006, 39, 8092–8101. doi:10.1021/ma061256d |
| 15. | Binder, W. H.; Kluger, C.; Straif, C. J.; Friedbacher, G. Macromolecules 2005, 38, 9405–9410. doi:10.1021/ma0518252 |
| 16. | Binder, W. H.; Kluger, C. Macromolecules 2004, 37, 9321–9330. doi:10.1021/ma0480087 |
| 17. |
Binder, W. H.; Lomoschitz, M.; Sachsenhofer, R.; Friedbacher, G. J. Nanomater. 2009. doi:10.1155/2009/613813.
Article ID 613813. |
© 2010 Adekunle et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)