Abstract
The valence isomerization of the all-carbon and heteroelement analogues of cyclohepta-1,3,5-triene into the corresponding bicyclo[4.1.0]hepta-2,4-dienes is reviewed to show the impact of the heteroatom on the stability of both valence isomers. The focus is on the parent systems and their synthetic applications.

Graphical Abstract
Introduction
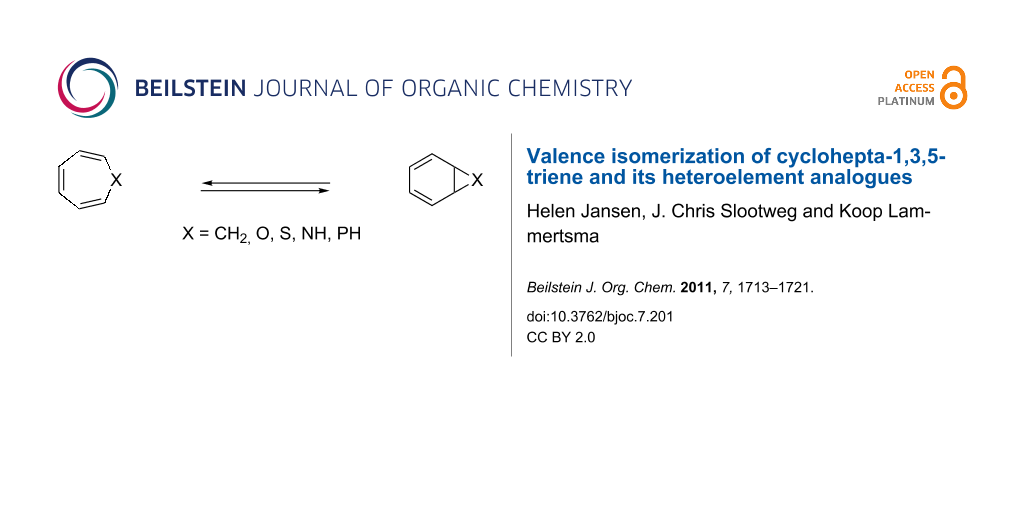
The valence isomerization of cyclohepta-1,3,5-triene (1) into bicyclo[4.1.0]hepta-2,4-diene (2) has captured the attention of chemists for over five decades [1,2]. This interest extended to the heterocyclic analogues 3–8, bearing one oxygen, sulfur or nitrogen atom, after the discovery of their biological importance (Scheme 1) [3,4]. The phosphane analogues 9 and 10 received far less attention, with their applicability as a phosphinidene (R–P) precursor being the most notable use [5-9].
![[1860-5397-7-201-i1]](/bjoc/content/inline/1860-5397-7-201-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Valence isomerization of cyclohepta-1,3,5-triene (1) and its heteroelement analogues.
Scheme 1: Valence isomerization of cyclohepta-1,3,5-triene (1) and its heteroelement analogues.
Reviewing the influence of the heteroatom on the cycloheptatriene–norcaradiene valence isomerization necessitates a brief overview of the parent all-carbon system. This section is followed by one in which experimental data on the oxepine, thiepine, 1H-azepine, and 1H-phosphepine valence isomerizations are compared with those obtained by theoretical calculations. Computational methods have the advantage that they enable reliable insight into the reaction energies and aromatic features of the parent isomers. In this brief review, only selected examples of substituted heteropines and their syntheses are given.
Review
Cycloheptatriene valence isomerization
Cyclohepta-1,3,5-triene (1), first isolated in 1883 [10], has a boat-shaped conformation as determined by electron diffraction [11] and microwave studies of the parent [12] and by an X-ray structure analysis of the derivative thujic acid [13,14]. These methods gave inconsistent α and β tilt angles (see Scheme 2 for a description of the bow (α) and stern (β) tilt angles) with those determined by electron diffraction standing out. Theoretical calculations at the B3LYP/6-311+G(d,p) level gave α and β angles of 52.9° and 25.4°, respectively [15-17], which are in reasonable harmony with those of the microwave and X-ray studies. Low temperature 1H NMR measurements showed that the slightly homoaromatic boat conformation is prone to undergo a degenerate ring flip via an antiaromatic C2v transition with a free energy barrier of 5.7 kcal·mol−1 in CBrF3 [18] and 6.3 kcal·mol−1 in CF2Cl2 [19-21].
![[1860-5397-7-201-i2]](/bjoc/content/inline/1860-5397-7-201-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Conformational ring inversions.
Scheme 2: Conformational ring inversions.
Cycloheptatriene is in equilibrium with bicyclo[4.1.0]hepta-2,4-diene (2) by means of a Woodward–Hoffmann symmetry-allowed disrotatory ring closure [22,23]. Although the equilibrium strongly favours the seven-membered ring, the presence of small quantities of the bicyclic isomer 2 was inferred by Diels–Alder trapping reactions [24]. In 1981, Ruben was the first to observe norcaradiene (2) directly, by employing low-temperature photolysis, and an activation barrier of 11 ± 2 kcal·mol−1 was determined for the formation of 2 from 1, with the product being 4 kcal·mol−1 less stable [25]. Strong electron-withdrawing groups at the methylene bridge influence the 1–2 equilibrium in favour of the norcaradiene isomer, as is the case for the thermally stable 7,7-dicyano-derivative [26,27]. At the B3LYP/6-311+G(d,p) level the geometry of the parent was shown to have a straighter bow (α = 65.8°) and flatter stern (β = 18.9°) as compared to cyclohepta-1,3,5-triene [14].
Besides the 1–2 interconversion, the C7H8 system is rich in rearrangements (Scheme 3). In 1957, Woods found that bicyclo[2.2.1]hepta-2,5-diene (12) converts to cycloheptatriene (1), which was postulated to proceed via diradical 11 and norcaradiene (2) [28]. Instead, pyrolysis of 1 yielded toluene, presumably through a [1,3]-H shift of the diradical [29]. Norcaradiene (2) can also undergo a [1,5]-carbon circumambulatory rearrangement (“walk”), as was discovered by Berson and Willcott in 1965 [30,31]. Although, this process should proceed with retention of the configuration according to the symmetry conservation rules, studies of chiral substituted cycloheptatrienes showed a preference for the “forbidden” path with inversion of configuration [32-35]. Finally, a suprafacial [1,5]-hydrogen shift with an activation energy of approximately 31 kcal·mol−1 was unveiled by a high-temperature NMR study (100–140 °C) of hydrogen isotopomers of cycloheptatriene (Scheme 3) [36-38].
![[1860-5397-7-201-i3]](/bjoc/content/inline/1860-5397-7-201-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Rearrangements of the parent cycloheptatriene 1 and norcaradiene 2.
Scheme 3: Rearrangements of the parent cycloheptatriene 1 and norcaradiene 2.
Valence isomerization of heteropines
Determining the conformations of the heteropines has been more of a challenge. Only the parent oxepine (3) is isolable at room temperature. NMR spectroscopy indicated a boat-shape structure with alternating C=C bonds for 3 [39,40], which was supported by single-crystal X-ray structure analyses of simple derivatives [41]. Table 1 also summarizes the relative energies obtained by high-level theoretical calculations for the parent heteropines and the corresponding bicyclic norcaradienes, and the barriers for their interconversion and ring inversion.
Table 1: Relative energies (in kcal·mol−1) of the norcaradienes (NCD) 2 (C), 4 (O), 6 (S), 8 (N), 10 (P), the cycloheptatrienes (CHT) 1 (C), 3 (O), 5 (S), 7 (N), 9 (P), their interconversion barriers, and the barriers for ring inversion of the monocycles.
| NCD | TS | CHT | TSinv | Methoda | Ref | |
|---|---|---|---|---|---|---|
| C (1,2) | 4 | 11b | 0.0 | ~6 | Exp. | [17,18,24] |
| O (3,4) | 0.0 | 9.1c | 1.7 | – | NMR | [38,39] |
| 0.0 | 7.0 | 0.1 | 3.5 | QCISD(T)/6-31G(d) | [40] | |
| S (5,6) | 0.0 | 20.5c | 7.0 | 7.3 | QCISD(T)/6-31G(d) | [40] |
| N (7,8) | 7.9 | 11.4b | 0.0 | ~3 | B3LYP/6-31G(d) | [41,42,44] |
| P (9,10) | 0.0 | 15.7 | 2.5 | 5.2 | B3LYP/6-311+G(d,p) | [7,45] |
aGibbs free energies for the experimental data (first two entries) and enthalpies for the computational data. bEquilibrium from CHT to NCD. cEquilibrium from NCD to CHT.
Although the boat form prevails for the monocyclic heteropines 1, 3 and 5, Cremer et al. showed that this represents an incomplete picture [43,44]. In fact, they are “perturbed” boats with at least 22% chair character, leading to an almost similar boat puckering for all. From the racemization of substituted benzene oxides (Scheme 2), the oxepine ring inversion barrier was estimated at 6.5 kcal·mol−1 at 135 K [45,46], which is similar to the 3.5 kcal·mol−1 calculated for the parent oxepine (3) at the QCISD(T)/6-31G(d) level [40]. The calculated barrier of 8.3 kcal·mol−1 for thiepine (5) is nearly twice as large, possibly due to the higher antiaromatic destabilization of the flattened thiepine ring [40], but the interconversion of the boat forms of azepine and phosphepine are about equally favourable, requiring 3.0 [41] and 5.2 kcal·mol−1 [7,47], respectively.
A question related to the valence isomerization is whether aromatic properties can be ascribed to the heteropines. Indeed, the monocyclic boat-shaped heteropines exhibit homoaromatic features by conjugative interaction of the triene unit through 1,6-overlap of 2p π-orbitals [41], as is the case for cyclohepta-1,3,5-triene (1) [19,20]. Through the use of nucleus-independent chemical shifts (NICS(1)) [48], it was shown that thiepine (−2.3 ppm) [49] and phenyl phosphepine (−4.8 ppm) [7] display aromatic character when compared to the well-known 6π-electron Hückel-aromatic tropylium cation [46], which has a NICS(1) value of −8.2 ppm. Adding electronegative substituents enhances the effect, and fully aromatic systems are obtained after complete fluorination of the heteropines (Figure 1) [50]. In contrast, the flattened transition structures for ring inversion of thiepine and phosphepine are indeed highly antiaromatic planar 8π-electron systems, with positive NICS(1) values of 19.3 [47] and 6.4 ppm [7], respectively. The inherent instability of thiepine (5) has been attributed to this effect [51,52].
![[1860-5397-7-201-1]](/bjoc/content/figures/1860-5397-7-201-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: NICS(0) values of fluorinated heteropines.
Figure 1: NICS(0) values of fluorinated heteropines.
Oxepine – benzene oxide
Oxepine (3) was isolated first by Vogel et al. using a double dehydrohalogenation of 1,2-dibromo-4,5-epoxycyclohexane [38,53], but is also accessible by epoxidation of Dewar benzene followed by photolytic or thermal ring expansion [54]. The molecular structure of the 2-tert-butoxycarbonyl oxepine showed a boat configuration with bow (α) and stern (β) fold angles of 56.5° and 26.0°, respectively [44], which differs little from the MP2/6-31G(d) geometry of the parent 3 (Cs symmetry; α = 58.3°, β = 30.8°), illustrating that the substituent hardly influences the geometry [40]. Oxepine (3) is more curved than cyclohepta-1,3,5-triene (1; α = 52.9°, β = 25.4°; same level of theory) [14].
Using 1H NMR spectroscopy, Vogel and Günther determined that 7-oxa-bicyclo[4.1.0]hepta-2,4-diene (4, benzene oxide; Scheme 4) is 1.7 kcal·mol−1 more stable than monocyclic 3 in apolar solvents [38,39], with an activation barrier for the conversion of 3 to 4 of 7.2 kcal·mol−1. Calculations at the QCISD(T)/6-31G(d) level confirm the bicyclic form to be the most stable isomer, albeit with an energy difference of a mere 0.1 kcal·mol−1 and a barrier to interconversion of 9.1 kcal·mol−1 [40]. By changing to more polar solvents, the oxepine isomerization equilibrium shifts further toward benzene oxide (more positive ΔG), suggesting that benzene oxide has the larger dipole moment. Methyl substitution at the 2- and 7-positions reverses the stability order, rendering the oxepine as the energetically favoured isomer due to the destabilizing eclipsing of the two methyl groups in benzene oxide (4) [38,40,51]. Thus, in contrast to the cycloheptatriene–norcaradiene (1–2) pair, the equilibrium constant for oxepine (3) and bicyclic benzene oxide (4) varies widely with solvent polarity and to some extent with temperature and substituents, making it possible to work with solutions highly enriched with either one or the other isomer [38,55]. The facile 3→4 valence isomerization [56-58], pioneered by the synthesis of 1,2-naphthalene oxide by Vogel and Klärner [1,59,60], is of considerable interest as arene oxides are intermediates in the oxidative metabolism of aromatic substrates [61-64]. In addition, also photo-oxidation of benzene creates this isomeric pair [65,66].
![[1860-5397-7-201-i4]](/bjoc/content/inline/1860-5397-7-201-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactivity of oxepine (3) and benzene oxide (4).
Scheme 4: Reactivity of oxepine (3) and benzene oxide (4).
Depicted in Scheme 4 are the most important reactions that the parent oxepine (3) and benzene oxide (4) can undergo. Irradiation of oxepine results in ring contraction yielding 2-oxabicyclo[2.3.0]hepta-3,6-diene (13) [38,51], while under thermal, photochemical or acidic conditions, the three-membered ring of bicyclic 4 opens, generating phenol [67,68], in analogy to the all-carbon norcaradiene (2), which gives toluene. In addition, 4 undergoes highly selective Diels–Alder reactions, such as with N-phenylmaleimide and dimethyl acetylenedicarboxylate, providing single anti-adducts (e.g., 14; Scheme 4) [61,69]. Theoretical calculations on model structures showed the anti cycloaddition to be the kinetically controlled path and the syn addition the thermodynamically favoured one [40].
Thiepine – benzene sulfide
The parent thiepine (5) is 7.0 kcal·mol−1 less stable than benzene sulfide (6). This energy difference is much larger than for the oxygen homologues, because three-membered rings accommodate sulfur better than oxygen [40]. Nonetheless, bicyclic 6 has never been isolated, probably due to the low activation barrier for sulfur extrusion [40,48,70], which occurs through a sequence of low-energy processes involving several sulfur-containing intermediates [71,72].
Thiepine (5) can be stabilized by Fe(CO)3 complexation (15; Figure 2) [73] or by decorating the seven-membered ring with substituents. The first isolated metal-free thiepine (16; Figure 2) was reported in 1974 by Reinhoudt and Kouwenhoven, who used electron-withdrawing groups to delocalize the π-electrons of the thiepine ring, but this species still eliminates sulfur at room temperature [74]. With the synthesis of the sterically shielded 2,7-di-tert-butylthiepine (17) (Figure 2), a relatively simple and thermally stable thiepine was obtained, allowing experimental studies of its chemical and physical properties [75]. A single-crystal X-ray analysis showed 17 to be less curved (α = 49.6° and β = 28.0°) [70] than the computed structure of cyclohepta-1,3,5-triene (1; α = 52.9°, β = 25.4°) [14]; The MP2/6-31G(d) optimized geometry of the parent thiepine (5) (α = 50.3° and β = 30.8°) [40] is similar to that of the molecular structure of 17 [76]. Benzannulation of the thiepine ring on both sides results in the thermally robust dibenzo[b,f]thiepines, which are of interest for their potent biological activity, illustrated by the psychosedative and antipsychotic properties of zotepine (18; Figure 2) [76-80].
![[1860-5397-7-201-2]](/bjoc/content/figures/1860-5397-7-201-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
1H-Azepine – benzene imine
The parent 1H-azepine (7) [81] was first generated in 1963 by Hafner, by the hydrolysis of ethyl-1H-azepine-N-carboxylate with potassium hydroxide and subsequent protonation [82]. Because 1H-azepine is highly unstable and rapidly undergoes a [1,3]-H shift to 3H-azepine, only an X-ray structure determination at −78 °C of an N-substituted derivative was reported by Vogel et al. 17 years later [83-85]. The molecular structure of N-(phenoxycarbonyl)azepine displays a rather shallow boat structure (α = 43.4° and β = 21.6°) [86], which is solely due to the N-substituent, as the CASSCF/3-21G optimized geometry showed a more curved β angle of 36.4° for the parent 7 [87].
Like the all-carbon analogues, the valence isomerization strongly favours the monocyclic form with an estimated preference of 7.9 kcal·mol−1 at the B3LYP/6-31G(d) level for the parent system (7→8) [42]. Also low temperature 1H and 13C NMR measurements on 19 display only small amounts of the bicyclic isomers 20 (Scheme 5) [79,88].
![[1860-5397-7-201-i5]](/bjoc/content/inline/1860-5397-7-201-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Valence isomerization of 1H-azepines.
Scheme 5: Valence isomerization of 1H-azepines.
The reluctance to form the bicyclic isomer dictates the reactivity of azepines, as they exhibit the characteristics of cyclic polyene chemistry, which is illustrated by the ability of the monocyclic isomer to undergo cycloadditions as a 2π (→21) [89], 4π (→22) [84,90], or 6π (→23, 24) [91,92] component (Scheme 6). In addition, azepine (7) rearranges photochemically to bicyclic 25 [93], and in the presence of an acid yields aniline derivatives 26 [94] in analogy to the cycloheptatriene and oxepine [95].
![[1860-5397-7-201-i6]](/bjoc/content/inline/1860-5397-7-201-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Like the thiepines, the benzannulated azepines have also received considerable attention due to their biological importance and pharmaceutical relevance [96]. For instance, 3H-3-benzazepin-2-amines 27 possess antihypertensive activity [97], and all tricyclic dibenzo[b,f]azepines (e.g., 28; Figure 3) bearing a basic side chain affect the central nervous system [98].
![[1860-5397-7-201-3]](/bjoc/content/figures/1860-5397-7-201-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Benzannulated azepines 27 and 28.
Figure 3: Benzannulated azepines 27 and 28.
1H-Phosphepine – benzene phosphane
Although the parent 1H-phosphepine (9) and its 2.5 kcal·mol−1 more-stable valence isomer benzene phosphane (10) have never been isolated [45], there is evidence for the existence of the parent phosphatropylium ion (29; Figure 4), which was generated in the gas phase by collision activation between PI3 and benzene [99]. P-phenyl substitution stabilizes the phosphanorcaradiene (ΔE = 4.8 kcal·mol−1), but this species has also never been observed experimentally [7]. The thermal instability of the phosphepines and their valence isomers is due to the facile decomposition of the bicyclic phosphanorcaradiene (10) into benzene and phosphinidene R–P [100]. However, the 7-membered ring can be stabilized by phosphorus oxidation (30; see Figure 4) [95], the introduction of bulky substituents at the 2 and 7 positions (31) [101], or benzannulation (e.g., 3H-benzophosphepine, 32) [7,102-107]. The single-crystal X-ray structure analysis of phenyl-substituted phosphepine 33 (Scheme 7) also showed a flattened-boat conformation (α = 40.5°, β = 28.2°) [5] compared to the metal-free parent structure (α = 48.3°, β = 27.8°), computed at the B3PW91/6-311+G(d,p) level [7].
![[1860-5397-7-201-4]](/bjoc/content/figures/1860-5397-7-201-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-7-201-i7]](/bjoc/content/inline/1860-5397-7-201-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Phosphinidene generation from metal-complexed benzophosphepine 33.
Scheme 7: Phosphinidene generation from metal-complexed benzophosphepine 33.
Also for the phosphepine system [108], benzannulation leads to interesting targets. Namely, the thermal lability of the transition-metal-complexed 3H-benzophosphepine 33 was explored by Lammertsma et al. for the synthesis of a variety of organophosphorus compounds by means of [1 + 2] cycloadditions of the in situ generated singlet phosphinidene 35 with olefins or acetylenes (Scheme 7) [5-9]. This approach has even lead to the detection of the transient phosphinidene species by employing electrospray ionization tandem mass spectrometry (ESIMS/MS); its gas-phase reactivity perfectly matches the well-established solution-phase chemistry [109]. Using these phosphinidenes [110,111] led to the synthesis of unique P-ligands for catalysis [112,113] as well as to attractive building blocks for the creation of P-functionalized polymers [114,115].
Conclusion
The valence isomerization of cyclohepta-1,3,5-triene into the parent norcaradiene, and of their corresponding heteroelement analogues, has been reviewed with a focus on the chemical and physical properties of these fascinating species. The presence of a heteroatom has an impact on the stability of the heteropines, of which to date only the parent oxepine has been isolated. The generation of these (transient) heterocycles allowed the development of a rich chemistry, which has been extensively explored using the full toolbox of physical organic chemistry.
References
-
Vogel, E. Angew. Chem., Int. Ed. 2011, 50, 4278–4287. doi:10.1002/anie.201101347
Return to citation in text: [1] [2] -
Maier, G. Angew. Chem., Int. Ed. Engl. 1967, 6, 402–413. doi:10.1002/anie.196704021
Return to citation in text: [1] -
Rodríguez-Hahn, L.; Esquivel, B.; Sánchez, A. A.; Cárdenas, J.; Tovar, O. G.; Soriano-García, M.; Toscano, A. J. Org. Chem. 1988, 53, 3933–3936. doi:10.1021/jo00252a010
Return to citation in text: [1] -
Güney, M.; Daştan, A.; Balci, M. Helv. Chim. Acta 2005, 88, 830–838. doi:10.1002/hlca.200590061
Return to citation in text: [1] -
Borst, M. L. G.; Bulo, R. E.; Winkel, C. W.; Gibney, D. J.; Ehlers, A. W.; Schakel, M.; Lutz, M.; Spek, A. L.; Lammertsma, K. J. Am. Chem. Soc. 2005, 127, 5800–5801. doi:10.1021/ja050817y
Return to citation in text: [1] [2] [3] -
Borst, M. L. G.; Bulo, R. E.; Gibney, D. J.; Alem, Y.; de Kanter, F. J. J.; Ehlers, A. W.; Schakel, M.; Lutz, M.; Spek, A. L.; Lammertsma, K. J. Am. Chem. Soc. 2005, 127, 16985–16999. doi:10.1021/ja054885w
Return to citation in text: [1] [2] -
Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Couzijn, E. P. A.; Ehlers, A. W.; Slootweg, J. C.; Schakel, M.; Krill, S.; Lutz, M.; Spek, A. L.; Lammertsma, K. Chem.–Eur. J. 2008, 14, 1499–1507. doi:10.1002/chem.200700958
Return to citation in text: [1] [2] -
Jansen, H.; Rosenthal, A. J.; Slootweg, J. C.; Ehlers, A. W.; Lutz, M.; Spek, A. L.; Lammertsma, K. Organometallics 2008, 27, 2868–2872. doi:10.1021/om800188h
Return to citation in text: [1] [2] -
Ladenburg, A. Liebigs Ann. Chem. 1883, 217, 74–149. doi:10.1002/jlac.18832170107
Return to citation in text: [1] -
Traetteberg, M. J. Am. Chem. Soc. 1964, 86, 4265–4270. doi:10.1021/ja01074a008
Return to citation in text: [1] -
Butcher, S. S. J. Chem. Phys. 1965, 42, 1833–1836.
Return to citation in text: [1] -
Reed, T. B.; Libscomb, W. N. Acta Crystallogr. 1953, 6, 108. doi:10.1107/S0365110X53000399
Suitable crystals for an accurate X-ray determination could not be obtained for cyclohepta-1,3,5-triene.
Return to citation in text: [1] -
Davis, R. E.; Tulinksy, A. Tetrahedron Lett. 1962, 3, 839–846.
However the derivative 7,7-dimethylcycloheptatriene-3-carboxylic acid did result in suitable crystals.
Return to citation in text: [1] [2] [3] [4] -
Chen, Z.; Jiao, H.; Wu, J. I.; Herges, R.; Zhang, S. B.; von Ragué Schleyer, P. J. Phys. Chem. A 2008, 112, 10586–10594. doi:10.1021/jp802496m
Return to citation in text: [1] -
Jarzeçki, A. A.; Gajewski, J.; Davidson, E. R. J. Am. Chem. Soc. 1999, 121, 6928–6935. doi:10.1021/ja984471l
See for similar computational geometrical studies α = 52.1°, β= 25.2° determined at the CASSCF(6,6)/6-31G(d) level.
Return to citation in text: [1] -
Scott, A. P.; Agranat, I.; Biedermann, P. U.; Riggs, N. V.; Radom, L. J. Org. Chem. 1997, 62, 2026–2038. doi:10.1021/jo962407l
α = 57.5°, β= 27.6° determined at the B-LYP/6-31G(d) level.
Return to citation in text: [1] [2] -
Jensen, F. R.; Smith, L. A. J. Am. Chem. Soc. 1964, 86, 956–957. doi:10.1021/ja01059a065
Return to citation in text: [1] [2] -
Anet, F. A. L. J. Am. Chem. Soc. 1964, 86, 458–460. doi:10.1021/ja01057a034
Return to citation in text: [1] [2] -
Nishinaga, T.; Izukawa, Y.; Komatsu, K. J. Phys. Org. Chem. 1998, 11, 475–477. doi:10.1002/(SICI)1099-1395(199807)11:7<475::AID-POC991>3.0.CO;2-O
See for determination of “Nuclear Independent Chemical Shift” (NICS(0), benzene-8.0 ppm) gives-4.2 ppm for 1.
Return to citation in text: [1] [2] -
Herges, R.; Geuenich, D. J. Phys. Chem. A 2001, 105, 3214–3220. doi:10.1021/jp0034426
Return to citation in text: [1] -
Maier, G. Angew. Chem., Int. Ed. Engl. 1967, 6, 812. doi:10.1002/anie.196708121
Return to citation in text: [1] -
Lowry, T. H.; Richardson, K. S. Mechanism and Theory in Organic Chemistry, 4th ed.; Harper and Row: London, 1990.
Return to citation in text: [1] -
Menzek, A.; Balci, M. Tetrahedron 1993, 49, 6071–6078. doi:10.1016/S0040-4020(01)87191-X
And references therein.
Return to citation in text: [1] [2] -
Rubin, M. B. J. Am. Chem. Soc. 1981, 103, 7791–7792. doi:10.1021/ja00416a018
Return to citation in text: [1] -
Hall, G. E.; Roberts, J. D. J. Am. Chem. Soc. 1971, 93, 2203–2207. doi:10.1021/ja00738a019
Return to citation in text: [1] -
Tang, T.-H.; Lew, C. S. Q.; Cui, Y.-P.; Capon, B.; Csizmadia, I. G. J. Mol. Struct.: THEOCHEM 1994, 305, 149–164. doi:10.1016/0166-1280(94)80150-9
Return to citation in text: [1] -
Woods, W. G. J. Org. Chem. 1958, 23, 110–112. doi:10.1021/jo01095a617
Return to citation in text: [1] -
Klump, K. N.; Chesick, J. P. J. Am. Chem. Soc. 1963, 85, 130–132. doi:10.1021/ja00885a003
Return to citation in text: [1] -
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1965, 87, 2751–2752. doi:10.1021/ja01090a037
Return to citation in text: [1] -
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1965, 87, 2752–2753. doi:10.1021/ja01090a038
Return to citation in text: [1] -
Klärner, F.-G. Angew. Chem., Int. Ed. Engl. 1974, 13, 268–270. doi:10.1002/anie.197402681
Return to citation in text: [1] -
Klärner, K.-G.; Yaşlak, S.; Wette, M. Chem. Ber. 1977, 110, 107–123. doi:10.1002/cber.19771100112
Return to citation in text: [1] -
Klärner, K.-G.; Yaşlak, S.; Wette, M. Chem. Ber. 1979, 112, 1168–1188. doi:10.1002/cber.19791120412
Return to citation in text: [1] -
Klärner, K.-G.; Brassel, B. J. Am. Chem. Soc. 1980, 102, 2469–2470. doi:10.1021/ja00527a062
Return to citation in text: [1] -
Ter Borg, A. P.; Kloosterziel, H.; Van Meurs, N. Proc. Chem. Soc., London 1962, 359.
Return to citation in text: [1] -
Weth, E.; Dreiding, A. S. Proc. Chem. Soc., London 1964, 59–60.
Return to citation in text: [1] -
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Vogel, E.; Böll, W. A.; Günther, H. Tetrahedron Lett. 1965, 6, 609–615. doi:10.1016/S0040-4039(00)90005-4
Return to citation in text: [1] [2] [3] -
Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] -
Pye, C. C.; Xidos, J. D.; Poirier, R. A.; Burnell, D. J. J. Phys. Chem. A 1997, 101, 3371–3376. doi:10.1021/jp9623498
Return to citation in text: [1] [2] [3] [4] -
Kassaee, M. Z.; Arshadi, S.; Haerizade, B. N.; Vessally, E. J. Mol. Struct.: THEOCHEM 2005, 731, 29–37. doi:10.1016/j.theochem.2005.02.087
Return to citation in text: [1] [2] -
Cremer, D.; Dick, B.; Christeu, D. J. Mol. Struct.: THEOCHEM 1984, 110, 277–291. doi:10.1016/0166-1280(84)80077-9
Return to citation in text: [1] -
Kao, J. J. Comput. Chem. 1988, 9, 905–923. doi:10.1002/jcc.540090812
Return to citation in text: [1] [2] [3] -
Jennings, W. B.; Rutherford, M.; Boyd, D. R.; Agarwal, S. K.; Sharma, N. D. Tetrahedron 1988, 44, 7551–7558. doi:10.1016/S0040-4020(01)86249-9
Return to citation in text: [1] [2] [3] -
Jennings, W. B.; Rutherford, M.; Agarwal, S. K.; Boyd, D. R.; Malone, J. F.; Kennedy, D. A. J. Chem. Soc., Chem. Commun. 1986, 970–972. doi:10.1039/C39860000970
Return to citation in text: [1] [2] -
Kassaee, M. Z.; Cheshmehkani, A.; Musavi, S. M.; Majdi, M.; Motamedi, E. J. Mol. Struct.: THEOCHEM 2008, 865, 73–78. doi:10.1016/j.theochem.2008.06.025
Return to citation in text: [1] [2] -
Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; von Ragué Schleyer, P. Chem. Rev. 2005, 105, 3842–3888. doi:10.1021/cr030088+
The NICS value is obtained from NMR calculations of a ghost atom (Bq) at the geometrical ring center (NICS(0)), but the value 1 Å above the ring (NICS(1)) is recommended, because a lack of σ shielding makes it a better measure of π-electron delocalization.
Return to citation in text: [1] [2] -
Kassaee, M. Z.; Musavi, S. M.; Momeni, M. R.; Shakib, F. A.; Ghambarian, M. J. Mol. Struct.: THEOCHEM 2008, 861, 117–121. doi:10.1016/j.theochem.2008.04.031
See for IAO-B3LYP/6-311G(d)//B3LYP/6-311G(d) level of theory.
Return to citation in text: [1] -
Karney, W. L.; Kastrup, C. J.; Oldfield, S. P.; Rzepa, H. S. J. Chem. Soc., Perkin Trans. 2 2002, 388–392. doi:10.1039/b111369k
Return to citation in text: [1] -
Dewar, M. J. S.; Trinajstić, N. J. Am. Chem. Soc. 1970, 92, 1453–1459. doi:10.1021/ja00709a001
Return to citation in text: [1] [2] [3] -
Hess, B. A., Jr.; Schaad, L. J. J. Am. Chem. Soc. 1973, 95, 3907–3912. doi:10.1021/ja00793a013
Return to citation in text: [1] -
Vogel, E.; Schubart, R.; Böll, W. A. Angew. Chem., Int. Ed. Engl. 1964, 3, 510. doi:10.1002/anie.196405101
Return to citation in text: [1] -
van Tamelen, E. E.; Carty, D. T. J. Am. Chem. Soc. 1967, 89, 3922–3923. doi:10.1021/ja00991a056
Return to citation in text: [1] -
Vogel, E.; Günther, H. Angew. Chem., Int. Ed. Engl. 1967, 6, 385–401. doi:10.1002/anie.196703851
Return to citation in text: [1] -
Belen’kll, L. I. Oxepanes and Oxepines. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 45–66. doi:10.1016/B978-008096518-5.00210-0
Return to citation in text: [1] -
Bock, C. W.; George, P.; Stezowski, J. J.; Glusker, J. P. Struct. Chem. 1989, 1, 33–39. doi:10.1007/BF00675782
Return to citation in text: [1] -
Hayes, D. M.; Nelson, S. D.; Garland, W. A.; Kollman, P. A. J. Am. Chem. Soc. 1980, 102, 1255–1262. doi:10.1021/ja00524a007
Return to citation in text: [1] -
Vogel, E.; Klärner, F.-G. Angew. Chem., Int. Ed. Engl. 1968, 7, 374–375. doi:10.1002/anie.196803742
Return to citation in text: [1] -
Klärner, F.-G.; Vogel, E. Angew. Chem., Int. Ed. Engl. 1973, 12, 840–841. doi:10.1002/anie.197308401
Return to citation in text: [1] -
Jerina, D. M.; Daly, J. W.; Witkop, B. In Biogenic Amines and Physiological Membranes in Drug Therapy, Part B; Biel, J. H.; Abood, L. G., Eds.; Marcel Dekker: New York, 1971; pp 413–476.
Return to citation in text: [1] [2] -
Daly, J. W.; Jerina, D. M.; Witkop, B. Experientia 1972, 28, 1129–1149. doi:10.1007/BF01946135
Return to citation in text: [1] -
Jerina, D. M.; Daly, J. W. Science 1974, 185, 573–582. doi:10.1126/science.185.4151.573
Return to citation in text: [1] -
Boyd, D. R.; Sharma, N. D. Chem. Soc. Rev. 1996, 25, 289–296. doi:10.1039/cs9962500289
Return to citation in text: [1] -
Klotz, B.; Barnes, I.; Becker, K. H.; Golding, B. T. J. Chem. Soc., Faraday Trans. 1997, 93, 1507–1516. doi:10.1039/a606152d
Return to citation in text: [1] -
Henderson, A. P.; Mutlu, E.; Leclercq, A.; Bleasdale, C.; Clegg, W.; Henderson, R. A.; Golding, B. T. Chem. Commun. 2002, 1956–1957. doi:10.1039/b205079j
Return to citation in text: [1] -
George, P.; Bock, C. W.; Glusker, J. P. J. Phys. Chem. 1990, 94, 8161–8168. doi:10.1021/j100384a034
In the presence of acid, protonation results in a carbocation intermediate and does not generate a radical species.
Return to citation in text: [1] -
Holovka, J. M.; Gardner, P. D. J. Am. Chem. Soc. 1967, 89, 6390–9391. doi:10.1021/ja01000a092
Return to citation in text: [1] -
Gillard, J. R.; Newlands, M. J.; Bridson, J. N.; Burnell, D. J. Can. J. Chem. 1991, 69, 1337–1343. doi:10.1139/v91-199
Return to citation in text: [1] -
Field, L.; Tuleen, D. L. Monocyclic Seven-Membered Rings Containing Sulfur. In Seven-Membered Heterocyclic Compounds Containing Oxygen and Sulfur; Rosowsky, A., Ed.; Wiley Interscience: New York, 1972; Chapter 10.
Return to citation in text: [1] [2] -
Miller, K. J.; Moschner, K. F.; Potts, K. T. J. Am. Chem. Soc. 1983, 105, 1705–1712. doi:10.1021/ja00345a001
Return to citation in text: [1] -
Gleiter, R.; Krennrich, G.; Cremer, D.; Yamamoto, K.; Murata, I. J. Am. Chem. Soc. 1985, 107, 6874–6879. doi:10.1021/ja00310a022
Return to citation in text: [1] -
Nishino, K.; Takagi, M.; Kawata, T.; Murata, I.; Inanaga, J.; Nakasuji, K. J. Am. Chem. Soc. 1991, 113, 5059–5060. doi:10.1021/ja00013a051
Return to citation in text: [1] -
Reinhoudt, D. N.; Kouwenhoven, C. G. Tetrahedron 1974, 30, 2093–2098. doi:10.1016/S0040-4020(01)97344-2
Return to citation in text: [1] -
Yamamoto, K.; Yamazaki, S.; Kohashi, Y.; Murata, I.; Kai, Y.; Kanehisa, N.; Miki, K.; Kasai, N. Tetrahedron Lett. 1982, 23, 3195–3198. doi:10.1016/S0040-4039(00)88594-9
Return to citation in text: [1] -
Yamamoto, K.; Yamazaki, S. Thiepanes and Thiepines. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 67–111. doi:10.1016/B978-008096518-5.00211-2
Return to citation in text: [1] [2] -
Ueda, I.; Sato, Y.; Maeno, S.; Umio, S. Chem. Pharm. Bull. 1978, 26, 3058–3070.
Return to citation in text: [1] -
Higashi, Y.; Momotani, Y.; Suzuki, E.; Kaku, T. Pharmacopsychiatry 1987, 20, 8–11. doi:10.1055/s-2007-1017124
Return to citation in text: [1] -
Schwan, A. L. Product Subclass 4: Benzothiepins and Selenium/Tellurium Analogues. In Science of Synthesis; Weinreb, S. M., Ed.; Thieme: Stuttgart, Germany, 2004; Vol. 17, pp 717–748.
Return to citation in text: [1] [2] -
Protiva, M. J. Heterocycl. Chem. 1996, 33, 497–521. doi:10.1002/jhet.5570330301
Return to citation in text: [1] -
Paquette, L. A. Angew. Chem., Int. Ed. Engl. 1971, 10, 11–20. doi:10.1002/anie.197100111
Return to citation in text: [1] -
Hafner, K. Angew. Chem., Int. Ed. Engl. 1964, 3, 165–173. doi:10.1002/anie.196401651
Return to citation in text: [1] -
Vogel, E.; Altenbach, H.-J.; Drossard, J.-M.; Schmickler, H.; Stegelmeier, H. Angew. Chem., Int. Ed. Engl. 1980, 19, 1016–1018. doi:10.1002/anie.198010161
Return to citation in text: [1] -
Smalley, R. K. Azepines. In Part 5: Small and Large Rings; Lwowski, W., Ed.; Comprehensive Heterocyclic Chemistry, Vol. 7; Elsevier Science Ltd.: Oxford, 1984; p 491.
Return to citation in text: [1] [2] -
Smalley, R. K. Azepines. In Part 5: Small and Large Rings; Lwowski, W., Ed.; Comprehensive Heterocyclic Chemistry, Vol. 7; Elsevier Science Ltd.: Oxford, 1984; p 543.
Return to citation in text: [1] -
Lindner, H. J.; von Gross, B. Chem. Ber. 1972, 105, 434–440. doi:10.1002/cber.19721050208
Return to citation in text: [1] -
Smith, D. A.; Bitar, J. J. Org. Chem. 1993, 58, 6–8. doi:10.1021/jo00053a003
Return to citation in text: [1] -
Prinzbach, H.; Babsch, H.; Fritz, H.; Hug, P. Tetrahedron Lett. 1977, 18, 1355–1358. doi:10.1016/S0040-4039(01)93042-4
Return to citation in text: [1] -
Paquette, L. A.; Kuhla, D. E.; Barrett, J. H.; Leichter, L. M. J. Org. Chem. 1969, 34, 2888–2896. doi:10.1021/jo01262a018
Return to citation in text: [1] -
van den Hende, J. H.; Kende, A. S. Chem. Commun. 1965, 384–385. doi:10.1039/c19650000384
Return to citation in text: [1] -
Paquette, L. A.; Barrett, J. H. J. Am. Chem. Soc. 1966, 88, 2590–2591. doi:10.1021/ja00963a041
Return to citation in text: [1] -
Paquette, L. A.; Barrett, J. H.; Kuhla, D. E. J. Am. Chem. Soc. 1969, 91, 3616–3624. doi:10.1021/ja01041a032
Return to citation in text: [1] -
Paquette, L. A.; Barrett, J. H. J. Am. Chem. Soc. 1966, 88, 1718–1722. doi:10.1021/ja00960a028
Return to citation in text: [1] -
Paquette, L. A.; Kuhla, D. E.; Barrett, J. H. J. Org. Chem. 1969, 34, 2879–2884. doi:10.1021/jo01262a016
Return to citation in text: [1] -
Le Count, D. J. Azepines and their Fused-ring Derivatives. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 45–66. doi:10.1016/B978-008096518-5.00209-4
See for more details on azepines.
Return to citation in text: [1] [2] -
Meigh, J.-P. K. Product Subclass 6: Benzazepines and Their Group 15 Analogues. In Science of Synthesis; Weinreb, S. M., Ed.; Thieme: Stuttgart, Germany, 2004; Vol. 17, pp 717–748.
See for more synthetic routes.
Return to citation in text: [1] -
Robey, R. L.; Copley-Merriman, C. R.; Phelps, M. A. J. Heterocycl. Chem. 1989, 26, 779–783. doi:10.1002/jhet.5570260350
Return to citation in text: [1] -
Steiner, G.; Franke, A.; Hädicke, E.; Lenke, D.; Teschendorf, H.-J.; Hofmann, H.-P.; Kreiskott, H.; Worstmann, W. J. Med. Chem. 1986, 29, 1877–1888. doi:10.1021/jm00160a015
Return to citation in text: [1] -
Muedas, C. A.; Schröder, D.; Sülzle, D.; Schwarz, H. J. Am. Chem. Soc. 1992, 114, 7582–7584. doi:10.1021/ja00045a052
Return to citation in text: [1] -
Märkl, G.; Schubert, H. Tetrahedron Lett. 1970, 11, 1273–1276. doi:10.1016/S0040-4039(01)91607-7
Return to citation in text: [1] -
Märkl, G.; Burger, W. Angew. Chem., Int. Ed. Engl. 1984, 23, 894–895. doi:10.1002/anie.198408941
Return to citation in text: [1] -
Märkl, G.; Burger, W. Tetrahedron Lett. 1983, 24, 2545–2548. doi:10.1016/S0040-4039(00)81977-2
Return to citation in text: [1] -
Kurita, J.; Shiratori, S.; Yasuike, S.; Tsuchiya, T. J. Chem. Soc., Chem. Commun. 1991, 1227–1228. doi:10.1039/C39910001227
Return to citation in text: [1] -
Yasuike, S.; Ohta, H.; Shiratori, S.; Kurita, J.; Tsuchiya, T. J. Chem. Soc., Chem. Commun. 1993, 1817–1819. doi:10.1039/C39930001817
Return to citation in text: [1] -
Segall, Y.; Shirin, E.; Granoth, I. Phosphorus Sulfur Relat. Elem. 1980, 8, 243–254. doi:10.1080/03086648008078196
Return to citation in text: [1] -
Yasuike, S.; Kiharada, T.; Kurita, J.; Tsuchiya, T. Chem. Commun. 1996, 2183–2184. doi:10.1039/cc9960002183
Return to citation in text: [1] -
Yasuike, S.; Kiharada, T.; Tsuchiya, T.; Kurita, J. Chem. Pharm. Bull. 2003, 51, 1283–1288. doi:10.1248/cpb.51.1283
Return to citation in text: [1] -
Pabel, M.; Wild, S. B. Rings containing Phosphorus. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 947–950. doi:10.1016/B978-008096518-5.00242-2
See for more details on phosphepines.
Return to citation in text: [1] -
Jansen, H.; Samuels, M. C.; Couzijn, E. P. A.; Slootweg, J. C.; Ehlers, A. W.; Chen, P.; Lammertsma, K. Chem.–Eur. J. 2010, 16, 1454–1458. doi:10.1002/chem.200902715
Return to citation in text: [1] -
Slootweg, J. C.; Lammertsma, K. Product Class 2: Phosphinidenes and termian Phosphinidene Complexes. In Science of Synthesis; Mathey, F., Ed.; Thieme: Stuttgart, Germany, 2009; Vol. 42, pp 15–36.
Return to citation in text: [1] -
Lammertsma, K. Top. Curr. Chem. 2003, 229, 95–119. doi:10.1007/b11152
Return to citation in text: [1] -
Mathey, F. Angew. Chem., Int. Ed. 2003, 42, 1578–1604. doi:10.1002/anie.200200557
Return to citation in text: [1] -
Le Floch, P. Coord. Chem. Rev. 2006, 250, 627–681. doi:10.1016/j.ccr.2005.04.032
Return to citation in text: [1] -
Noonan, K. J. T.; Gates, D. P. Angew. Chem., Int. Ed. 2006, 45, 7271–7274. doi:10.1002/anie.200602955
Return to citation in text: [1] -
Gates, D. P. Top. Curr. Chem. 2005, 250, 107–126. doi:10.1007/b100983
Return to citation in text: [1]
| 81. | Paquette, L. A. Angew. Chem., Int. Ed. Engl. 1971, 10, 11–20. doi:10.1002/anie.197100111 |
| 82. | Hafner, K. Angew. Chem., Int. Ed. Engl. 1964, 3, 165–173. doi:10.1002/anie.196401651 |
| 76. | Yamamoto, K.; Yamazaki, S. Thiepanes and Thiepines. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 67–111. doi:10.1016/B978-008096518-5.00211-2 |
| 76. | Yamamoto, K.; Yamazaki, S. Thiepanes and Thiepines. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 67–111. doi:10.1016/B978-008096518-5.00211-2 |
| 77. | Ueda, I.; Sato, Y.; Maeno, S.; Umio, S. Chem. Pharm. Bull. 1978, 26, 3058–3070. |
| 78. | Higashi, Y.; Momotani, Y.; Suzuki, E.; Kaku, T. Pharmacopsychiatry 1987, 20, 8–11. doi:10.1055/s-2007-1017124 |
| 79. | Schwan, A. L. Product Subclass 4: Benzothiepins and Selenium/Tellurium Analogues. In Science of Synthesis; Weinreb, S. M., Ed.; Thieme: Stuttgart, Germany, 2004; Vol. 17, pp 717–748. |
| 80. | Protiva, M. J. Heterocycl. Chem. 1996, 33, 497–521. doi:10.1002/jhet.5570330301 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 79. | Schwan, A. L. Product Subclass 4: Benzothiepins and Selenium/Tellurium Analogues. In Science of Synthesis; Weinreb, S. M., Ed.; Thieme: Stuttgart, Germany, 2004; Vol. 17, pp 717–748. |
| 88. | Prinzbach, H.; Babsch, H.; Fritz, H.; Hug, P. Tetrahedron Lett. 1977, 18, 1355–1358. doi:10.1016/S0040-4039(01)93042-4 |
| 42. | Kassaee, M. Z.; Arshadi, S.; Haerizade, B. N.; Vessally, E. J. Mol. Struct.: THEOCHEM 2005, 731, 29–37. doi:10.1016/j.theochem.2005.02.087 |
| 83. | Vogel, E.; Altenbach, H.-J.; Drossard, J.-M.; Schmickler, H.; Stegelmeier, H. Angew. Chem., Int. Ed. Engl. 1980, 19, 1016–1018. doi:10.1002/anie.198010161 |
| 84. | Smalley, R. K. Azepines. In Part 5: Small and Large Rings; Lwowski, W., Ed.; Comprehensive Heterocyclic Chemistry, Vol. 7; Elsevier Science Ltd.: Oxford, 1984; p 491. |
| 85. | Smalley, R. K. Azepines. In Part 5: Small and Large Rings; Lwowski, W., Ed.; Comprehensive Heterocyclic Chemistry, Vol. 7; Elsevier Science Ltd.: Oxford, 1984; p 543. |
| 86. | Lindner, H. J.; von Gross, B. Chem. Ber. 1972, 105, 434–440. doi:10.1002/cber.19721050208 |
| 94. | Paquette, L. A.; Kuhla, D. E.; Barrett, J. H. J. Org. Chem. 1969, 34, 2879–2884. doi:10.1021/jo01262a016 |
| 95. |
Le Count, D. J. Azepines and their Fused-ring Derivatives. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 45–66. doi:10.1016/B978-008096518-5.00209-4
See for more details on azepines. |
| 91. | Paquette, L. A.; Barrett, J. H. J. Am. Chem. Soc. 1966, 88, 2590–2591. doi:10.1021/ja00963a041 |
| 92. | Paquette, L. A.; Barrett, J. H.; Kuhla, D. E. J. Am. Chem. Soc. 1969, 91, 3616–3624. doi:10.1021/ja01041a032 |
| 93. | Paquette, L. A.; Barrett, J. H. J. Am. Chem. Soc. 1966, 88, 1718–1722. doi:10.1021/ja00960a028 |
| 89. | Paquette, L. A.; Kuhla, D. E.; Barrett, J. H.; Leichter, L. M. J. Org. Chem. 1969, 34, 2888–2896. doi:10.1021/jo01262a018 |
| 112. | Mathey, F. Angew. Chem., Int. Ed. 2003, 42, 1578–1604. doi:10.1002/anie.200200557 |
| 113. | Le Floch, P. Coord. Chem. Rev. 2006, 250, 627–681. doi:10.1016/j.ccr.2005.04.032 |
| 84. | Smalley, R. K. Azepines. In Part 5: Small and Large Rings; Lwowski, W., Ed.; Comprehensive Heterocyclic Chemistry, Vol. 7; Elsevier Science Ltd.: Oxford, 1984; p 491. |
| 90. | van den Hende, J. H.; Kende, A. S. Chem. Commun. 1965, 384–385. doi:10.1039/c19650000384 |
| 110. | Slootweg, J. C.; Lammertsma, K. Product Class 2: Phosphinidenes and termian Phosphinidene Complexes. In Science of Synthesis; Mathey, F., Ed.; Thieme: Stuttgart, Germany, 2009; Vol. 42, pp 15–36. |
| 111. | Lammertsma, K. Top. Curr. Chem. 2003, 229, 95–119. doi:10.1007/b11152 |
| 114. | Noonan, K. J. T.; Gates, D. P. Angew. Chem., Int. Ed. 2006, 45, 7271–7274. doi:10.1002/anie.200602955 |
| 115. | Gates, D. P. Top. Curr. Chem. 2005, 250, 107–126. doi:10.1007/b100983 |
| 109. | Jansen, H.; Samuels, M. C.; Couzijn, E. P. A.; Slootweg, J. C.; Ehlers, A. W.; Chen, P.; Lammertsma, K. Chem.–Eur. J. 2010, 16, 1454–1458. doi:10.1002/chem.200902715 |
| 98. | Steiner, G.; Franke, A.; Hädicke, E.; Lenke, D.; Teschendorf, H.-J.; Hofmann, H.-P.; Kreiskott, H.; Worstmann, W. J. Med. Chem. 1986, 29, 1877–1888. doi:10.1021/jm00160a015 |
| 45. | Jennings, W. B.; Rutherford, M.; Boyd, D. R.; Agarwal, S. K.; Sharma, N. D. Tetrahedron 1988, 44, 7551–7558. doi:10.1016/S0040-4020(01)86249-9 |
| 96. |
Meigh, J.-P. K. Product Subclass 6: Benzazepines and Their Group 15 Analogues. In Science of Synthesis; Weinreb, S. M., Ed.; Thieme: Stuttgart, Germany, 2004; Vol. 17, pp 717–748.
See for more synthetic routes. |
| 97. | Robey, R. L.; Copley-Merriman, C. R.; Phelps, M. A. J. Heterocycl. Chem. 1989, 26, 779–783. doi:10.1002/jhet.5570260350 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 38. |
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues. |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 51. | Dewar, M. J. S.; Trinajstić, N. J. Am. Chem. Soc. 1970, 92, 1453–1459. doi:10.1021/ja00709a001 |
| 38. |
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues. |
| 39. | Vogel, E.; Böll, W. A.; Günther, H. Tetrahedron Lett. 1965, 6, 609–615. doi:10.1016/S0040-4039(00)90005-4 |
| 1. | Vogel, E. Angew. Chem., Int. Ed. 2011, 50, 4278–4287. doi:10.1002/anie.201101347 |
| 2. | Maier, G. Angew. Chem., Int. Ed. Engl. 1967, 6, 402–413. doi:10.1002/anie.196704021 |
| 11. | Traetteberg, M. J. Am. Chem. Soc. 1964, 86, 4265–4270. doi:10.1021/ja01074a008 |
| 67. |
George, P.; Bock, C. W.; Glusker, J. P. J. Phys. Chem. 1990, 94, 8161–8168. doi:10.1021/j100384a034
In the presence of acid, protonation results in a carbocation intermediate and does not generate a radical species. |
| 68. | Holovka, J. M.; Gardner, P. D. J. Am. Chem. Soc. 1967, 89, 6390–9391. doi:10.1021/ja01000a092 |
| 10. | Ladenburg, A. Liebigs Ann. Chem. 1883, 217, 74–149. doi:10.1002/jlac.18832170107 |
| 5. | Borst, M. L. G.; Bulo, R. E.; Winkel, C. W.; Gibney, D. J.; Ehlers, A. W.; Schakel, M.; Lutz, M.; Spek, A. L.; Lammertsma, K. J. Am. Chem. Soc. 2005, 127, 5800–5801. doi:10.1021/ja050817y |
| 6. | Borst, M. L. G.; Bulo, R. E.; Gibney, D. J.; Alem, Y.; de Kanter, F. J. J.; Ehlers, A. W.; Schakel, M.; Lutz, M.; Spek, A. L.; Lammertsma, K. J. Am. Chem. Soc. 2005, 127, 16985–16999. doi:10.1021/ja054885w |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 8. | Couzijn, E. P. A.; Ehlers, A. W.; Slootweg, J. C.; Schakel, M.; Krill, S.; Lutz, M.; Spek, A. L.; Lammertsma, K. Chem.–Eur. J. 2008, 14, 1499–1507. doi:10.1002/chem.200700958 |
| 9. | Jansen, H.; Rosenthal, A. J.; Slootweg, J. C.; Ehlers, A. W.; Lutz, M.; Spek, A. L.; Lammertsma, K. Organometallics 2008, 27, 2868–2872. doi:10.1021/om800188h |
| 65. | Klotz, B.; Barnes, I.; Becker, K. H.; Golding, B. T. J. Chem. Soc., Faraday Trans. 1997, 93, 1507–1516. doi:10.1039/a606152d |
| 66. | Henderson, A. P.; Mutlu, E.; Leclercq, A.; Bleasdale, C.; Clegg, W.; Henderson, R. A.; Golding, B. T. Chem. Commun. 2002, 1956–1957. doi:10.1039/b205079j |
| 3. | Rodríguez-Hahn, L.; Esquivel, B.; Sánchez, A. A.; Cárdenas, J.; Tovar, O. G.; Soriano-García, M.; Toscano, A. J. Org. Chem. 1988, 53, 3933–3936. doi:10.1021/jo00252a010 |
| 4. | Güney, M.; Daştan, A.; Balci, M. Helv. Chim. Acta 2005, 88, 830–838. doi:10.1002/hlca.200590061 |
| 38. |
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues. |
| 51. | Dewar, M. J. S.; Trinajstić, N. J. Am. Chem. Soc. 1970, 92, 1453–1459. doi:10.1021/ja00709a001 |
| 18. | Jensen, F. R.; Smith, L. A. J. Am. Chem. Soc. 1964, 86, 956–957. doi:10.1021/ja01059a065 |
| 1. | Vogel, E. Angew. Chem., Int. Ed. 2011, 50, 4278–4287. doi:10.1002/anie.201101347 |
| 59. | Vogel, E.; Klärner, F.-G. Angew. Chem., Int. Ed. Engl. 1968, 7, 374–375. doi:10.1002/anie.196803742 |
| 60. | Klärner, F.-G.; Vogel, E. Angew. Chem., Int. Ed. Engl. 1973, 12, 840–841. doi:10.1002/anie.197308401 |
| 15. | Chen, Z.; Jiao, H.; Wu, J. I.; Herges, R.; Zhang, S. B.; von Ragué Schleyer, P. J. Phys. Chem. A 2008, 112, 10586–10594. doi:10.1021/jp802496m |
| 16. |
Jarzeçki, A. A.; Gajewski, J.; Davidson, E. R. J. Am. Chem. Soc. 1999, 121, 6928–6935. doi:10.1021/ja984471l
See for similar computational geometrical studies α = 52.1°, β= 25.2° determined at the CASSCF(6,6)/6-31G(d) level. |
| 17. |
Scott, A. P.; Agranat, I.; Biedermann, P. U.; Riggs, N. V.; Radom, L. J. Org. Chem. 1997, 62, 2026–2038. doi:10.1021/jo962407l
α = 57.5°, β= 27.6° determined at the B-LYP/6-31G(d) level. |
| 61. | Jerina, D. M.; Daly, J. W.; Witkop, B. In Biogenic Amines and Physiological Membranes in Drug Therapy, Part B; Biel, J. H.; Abood, L. G., Eds.; Marcel Dekker: New York, 1971; pp 413–476. |
| 62. | Daly, J. W.; Jerina, D. M.; Witkop, B. Experientia 1972, 28, 1129–1149. doi:10.1007/BF01946135 |
| 63. | Jerina, D. M.; Daly, J. W. Science 1974, 185, 573–582. doi:10.1126/science.185.4151.573 |
| 64. | Boyd, D. R.; Sharma, N. D. Chem. Soc. Rev. 1996, 25, 289–296. doi:10.1039/cs9962500289 |
| 13. |
Reed, T. B.; Libscomb, W. N. Acta Crystallogr. 1953, 6, 108. doi:10.1107/S0365110X53000399
Suitable crystals for an accurate X-ray determination could not be obtained for cyclohepta-1,3,5-triene. |
| 14. |
Davis, R. E.; Tulinksy, A. Tetrahedron Lett. 1962, 3, 839–846.
However the derivative 7,7-dimethylcycloheptatriene-3-carboxylic acid did result in suitable crystals. |
| 38. |
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues. |
| 55. | Vogel, E.; Günther, H. Angew. Chem., Int. Ed. Engl. 1967, 6, 385–401. doi:10.1002/anie.196703851 |
| 56. | Belen’kll, L. I. Oxepanes and Oxepines. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 45–66. doi:10.1016/B978-008096518-5.00210-0 |
| 57. | Bock, C. W.; George, P.; Stezowski, J. J.; Glusker, J. P. Struct. Chem. 1989, 1, 33–39. doi:10.1007/BF00675782 |
| 58. | Hayes, D. M.; Nelson, S. D.; Garland, W. A.; Kollman, P. A. J. Am. Chem. Soc. 1980, 102, 1255–1262. doi:10.1021/ja00524a007 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 48. |
Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; von Ragué Schleyer, P. Chem. Rev. 2005, 105, 3842–3888. doi:10.1021/cr030088+
The NICS value is obtained from NMR calculations of a ghost atom (Bq) at the geometrical ring center (NICS(0)), but the value 1 Å above the ring (NICS(1)) is recommended, because a lack of σ shielding makes it a better measure of π-electron delocalization. |
| 70. | Field, L.; Tuleen, D. L. Monocyclic Seven-Membered Rings Containing Sulfur. In Seven-Membered Heterocyclic Compounds Containing Oxygen and Sulfur; Rosowsky, A., Ed.; Wiley Interscience: New York, 1972; Chapter 10. |
| 61. | Jerina, D. M.; Daly, J. W.; Witkop, B. In Biogenic Amines and Physiological Membranes in Drug Therapy, Part B; Biel, J. H.; Abood, L. G., Eds.; Marcel Dekker: New York, 1971; pp 413–476. |
| 69. | Gillard, J. R.; Newlands, M. J.; Bridson, J. N.; Burnell, D. J. Can. J. Chem. 1991, 69, 1337–1343. doi:10.1139/v91-199 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 70. | Field, L.; Tuleen, D. L. Monocyclic Seven-Membered Rings Containing Sulfur. In Seven-Membered Heterocyclic Compounds Containing Oxygen and Sulfur; Rosowsky, A., Ed.; Wiley Interscience: New York, 1972; Chapter 10. |
| 14. |
Davis, R. E.; Tulinksy, A. Tetrahedron Lett. 1962, 3, 839–846.
However the derivative 7,7-dimethylcycloheptatriene-3-carboxylic acid did result in suitable crystals. |
| 74. | Reinhoudt, D. N.; Kouwenhoven, C. G. Tetrahedron 1974, 30, 2093–2098. doi:10.1016/S0040-4020(01)97344-2 |
| 75. | Yamamoto, K.; Yamazaki, S.; Kohashi, Y.; Murata, I.; Kai, Y.; Kanehisa, N.; Miki, K.; Kasai, N. Tetrahedron Lett. 1982, 23, 3195–3198. doi:10.1016/S0040-4039(00)88594-9 |
| 71. | Miller, K. J.; Moschner, K. F.; Potts, K. T. J. Am. Chem. Soc. 1983, 105, 1705–1712. doi:10.1021/ja00345a001 |
| 72. | Gleiter, R.; Krennrich, G.; Cremer, D.; Yamamoto, K.; Murata, I. J. Am. Chem. Soc. 1985, 107, 6874–6879. doi:10.1021/ja00310a022 |
| 73. | Nishino, K.; Takagi, M.; Kawata, T.; Murata, I.; Inanaga, J.; Nakasuji, K. J. Am. Chem. Soc. 1991, 113, 5059–5060. doi:10.1021/ja00013a051 |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 45. | Jennings, W. B.; Rutherford, M.; Boyd, D. R.; Agarwal, S. K.; Sharma, N. D. Tetrahedron 1988, 44, 7551–7558. doi:10.1016/S0040-4020(01)86249-9 |
| 43. | Cremer, D.; Dick, B.; Christeu, D. J. Mol. Struct.: THEOCHEM 1984, 110, 277–291. doi:10.1016/0166-1280(84)80077-9 |
| 44. | Kao, J. J. Comput. Chem. 1988, 9, 905–923. doi:10.1002/jcc.540090812 |
| 45. | Jennings, W. B.; Rutherford, M.; Boyd, D. R.; Agarwal, S. K.; Sharma, N. D. Tetrahedron 1988, 44, 7551–7558. doi:10.1016/S0040-4020(01)86249-9 |
| 46. | Jennings, W. B.; Rutherford, M.; Agarwal, S. K.; Boyd, D. R.; Malone, J. F.; Kennedy, D. A. J. Chem. Soc., Chem. Commun. 1986, 970–972. doi:10.1039/C39860000970 |
| 48. |
Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; von Ragué Schleyer, P. Chem. Rev. 2005, 105, 3842–3888. doi:10.1021/cr030088+
The NICS value is obtained from NMR calculations of a ghost atom (Bq) at the geometrical ring center (NICS(0)), but the value 1 Å above the ring (NICS(1)) is recommended, because a lack of σ shielding makes it a better measure of π-electron delocalization. |
| 49. |
Kassaee, M. Z.; Musavi, S. M.; Momeni, M. R.; Shakib, F. A.; Ghambarian, M. J. Mol. Struct.: THEOCHEM 2008, 861, 117–121. doi:10.1016/j.theochem.2008.04.031
See for IAO-B3LYP/6-311G(d)//B3LYP/6-311G(d) level of theory. |
| 41. | Pye, C. C.; Xidos, J. D.; Poirier, R. A.; Burnell, D. J. J. Phys. Chem. A 1997, 101, 3371–3376. doi:10.1021/jp9623498 |
| 19. | Anet, F. A. L. J. Am. Chem. Soc. 1964, 86, 458–460. doi:10.1021/ja01057a034 |
| 20. |
Nishinaga, T.; Izukawa, Y.; Komatsu, K. J. Phys. Org. Chem. 1998, 11, 475–477. doi:10.1002/(SICI)1099-1395(199807)11:7<475::AID-POC991>3.0.CO;2-O
See for determination of “Nuclear Independent Chemical Shift” (NICS(0), benzene-8.0 ppm) gives-4.2 ppm for 1. |
| 41. | Pye, C. C.; Xidos, J. D.; Poirier, R. A.; Burnell, D. J. J. Phys. Chem. A 1997, 101, 3371–3376. doi:10.1021/jp9623498 |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 47. | Kassaee, M. Z.; Cheshmehkani, A.; Musavi, S. M.; Majdi, M.; Motamedi, E. J. Mol. Struct.: THEOCHEM 2008, 865, 73–78. doi:10.1016/j.theochem.2008.06.025 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 46. | Jennings, W. B.; Rutherford, M.; Agarwal, S. K.; Boyd, D. R.; Malone, J. F.; Kennedy, D. A. J. Chem. Soc., Chem. Commun. 1986, 970–972. doi:10.1039/C39860000970 |
| 50. | Karney, W. L.; Kastrup, C. J.; Oldfield, S. P.; Rzepa, H. S. J. Chem. Soc., Perkin Trans. 2 2002, 388–392. doi:10.1039/b111369k |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 14. |
Davis, R. E.; Tulinksy, A. Tetrahedron Lett. 1962, 3, 839–846.
However the derivative 7,7-dimethylcycloheptatriene-3-carboxylic acid did result in suitable crystals. |
| 54. | van Tamelen, E. E.; Carty, D. T. J. Am. Chem. Soc. 1967, 89, 3922–3923. doi:10.1021/ja00991a056 |
| 51. | Dewar, M. J. S.; Trinajstić, N. J. Am. Chem. Soc. 1970, 92, 1453–1459. doi:10.1021/ja00709a001 |
| 52. | Hess, B. A., Jr.; Schaad, L. J. J. Am. Chem. Soc. 1973, 95, 3907–3912. doi:10.1021/ja00793a013 |
| 38. |
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues. |
| 53. | Vogel, E.; Schubart, R.; Böll, W. A. Angew. Chem., Int. Ed. Engl. 1964, 3, 510. doi:10.1002/anie.196405101 |
| 47. | Kassaee, M. Z.; Cheshmehkani, A.; Musavi, S. M.; Majdi, M.; Motamedi, E. J. Mol. Struct.: THEOCHEM 2008, 865, 73–78. doi:10.1016/j.theochem.2008.06.025 |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 102. | Märkl, G.; Burger, W. Tetrahedron Lett. 1983, 24, 2545–2548. doi:10.1016/S0040-4039(00)81977-2 |
| 103. | Kurita, J.; Shiratori, S.; Yasuike, S.; Tsuchiya, T. J. Chem. Soc., Chem. Commun. 1991, 1227–1228. doi:10.1039/C39910001227 |
| 104. | Yasuike, S.; Ohta, H.; Shiratori, S.; Kurita, J.; Tsuchiya, T. J. Chem. Soc., Chem. Commun. 1993, 1817–1819. doi:10.1039/C39930001817 |
| 105. | Segall, Y.; Shirin, E.; Granoth, I. Phosphorus Sulfur Relat. Elem. 1980, 8, 243–254. doi:10.1080/03086648008078196 |
| 106. | Yasuike, S.; Kiharada, T.; Kurita, J.; Tsuchiya, T. Chem. Commun. 1996, 2183–2184. doi:10.1039/cc9960002183 |
| 107. | Yasuike, S.; Kiharada, T.; Tsuchiya, T.; Kurita, J. Chem. Pharm. Bull. 2003, 51, 1283–1288. doi:10.1248/cpb.51.1283 |
| 5. | Borst, M. L. G.; Bulo, R. E.; Winkel, C. W.; Gibney, D. J.; Ehlers, A. W.; Schakel, M.; Lutz, M.; Spek, A. L.; Lammertsma, K. J. Am. Chem. Soc. 2005, 127, 5800–5801. doi:10.1021/ja050817y |
| 95. |
Le Count, D. J. Azepines and their Fused-ring Derivatives. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 45–66. doi:10.1016/B978-008096518-5.00209-4
See for more details on azepines. |
| 101. | Märkl, G.; Burger, W. Angew. Chem., Int. Ed. Engl. 1984, 23, 894–895. doi:10.1002/anie.198408941 |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 100. | Märkl, G.; Schubert, H. Tetrahedron Lett. 1970, 11, 1273–1276. doi:10.1016/S0040-4039(01)91607-7 |
| 99. | Muedas, C. A.; Schröder, D.; Sülzle, D.; Schwarz, H. J. Am. Chem. Soc. 1992, 114, 7582–7584. doi:10.1021/ja00045a052 |
| 14. |
Davis, R. E.; Tulinksy, A. Tetrahedron Lett. 1962, 3, 839–846.
However the derivative 7,7-dimethylcycloheptatriene-3-carboxylic acid did result in suitable crystals. |
| 26. | Hall, G. E.; Roberts, J. D. J. Am. Chem. Soc. 1971, 93, 2203–2207. doi:10.1021/ja00738a019 |
| 27. | Tang, T.-H.; Lew, C. S. Q.; Cui, Y.-P.; Capon, B.; Csizmadia, I. G. J. Mol. Struct.: THEOCHEM 1994, 305, 149–164. doi:10.1016/0166-1280(94)80150-9 |
| 22. | Maier, G. Angew. Chem., Int. Ed. Engl. 1967, 6, 812. doi:10.1002/anie.196708121 |
| 23. | Lowry, T. H.; Richardson, K. S. Mechanism and Theory in Organic Chemistry, 4th ed.; Harper and Row: London, 1990. |
| 5. | Borst, M. L. G.; Bulo, R. E.; Winkel, C. W.; Gibney, D. J.; Ehlers, A. W.; Schakel, M.; Lutz, M.; Spek, A. L.; Lammertsma, K. J. Am. Chem. Soc. 2005, 127, 5800–5801. doi:10.1021/ja050817y |
| 6. | Borst, M. L. G.; Bulo, R. E.; Gibney, D. J.; Alem, Y.; de Kanter, F. J. J.; Ehlers, A. W.; Schakel, M.; Lutz, M.; Spek, A. L.; Lammertsma, K. J. Am. Chem. Soc. 2005, 127, 16985–16999. doi:10.1021/ja054885w |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 8. | Couzijn, E. P. A.; Ehlers, A. W.; Slootweg, J. C.; Schakel, M.; Krill, S.; Lutz, M.; Spek, A. L.; Lammertsma, K. Chem.–Eur. J. 2008, 14, 1499–1507. doi:10.1002/chem.200700958 |
| 9. | Jansen, H.; Rosenthal, A. J.; Slootweg, J. C.; Ehlers, A. W.; Lutz, M.; Spek, A. L.; Lammertsma, K. Organometallics 2008, 27, 2868–2872. doi:10.1021/om800188h |
| 24. |
Menzek, A.; Balci, M. Tetrahedron 1993, 49, 6071–6078. doi:10.1016/S0040-4020(01)87191-X
And references therein. |
| 7. | Jansen, H.; Slootweg, J. C.; Ehlers, A. W.; Lammertsma, K. Organometallics 2010, 29, 6653–6659. doi:10.1021/om1004379 |
| 19. | Anet, F. A. L. J. Am. Chem. Soc. 1964, 86, 458–460. doi:10.1021/ja01057a034 |
| 20. |
Nishinaga, T.; Izukawa, Y.; Komatsu, K. J. Phys. Org. Chem. 1998, 11, 475–477. doi:10.1002/(SICI)1099-1395(199807)11:7<475::AID-POC991>3.0.CO;2-O
See for determination of “Nuclear Independent Chemical Shift” (NICS(0), benzene-8.0 ppm) gives-4.2 ppm for 1. |
| 21. | Herges, R.; Geuenich, D. J. Phys. Chem. A 2001, 105, 3214–3220. doi:10.1021/jp0034426 |
| 108. |
Pabel, M.; Wild, S. B. Rings containing Phosphorus. In Seven-membered and Larger Rings and Fused Derivatives; Newkome, G. R., Ed.; Comprehensive Heterocyclic Chemistry II, Vol. 9; Elsevier Science Ltd.: Oxford, 1996; pp 947–950. doi:10.1016/B978-008096518-5.00242-2
See for more details on phosphepines. |
| 32. | Klärner, F.-G. Angew. Chem., Int. Ed. Engl. 1974, 13, 268–270. doi:10.1002/anie.197402681 |
| 33. | Klärner, K.-G.; Yaşlak, S.; Wette, M. Chem. Ber. 1977, 110, 107–123. doi:10.1002/cber.19771100112 |
| 34. | Klärner, K.-G.; Yaşlak, S.; Wette, M. Chem. Ber. 1979, 112, 1168–1188. doi:10.1002/cber.19791120412 |
| 35. | Klärner, K.-G.; Brassel, B. J. Am. Chem. Soc. 1980, 102, 2469–2470. doi:10.1021/ja00527a062 |
| 29. | Klump, K. N.; Chesick, J. P. J. Am. Chem. Soc. 1963, 85, 130–132. doi:10.1021/ja00885a003 |
| 30. | Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1965, 87, 2751–2752. doi:10.1021/ja01090a037 |
| 31. | Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1965, 87, 2752–2753. doi:10.1021/ja01090a038 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 41. | Pye, C. C.; Xidos, J. D.; Poirier, R. A.; Burnell, D. J. J. Phys. Chem. A 1997, 101, 3371–3376. doi:10.1021/jp9623498 |
| 42. | Kassaee, M. Z.; Arshadi, S.; Haerizade, B. N.; Vessally, E. J. Mol. Struct.: THEOCHEM 2005, 731, 29–37. doi:10.1016/j.theochem.2005.02.087 |
| 44. | Kao, J. J. Comput. Chem. 1988, 9, 905–923. doi:10.1002/jcc.540090812 |
| 38. |
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues. |
| 39. | Vogel, E.; Böll, W. A.; Günther, H. Tetrahedron Lett. 1965, 6, 609–615. doi:10.1016/S0040-4039(00)90005-4 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
| 41. | Pye, C. C.; Xidos, J. D.; Poirier, R. A.; Burnell, D. J. J. Phys. Chem. A 1997, 101, 3371–3376. doi:10.1021/jp9623498 |
| 17. |
Scott, A. P.; Agranat, I.; Biedermann, P. U.; Riggs, N. V.; Radom, L. J. Org. Chem. 1997, 62, 2026–2038. doi:10.1021/jo962407l
α = 57.5°, β= 27.6° determined at the B-LYP/6-31G(d) level. |
| 18. | Jensen, F. R.; Smith, L. A. J. Am. Chem. Soc. 1964, 86, 956–957. doi:10.1021/ja01059a065 |
| 24. |
Menzek, A.; Balci, M. Tetrahedron 1993, 49, 6071–6078. doi:10.1016/S0040-4020(01)87191-X
And references therein. |
| 36. | Ter Borg, A. P.; Kloosterziel, H.; Van Meurs, N. Proc. Chem. Soc., London 1962, 359. |
| 37. | Weth, E.; Dreiding, A. S. Proc. Chem. Soc., London 1964, 59–60. |
| 38. |
Berson, J. A.; Willcott, M. R., III. J. Am. Chem. Soc. 1966, 88, 2494–2502. doi:10.1021/ja00963a025
See for the methyl-substituted analogues. |
| 39. | Vogel, E.; Böll, W. A.; Günther, H. Tetrahedron Lett. 1965, 6, 609–615. doi:10.1016/S0040-4039(00)90005-4 |
| 40. | Günther, H. Tetrahedron Lett. 1965, 6, 4085–4090. doi:10.1016/S0040-4039(01)99570-X |
© 2011 Jansen et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)