Abstract
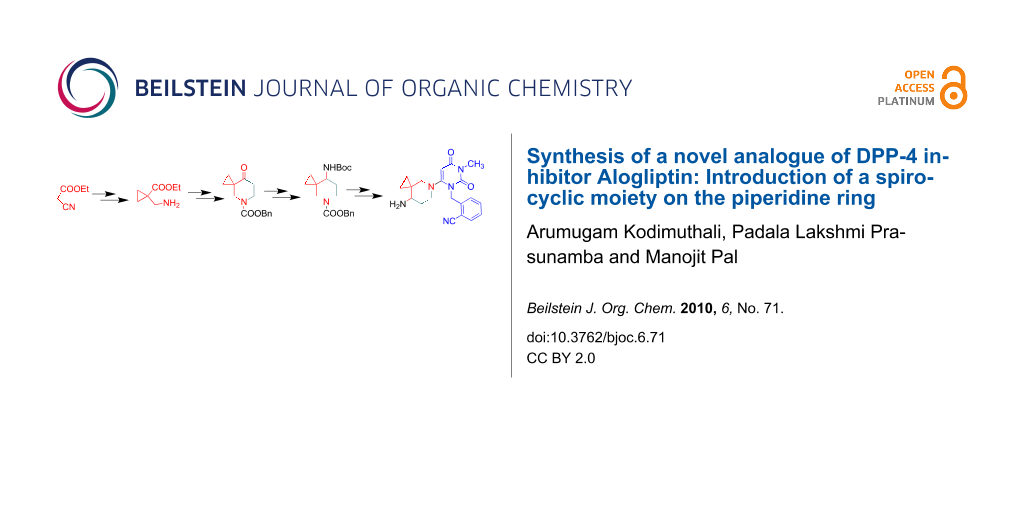
We report the synthesis of a novel analogue of Alogliptin via condensation of two key intermediates one of which is an aminopiperidine derivative bearing a spirocyclic ring on the piperidine moiety. Preparation of the aminopiperidine intermediate was carried out by constructing the cyclopropyl ring prior to assembling the piperidine ring.

Graphical Abstract
Introduction
Inhibition of dipeptidyl peptidase-4 (DPP-4; CD26; E.C. 3.4.14.5) by small molecules has emerged as one of the key approaches for the treatment of type-2 diabetes [1-5]. DPP-4, a member of the prolyl oligopeptidase family of serine protease, cleaves the N-terminal dipeptide from peptides with proline or alanine in the second position. A large number of DPP-4 inhibitors have been reported in the literature [6,7] including NVP-LAF237 (Vildagliptin), MK-0431 (Sitagliptin) and BMS-477118 (Saxagliptin). Presently, Sitagliptin is available for clinical use and Vildagliptin has been launched in Europe only. Several other inhibitors are in various stages of development. For example, Alogliptin or (2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]benzonitrile) (A, Figure 1), a potent (IC50 < 10 nM) and selective inhibitor (selectivity > 10,000 over DPP-8 and 9) is currently being evaluated in phase 3 clinical trials [8-10]. This compound was identified by replacing the quinazolinone moiety of another inhibitor B with a pyrimidine dione (Figure 1) [8]. All these inhibitors that belong to the non-peptidomimetic class contain an aminopiperidinyl moiety which interacts with the DPP-4 enzyme through salt bridges. As part of our ongoing drug discovery program we sought to prepare a novel DPP-4 inhibitor of structure C (Figure 1) bearing a spirocyclic ring on the piperidinyl moiety. Our aim was to study the effect of this structurally modified piperidinyl moiety on the interaction of C with DPP-4 enzyme. To the best of our knowledge this type of Alogliptin analogue has not yet been explored. Herein we report an efficient synthesis of compound C starting from readily available starting materials and reagents.
![[1860-5397-6-71-1]](/bjoc/content/figures/1860-5397-6-71-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Design of a new DPP-4 inhibitor (C) based on Alogliptin (A) and other inhibitors (B).
Figure 1: Design of a new DPP-4 inhibitor (C) based on Alogliptin (A) and other inhibitors (B).
Results and Discussion
A retro synthetic analysis of compound C indicates that the key step would involve the condensation of two key intermediates, i.e., the piperidinyl derivative 9 and the appropriately substituted 6-chloro uracil derivative 11 (Figure 2). The intermediate 9 in turn could be synthesized via functional group manipulations of the spiro derivative 9a. Preparation of 9a can be carried out following two strategies e.g. construction of cyclopropyl ring on piperidine moiety (Strategy 1) or vice versa (Strategy 2). While the use of first strategy for the preparation of 9a has been reported in the literature [11], application of other strategy is not common. Initially, we attempted to synthesize the compound 9a from N-Boc protected piperidone 9b (PG = Boc) by the reported method [11] based on C-alkylation. Surprisingly, introduction of a cyclopropyl group at the position α to the piperidine carbonyl group with 2-chloroethyl dimethyl sulphonium iodide did not work well in our hands. Other methods, e.g., the use of 1,2-dibromoethane in the presence of various bases such as NaH, CH3ONa or KO t-Bu at different temperatures was also examined but failed to afford the desired product. This prompted us to develop an alternative but more effective method based on strategy 2 (Figure 2) for the synthesis of compound 9 (R = Boc). Apart from our own requirements, we noted that substituted piperidine derivatives containing spirocyclic ring have a wide range of therapeutic effects, namely, as Angiotensin Converting Enzyme (ACE) inhibitors [11], antibacterial agents [12-15], Janus kinase 3 (JAK3) inhibitors [16], Calcitonin-Gene Related Peptide (CGRP) modulators [17] etc. Our synthesis of compound 9 (R = Boc) is shown in Scheme 1.
![[1860-5397-6-71-2]](/bjoc/content/figures/1860-5397-6-71-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Strategy to prepare compound C (PG = protecting group).
Figure 2: Strategy to prepare compound C (PG = protecting group).
![[1860-5397-6-71-i1]](/bjoc/content/inline/1860-5397-6-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (i) EtONa, BrCH2CH2Br, EtOH, reflux, 3.5 h (70% yield); (ii) Pd/C, MeOH, H2, RT, 4 h (93% yield); (iii) CH2=CHCOOMe, THF, RT, 3 h (74% yield); (iv) ClCOOBn, TEA, DCM, 0–5 °C, 3 h (80% yield); (v) NaH, DMF, RT, 7 h (86% yield); (vi) NaCl, DMSO, H2O, 110 °C, 7 h (76% yield); (vii) NH3 in EtOH, Ti(OPr)4, NaBH4, 12 h (67% yield); (viii) (Boc)2O, TEA, DCM, 0 °C, 2 h (70% yield); (ix) Pd/C, MeOH, H2, RT, 4 h (85% yield).
Scheme 1: Reagents and conditions: (i) EtONa, BrCH2CH2Br, EtOH, reflux, 3.5 h (70% yield); (ii) Pd/C, MeOH, H2...
Commercially available ethyl cyanoacetate and 1,2-dibromoethane was used to construct the appropriately functionalized cyclopropyl ring (step i, Scheme 1) via a conventional C–C bond forming reaction. The reaction proceeded well to afford the functionalized cyclopropane derivative 1 as a result of the formation of two C–C bonds in a single step. Selective reduction of the cyano group of the resulting cyanoester 1 afforded the corresponding amine 2 (step ii, Scheme 1) which on Michael addition to methyl acrylate followed by N-protection furnished the diester 4 (steps iii and iv, Scheme 1). The N-protection step was necessary to avoid the undesirable participation of the NH group in subsequent steps. A facile base mediated intramolecular cyclization (Dieckmann reaction) [18] of compound 4 provided the key piperidin-4-one ester 5 which on decarboxylation under Krapcho conditions [19] afforded the desired compound 6 (steps v and vi, Scheme 1). Reductive amination of the ketone followed by Boc protection of the resulting amine 7 provided the compound 8 (steps vii and viii, Scheme 1). Finally, deprotection of the secondary amine of compound 8 (step ix, Scheme 1) afforded the target intermediate 9 which was used directly in the next step. It is worth noting that although a shorter synthesis of compound 6 or similar derivative could be achieved starting from N-Boc protected piperidin-4-one [11,20], its preparation from ethyl cyclopropanecarboxylate however, requires a longer synthetic route or the tedious preparation of suitable starting materials [21,22].
With the piperidinyl intermediate 9 in hand, we then focused our attention on the other intermediate 11, the preparation of which has been previously documented [23]. The reaction of commercially available 6-chlorouracil with 2-(bromomethyl)benzonitrile in the presence of NaH afforded the N-alkylated product 10 which on methylation with methyl iodide provided the target intermediate 11 (steps i and ii, Scheme 2). The chloro compound 11 was then reacted with the amine 9 in the presence of NaHCO3 in a sealed tube to give the expected coupled product 12 (step iii, Scheme 2). Finally, N-deprotection of compound 12 using trifluoroacetic acid (TFA) gave the target compound C as the TFA salt. The 1H NMR spectra of compound C indicated the presence of a spiro cyclopropyl ring: the four cyclopropyl protons appeared as a series of four multiplets in the region δ 0.15–0.25, 0.30–0.40, 0.50–0.60 and 0.65–0.75, the vinylic proton of pyrimidine-2,4-dione moiety appeared as a singlet at δ 5.36, whilst two doublets at δ 5.11 and 5.23 were indicative of the benzylic methylene hydrogen atoms. This in conjunction with IR bands at 2230 and 1642 cm−1 for CN and amide carbonyl group, respectively, characterized compound C.
![[1860-5397-6-71-i2]](/bjoc/content/inline/1860-5397-6-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: (i) NaH, LiBr, DMF - DMSO, 12 h (55% yield); (ii) NaH, LiBr, CH3I, DMF - THF, RT, 12 h (65% yield); (iii) NaHCO3, DMSO, 100 °C, 2 h (40% yield); (iv) TFA, THF, RT, 2.5 h (85% yield).
Scheme 2: Reagents and conditions: (i) NaH, LiBr, DMF - DMSO, 12 h (55% yield); (ii) NaH, LiBr, CH3I, DMF - T...
Compound C was then tested for its ability to inhibit DPP-4 enzyme in vitro at three concentrations, e.g., 1.0, 5.0 and 10.0 µM [24]. While significant inhibition of DPP-4 was observed at these concentrations, compound C however, was found to be less potent than Alogliptin. Based on the interaction [8] of compound B with the active site of DPP-4 (Figure 1) it was predicted that (i) the cyanobenzyl group of compound C would fill the S1 pocket (formed by V656, Y631, Y662, W659, Y666, and V711) and interact with Arg125, (ii) the carbonyl at C-2 would provide an important hydrogen bond to the backbone NH of Tyr631 (Figure 3). Although the cyclopropyl ring could potentially occupy the nearby empty space to enhance the hydrophobic interactions with DPP-4, the required orientation of the aminopiperidine motif of C to form an effective salt bridge to Glu205/Gul206 was probably perturbed. This could be the reason for observed lower potency of C in comparison to Alogliptin in vitro. Nevertheless, to gain further insight we plan to conduct Structure-Activity-Relationship (SAR) studies for this class of compound that would eventually help to identify a potential lead possessing favorable pharmacological properties.
![[1860-5397-6-71-3]](/bjoc/content/figures/1860-5397-6-71-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Predicted interactions of compound C with DPP-4.
Figure 3: Predicted interactions of compound C with DPP-4.
Conclusion
In conclusion, we have developed a route to an aminopiperidine derivative bearing a spirocyclic ring on the piperidine moiety by constructing the cyclopropyl ring prior to assembling the piperidine ring. All steps involved in the present synthesis are simple and based on conventional methods that do not require the use of expensive reagents or catalysts. We have demonstrated the utility of this aminopiperidine derivative for the first time in the preparation of a novel analogue of a potent DPP-4 inhibitor Alogliptin. We believe that the present synthesis could find application in the preparation of diverse aminopiperidine derivatives thereby facilitating the synthesis of novel compounds of potential biological interest.
Supporting Information
| Supporting Information File 1: Experimental procedures and spectral data | ||
| Format: PDF | Size: 172.3 KB | Download |
References
-
Weber, A. E. J. Med. Chem. 2004, 47, 4135–4141. doi:10.1021/jm030628v
Return to citation in text: [1] -
Augustyns, K.; Van der Veken, P.; Senten, K.; Haemers, A. Curr. Med. Chem. 2005, 12, 971–998. doi:10.2174/0929867053507298
Return to citation in text: [1] -
Augustyns, K.; Van der Veken, P.; Haemers, A. Expert Opin. Ther. Pat. 2005, 15, 1387–1407. doi:10.1517/13543776.15.10.1387
Return to citation in text: [1] -
von Geldern, T. W.; Trevillyan, J. M. Drug Dev. Res. 2006, 67, 627–642. doi:10.1002/ddr.20138
Return to citation in text: [1] -
Van der Veken, P.; Haemers, A.; Augustyns, K. Curr. Top. Med. Chem. 2007, 7, 621–635.
Return to citation in text: [1] -
Havale, S. H.; Pal, M. Bioorg. Med. Chem. 2009, 17, 1783–1802. doi:10.1016/j.bmc.2009.01.061
Return to citation in text: [1] -
Gupta, R.; Walunj, S. S.; Tokala, R. K.; Parsa, K. V. L.; Singh, S. K.; Pal, M. Curr. Drug Targets 2009, 10, 71–87. doi:10.2174/138945009787122860
Return to citation in text: [1] -
Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L. J. Med. Chem. 2007, 50, 2297–2300. doi:10.1021/jm070104l
Return to citation in text: [1] [2] [3] -
Feng, J.; Gwaltney, S. L.; Stafford, J. A.; Zhang, Z.; Elder, B. J.; Isbester, P. K.; Palmer, G. J.; Salsbury, J. S.; Ulysse, L. WO Patent Application. 2007035629, 2007.
Return to citation in text: [1] -
Feng, J.; Gwaltney, S. L.; Stafford, J. A.; Zhang, Z.; Brodluahrer, P. R.; Elder, B.; Isbester, P.; Fornicola, R. S.; Kisanga, P. B.; Mobele, B. I.; Palmer, G. J.; Reeve, M. M.; Salsbury, J. S.; Ulysse, L. G.; Venkatraman, S. U.S. Patent Application. 20090275750 A1, Nov 5, 2009.
Return to citation in text: [1] -
Benzencon, O.; Bur, D.; Fischli, W.; Remen, L.; Richard-Bildstein, S.; Sifferlen, T.; Weller, T. U.S. Patent Application. 20080234305 A1, Sept 25, 2008.
Return to citation in text: [1] [2] [3] [4] -
Hayakawa, I.; Atarashi, S.; Imamura, M.; Kimura, Y. European Patent Application. 0357047 A1, Aug 30, 1989.
Return to citation in text: [1] -
Toshifumi, A.; Tutomu, E.; Tatsuru, S.; Sadahiro, S.; Keiichi, H.; Naoki, O.; Toshiaki, T. U.S. Patent Application. 6218548 B1, April 17, 2001.
Return to citation in text: [1] -
Keiji, N.; Makoto, M.; Tatsuru, S.; Yuichiro, T.; Toshifumi, A. U.S. Patent Application. 20040019223 A1, Jan 29, 2004.
Return to citation in text: [1] -
Bird, P.; Ellsworth, E. L.; Nguyen, D. Q.; Sanchez, J. P.; Showalter, H. D. H.; Singh, R.; Stier, M. A.; Tran, T. P.; Watson, B. M.; Yip, J. U.S. Patent Application. 20060183762 A1, Aug 17, 2006.
Return to citation in text: [1] -
Inoue, T.; Tanaka, A.; Nakai, K.; Sasaki, H.; Takahashi, F.; Shirakami, S.; Hatanaka, K.; Nakajima, J.; Mukoyoshi, K.; Hamaguchi, H.; Kunikawa, S.; Higashi, Y. WO Patent Application. 2007077949, July 12, 2007.
Return to citation in text: [1] -
Bakthavatchalam, R.; Capitosti, S. M.; Hutchison, A. J.; LI, G.; Peterson, J. M.; Cheng, C. S. WO Patent Application. 2008060568, May 22, 2008.
Return to citation in text: [1] -
Schaefer, J. P.; Bloomfield, J. J. Org. React. 1967, 15, 1–203.
Return to citation in text: [1] -
Krapcho, A. P. Synthesis 1982, 805–822. doi:10.1055/s-1982-29953
Return to citation in text: [1] -
Remeň, L.; Bezençon, O.; Richard-Bildstein, S.; Bur, D.; Prade, L.; Corminboeuf, O.; Boss, C.; Grisostomi, C.; Sifferlen, T.; Strickner, P.; Hess, P.; Delahaye, S.; Treiber, A.; Weller, T.; Binkert, C.; Steiner, B.; Fischli, W. Bioorg. Med. Chem. Lett. 2009, 19, 6762–6765. doi:10.1016/j.bmcl.2009.09.104
Return to citation in text: [1] -
Brandi, A.; Cordero, F. M.; Sarlo, F. D.; Goti, A.; Guarna, A. Synlett 1993, 1–8. doi:10.1055/s-1993-22329
Return to citation in text: [1] -
Cordero, F. M.; Sarlo, F. D.; Brandi, A. Monatsh. Chem. 2004, 135, 649–669. doi:10.1007/s00706-003-0150-x
Return to citation in text: [1] -
Feng, J.; Gwaltney, S. L.; Stafford, J. A.; Zhang, Z.; Elder, B. J.; Isbester, P. K.; Palmer, G. J.; Salsbury, J. S.; Ulysse, L. G. U.S. Patent Application. 20080188501, Aug 7, 2008.
Return to citation in text: [1] -
Ten nanogram of recombinant human DPP-4 was incubated with different concentrations of compound C for 15 min at 30 °C. DPP-4 enzyme reaction was initiated with 50 mM of Gly-Pro-AMC. Fluorescence was measured after 30 min of incubation (see Supporting Information File 1 for further details).
Return to citation in text: [1]
| 8. | Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L. J. Med. Chem. 2007, 50, 2297–2300. doi:10.1021/jm070104l |
| 1. | Weber, A. E. J. Med. Chem. 2004, 47, 4135–4141. doi:10.1021/jm030628v |
| 2. | Augustyns, K.; Van der Veken, P.; Senten, K.; Haemers, A. Curr. Med. Chem. 2005, 12, 971–998. doi:10.2174/0929867053507298 |
| 3. | Augustyns, K.; Van der Veken, P.; Haemers, A. Expert Opin. Ther. Pat. 2005, 15, 1387–1407. doi:10.1517/13543776.15.10.1387 |
| 4. | von Geldern, T. W.; Trevillyan, J. M. Drug Dev. Res. 2006, 67, 627–642. doi:10.1002/ddr.20138 |
| 5. | Van der Veken, P.; Haemers, A.; Augustyns, K. Curr. Top. Med. Chem. 2007, 7, 621–635. |
| 11. | Benzencon, O.; Bur, D.; Fischli, W.; Remen, L.; Richard-Bildstein, S.; Sifferlen, T.; Weller, T. U.S. Patent Application. 20080234305 A1, Sept 25, 2008. |
| 23. | Feng, J.; Gwaltney, S. L.; Stafford, J. A.; Zhang, Z.; Elder, B. J.; Isbester, P. K.; Palmer, G. J.; Salsbury, J. S.; Ulysse, L. G. U.S. Patent Application. 20080188501, Aug 7, 2008. |
| 8. | Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L. J. Med. Chem. 2007, 50, 2297–2300. doi:10.1021/jm070104l |
| 24. | Ten nanogram of recombinant human DPP-4 was incubated with different concentrations of compound C for 15 min at 30 °C. DPP-4 enzyme reaction was initiated with 50 mM of Gly-Pro-AMC. Fluorescence was measured after 30 min of incubation (see Supporting Information File 1 for further details). |
| 8. | Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L. J. Med. Chem. 2007, 50, 2297–2300. doi:10.1021/jm070104l |
| 9. | Feng, J.; Gwaltney, S. L.; Stafford, J. A.; Zhang, Z.; Elder, B. J.; Isbester, P. K.; Palmer, G. J.; Salsbury, J. S.; Ulysse, L. WO Patent Application. 2007035629, 2007. |
| 10. | Feng, J.; Gwaltney, S. L.; Stafford, J. A.; Zhang, Z.; Brodluahrer, P. R.; Elder, B.; Isbester, P.; Fornicola, R. S.; Kisanga, P. B.; Mobele, B. I.; Palmer, G. J.; Reeve, M. M.; Salsbury, J. S.; Ulysse, L. G.; Venkatraman, S. U.S. Patent Application. 20090275750 A1, Nov 5, 2009. |
| 11. | Benzencon, O.; Bur, D.; Fischli, W.; Remen, L.; Richard-Bildstein, S.; Sifferlen, T.; Weller, T. U.S. Patent Application. 20080234305 A1, Sept 25, 2008. |
| 20. | Remeň, L.; Bezençon, O.; Richard-Bildstein, S.; Bur, D.; Prade, L.; Corminboeuf, O.; Boss, C.; Grisostomi, C.; Sifferlen, T.; Strickner, P.; Hess, P.; Delahaye, S.; Treiber, A.; Weller, T.; Binkert, C.; Steiner, B.; Fischli, W. Bioorg. Med. Chem. Lett. 2009, 19, 6762–6765. doi:10.1016/j.bmcl.2009.09.104 |
| 6. | Havale, S. H.; Pal, M. Bioorg. Med. Chem. 2009, 17, 1783–1802. doi:10.1016/j.bmc.2009.01.061 |
| 7. | Gupta, R.; Walunj, S. S.; Tokala, R. K.; Parsa, K. V. L.; Singh, S. K.; Pal, M. Curr. Drug Targets 2009, 10, 71–87. doi:10.2174/138945009787122860 |
| 21. | Brandi, A.; Cordero, F. M.; Sarlo, F. D.; Goti, A.; Guarna, A. Synlett 1993, 1–8. doi:10.1055/s-1993-22329 |
| 22. | Cordero, F. M.; Sarlo, F. D.; Brandi, A. Monatsh. Chem. 2004, 135, 649–669. doi:10.1007/s00706-003-0150-x |
| 16. | Inoue, T.; Tanaka, A.; Nakai, K.; Sasaki, H.; Takahashi, F.; Shirakami, S.; Hatanaka, K.; Nakajima, J.; Mukoyoshi, K.; Hamaguchi, H.; Kunikawa, S.; Higashi, Y. WO Patent Application. 2007077949, July 12, 2007. |
| 12. | Hayakawa, I.; Atarashi, S.; Imamura, M.; Kimura, Y. European Patent Application. 0357047 A1, Aug 30, 1989. |
| 13. | Toshifumi, A.; Tutomu, E.; Tatsuru, S.; Sadahiro, S.; Keiichi, H.; Naoki, O.; Toshiaki, T. U.S. Patent Application. 6218548 B1, April 17, 2001. |
| 14. | Keiji, N.; Makoto, M.; Tatsuru, S.; Yuichiro, T.; Toshifumi, A. U.S. Patent Application. 20040019223 A1, Jan 29, 2004. |
| 15. | Bird, P.; Ellsworth, E. L.; Nguyen, D. Q.; Sanchez, J. P.; Showalter, H. D. H.; Singh, R.; Stier, M. A.; Tran, T. P.; Watson, B. M.; Yip, J. U.S. Patent Application. 20060183762 A1, Aug 17, 2006. |
| 11. | Benzencon, O.; Bur, D.; Fischli, W.; Remen, L.; Richard-Bildstein, S.; Sifferlen, T.; Weller, T. U.S. Patent Application. 20080234305 A1, Sept 25, 2008. |
| 11. | Benzencon, O.; Bur, D.; Fischli, W.; Remen, L.; Richard-Bildstein, S.; Sifferlen, T.; Weller, T. U.S. Patent Application. 20080234305 A1, Sept 25, 2008. |
| 17. | Bakthavatchalam, R.; Capitosti, S. M.; Hutchison, A. J.; LI, G.; Peterson, J. M.; Cheng, C. S. WO Patent Application. 2008060568, May 22, 2008. |
© 2010 Kodimuthali et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)