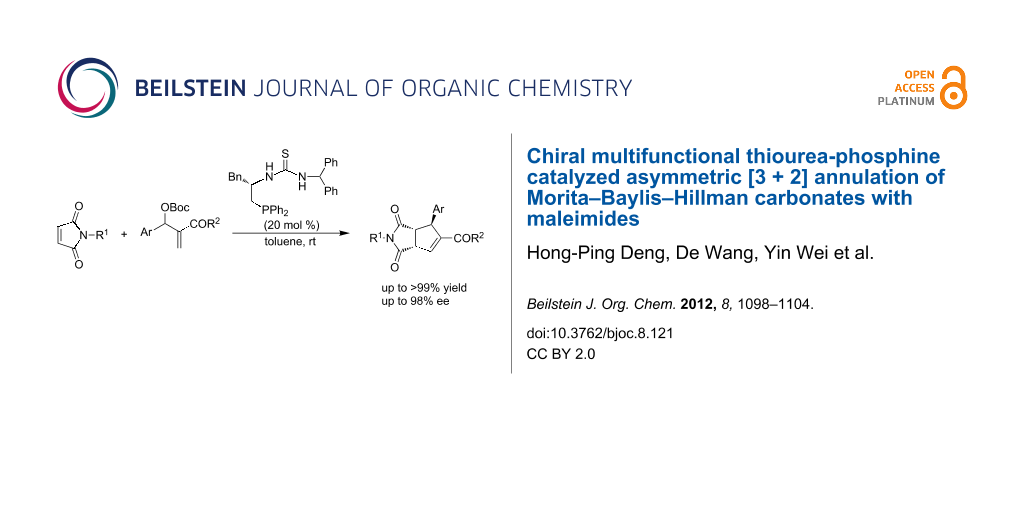
Abstract
We have developed a multifunctional thiourea-phosphine catalyzed asymmetric [3 + 2] annulation of Morita–Baylis–Hillman (MBH) carbonates with maleimides, which can efficiently construct functionalized cyclopentenes bearing three contiguous stereocenters in moderate to excellent yields and excellent diastereo- and enantioselectivities. A plausible mechanism has been also proposed on the basis of control experiments and previous literature.

Graphical Abstract
Introduction
Highly functionalized cyclopentene derivatives are important subunits in a variety of biologically active molecules and have attracted the broad attention of synthetic and pharmaceutical chemists [1,2]. Among numerous synthetic approaches, phosphine-mediated [3 + 2] annulation of electron-deficient olefins is an efficient method to construct this interesting structural motif [3-10]. According to the pioneering work of Lu [11-20], all phosphine-mediated [3 + 2] annulations proceeded through an important intermediate “1,3-dipolar synthon”. The formation of a 1,3-dipolar synthon by using a catalytic amount of phosphines have been directed toward the following two paths: phosphines attack the middle carbon atom of allenes to produce the 1,3-dipolar synthon (Scheme 1, reaction 1), and phosphines add to the β-position of MBH carbonate to remove carbon dioxide and tert-butanol, affording the 1,3-dipolar synthon (Scheme 1, reaction 2). Concerning the asymmetric [3 + 2] annulation catalyzed by chiral phosphines, Zhang and co-workers first reported the asymmetric [3 + 2] annulation of allenoates with acrylates catalyzed by a bicyclic chiral phosphine in 1997 [21]. Moreover, Fu [22-24], Marinetti [25-28], Lu [29-31] and other researchers [32-36] have also developed asymmetric [3 + 2] annulations of allenoates to give the corresponding cyclopentene derivatives in good yields with excellent enantioselectivities [37].
![[1860-5397-8-121-i1]](/bjoc/content/inline/1860-5397-8-121-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Paths to the formation of 1,3-dipolar synthons by using a catalytic amount of phosphines.
Scheme 1: Paths to the formation of 1,3-dipolar synthons by using a catalytic amount of phosphines.
On the other hand, some examples of phosphine-catalyzed [3 + 2] annulation of MBH carbonates with electron-deficient alkenes have been reported in the literature recently. Lu and co-workers first disclosed the formation of 1,3-dipoles through phosphine-catalyzed MBH carbonates under mild conditions, which can efficiently react with various electron-deficient olefins to afford the desired cyclic products in good yields along with high regioselectivities, through intra- and intermolecular [3 + n] annulations [38-47]. To the best of our knowledge, there is little in the literature regarding the asymmetric version of this reaction. In 2010, Tang and co-workers reported the first example of asymmetric intramolecular [3 + 2] annulations of MBH carbonates tethered with another electron-deficient olefin in the presence of spirobiindane-based chiral phosphines, giving the corresponding cycloadducts in good yields along with high ee values [48]. Barbas and co-workers first reported asymmetric intermolecular [3 + 2] cycloaddition of MBH carbonates with methyleneindolinones to afford the corresponding spirocyclopentaneoxindoles in good yields and high ee values in 2011 [49]. Moreover, Lu and co-workers have recently explored a series of thiourea-phosphine catalysts derived from L-threonine, which are effective catalysts in the [3 + 2] annulation of MBH carbonates with isatylidene malononitriles to give the desired products in high yields with high enantioselectivities [50]. Furthermore, our group recently also synthesized a series of L-phenylalanine-derived multifunctional thiourea-phosphine catalysts and used them in the [3 + 2] annulation of MBH carbonates with trifluoroethylidenemalonates to give the cycloadducts in high yields along with excellent diastereo- and enantioselectivities [51]. Herein we wish to report the development of a multifunctional thiourea-phosphine catalyzed asymmetric [3 + 2] annulation of MBH carbonates with maleimides, which can efficiently construct functionalized cyclopentene derivatives bearing three contiguous stereocenters in moderate to excellent yields along with excellent diastereo- and enantioselectivities.
Results and Discussion
In our previous work, it was shown that chiral multifunctional thiourea-phosphines were excellent catalysts for the asymmetric aza-MBH reaction, asymmetric allylic substitution of MBH adducts, and asymmetric [3 + 2] annulation of MBH carbonates with electron-deficient olefins [52-56]. Hence, we initially used multifunctional thiourea-phosphine (TP), which could be easily prepared from L-phenylalanine in four steps [57], as a catalyst to investigate the asymmetric [3 + 2] annulation of N-phenylmaleimide (1a) with MBH carbonate 2a in toluene at room temperature. We were pleased to find that the corresponding highly functionalized cyclopentene 3a was obtained in 54% yield with 96% ee after 24 h (Table 1, entry 1). The examination of solvent effects revealed that toluene is the best solvent (Table 1, entries 2–4). Increasing the ratio of 1a/2a from 1:1.3 to 2:1 afforded 3a in 74% yield and 96% ee, while continuously increasing the ratio of 1a/2a to 4:1 gave 3a in lower yield (Table 1, entries 5 and 6).
Table 1: Optimization of the reaction conditions for asymmetric [3 + 2] annulation of MBH carbonates and maleimides.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-121-i4.svg?max-width=637&scale=1.0)
|
||||
| entrya | 1a/2a | solvent | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 1/1.3 | toluene | 54 | 96 |
| 2 | 1/1.3 | CHCl3 | 54 | 96 |
| 3 | 1/1.3 | THF | 25 | 90 |
| 4 | 1/1.3 | CH3CN | 12 | 43 |
| 5d | 2/1 | toluene | 74 | 96 |
| 6e | 4/1 | toluene | 62 | 96 |
aReactions were performed with 1a (0.1 mmol) and 2a (0.13 mmol) in toluene (1.0 mL) at room temperature for 24 h. bIsolated yield of major isomer. cDetermined by chiral HPLC. dReaction was performed with 1a (0.2 mmol) and 2a (0.1 mmol) in toluene (1.0 mL) at room temperature for 24 h. eReaction was performed with 1a (0.4 mmol) and 2a (0.1 mmol) in toluene (1.0 mL) at room temperature for 24 h.
Having identified the optimal reaction conditions, we next set out to examine the scope and limitations of this asymmetric [3 + 2] annulation of maleimides with MBH carbonates, and the results are summarized in Table 2. All of the reactions proceeded smoothly under the optimal conditions, providing annulation products with good to excellent diastereo- and enantioselectivities. Using MBH carbonate 2a as a substrate, we examined its reaction with maleimides 1b–1e and found that the reactions proceeded smoothly to give the corresponding products 3b–3e in excellent yields along with excellent ee values (Table 2, entries 2–5). Taking N-benzhydrylmaleimide (1d) as a substrate, we found that substrates with an electron-withdrawing substituent on the aromatic ring of MBH carbonates 2 produced 3f and 3g in good yields and excellent ee values (Table 2, entries 6 and 7), and MBH carbonate 2d gave the corresponding annulation product 3h in 3:1 dr value with 74% yield of major isomer and 97% ee (Table 2, entry 8). A variety of MBH carbonates 2 having either electron-donating or -withdrawing groups as substituents at the para- and meta-position of the benzene ring and N-methylmaleimide (1b) underwent this asymmetric [3 + 2] annulation smoothly, affording the corresponding products 3 in moderate to excellent yields with excellent ee values upon lengthening of the reaction time (Table 2, entries 9–11, 13–15). Substrate 2k, incorporating a heteroaromatic group, could also react with 1b under the same reaction conditions, giving the corresponding cyclopentene 3p in 69% yield with 96% ee, and substrates 2l and 2m derived from methyl vinyl ketone (MVK) afforded the corresponding annulation products 3q and 3r in excellent yields and ee values under the standard conditions (Table 2, entries 16–18). The absolute configuration of the major product 3r was determined as (1S,2R,3S) by X-ray crystal structure. Its ORTEP plot is shown in Figure 1 and the corresponding CIF data are presented in the Supporting Information File 2 [58]. As for MBH carbonate 2g, having a chlorine substituent at the ortho-position of the aromatic ring, this produced 3l in lower yield and ee value, perhaps due to the steric effect; reaction of MBH carbonate 2n derived from isobutyraldehyde did not occur (entries 12 and 19). When we utilized dimethyl maleate or dimethyl fumarate instead of maleimides, it was found that the reactions could not be performed either.
Table 2: Substrate scope of asymmetric [3 + 2] annulation.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-121-i5.svg?max-width=637&scale=1.0)
|
|||||
| entrya | R1 | R2 | EWG | yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | 1a, Ph | 2a, 4-NO2C6H4 | CO2Et | 3a, 74 | 96 |
| 2 | 1b, Me | 2a, 4-NO2C6H4 | CO2Et | 3b, 99 | 98 |
| 3 | 1c, Bn | 2a, 4-NO2C6H4 | CO2Et | 3c, 86 | 94 |
| 4 | 1d, benzhydryl | 2a, 4-NO2C6H4 | CO2Et | 3d, >99 | 98 |
| 5 | 1e, 1-methylnaphthyl | 2a, 4-NO2C6H4 | CO2Et | 3e, 91 | 96 |
| 6 | 1d, benzhydryl | 2b, 3-NO2C6H4 | CO2Et | 3f, 87 | 94 |
| 7 | 1d, benzhydryl | 2c, 4-CNC6H4 | CO2Et | 3g, 84 | 96 |
| 8d,e | 1d, benzhydryl | 2d, 4-BrC6H4 | CO2Et | 3h, 74 | 97 |
| 9e | 1b, Me | 2d, 4-BrC6H4 | CO2Et | 3i, 79 | 95 |
| 10e | 1b, Me | 2e, 4-ClC6H4 | CO2Et | 3j, 90 | 96 |
| 11e | 1b, Me | 2f, 3-ClC6H4 | CO2Et | 3k, 81 | 97 |
| 12f | 1b, Me | 2g, 2-ClC6H4 | CO2Et | 3l, 39 | 73 |
| 13f | 1b, Me | 2h, C6H5 | CO2Et | 3m, 74 | 94 |
| 14f | 1b, Me | 2i, 4-MeC6H4 | CO2Et | 3n, 64 | 96 |
| 15f | 1b, Me | 2j, 4-MeOC6H4 | CO2Et | 3o, 55 | 98 |
| 16f | 1b, Me | 2k, 2-furyl | CO2Et | 3p, 69 | 96 |
| 17 | 1b, Me | 2l, 4-NO2C6H4 | COMe | 3q, >99 | 97 |
| 18 | 1b, Me | 2m, 4-NO2C6H4 | COMe | 3r, 92 | 98 |
| 19f | 1b, Me | 2n, (CH3)2CH | CO2Et | – | – |
aReactions were performed with 1 (0.2 mmol), 2 (0.1 mmol) in toluene (1.0 mL) at room temperature for 24 h. bIsolated yield of major isomer. cDetermined by chiral HPLC. ddr = 3:1. eReaction was performed for 36 h. fReaction was performed for 48 h.
![[1860-5397-8-121-1]](/bjoc/content/figures/1860-5397-8-121-1.png?scale=2.0&max-width=1024&background=FFFFFF)
As aforementioned, increasing the ratio of 1a/2a from 1:1.3 to 2:1 obtained the highest yield. In order to explain this finding, three control experiments were performed in CDCl3 and the results were studied by using 31P NMR spectroscopy (Figure 2, also see Figure SI-1 in the Supporting Information File 1). The NMR studies revealed that catalyst TP seems to react with N-phenylmaleimide (1a) to form a new species (Figure 2, spectra b), which costs some maleimide 1a. Thus, the excess 1a has to be added in order to make the reaction complete. This is why increasing the ratio of 1a/2a raised the yield of product 3a (Table 1, entry 5). On the other hand, if too much maleimide 1a is used in this reaction, it will waste some of the catalyst TP, leading to a decrease in the yield of product 3a (Table 1, entry 6). The allylic phosphorus ylide species can be recognized in spectra c from the combination of 2d and TP (1:1) (Figure 2, also see Figure SI-1 in the Supporting Information File 1) [59].
![[1860-5397-8-121-2]](/bjoc/content/figures/1860-5397-8-121-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 31P NMR spectra (161.9 MHz, CDCl3) of control experiments.
Figure 2: 31P NMR spectra (161.9 MHz, CDCl3) of control experiments.
As reported before [15,51], TP attacks MBH carbonate to afford allylic phosphorus ylide I, which attacked the maleimide to produce intermediate II (Scheme 2). Since there is a steric effect between the phenyl group and the benzyl group in intermediate II-B, allylic phosphorus ylide I using its Si-face to attack maleimide is favored (intermediate II-A). Undergoing Michael addition and elimination of catalyst along with the double-bond formation, intermediate I affords the corresponding highly functionalized cyclopentene product and completes the catalytic cycle. The control experiments demonstrated that catalyst TP can react with maleimide, affording an unidentified complex and partially wasting maleimide and catalyst (spectra d in Figure 2). Hypothetically, the activity of the in situ generated allylic phosphorous ylide I is crucial for the yield of product. If the activity of phosphorous ylide is not high enough, it may deprotonate the NH proton in TP, which will cause the catalyst to lose activity (See Table 2, entries 12–16). In addition, if the phosphorous ylide is not active enough to react with maleimide, maleimide can directly react with the catalyst to form the other unidentified species as indicated in the 31P NMR spectroscopy (spectra b in Figure 2), which may retard the desired catalytic cycle.
![[1860-5397-8-121-i2]](/bjoc/content/inline/1860-5397-8-121-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
To illustrate the synthetic utility of these products 3 obtained from the above asymmetric [3 + 2] annulation, the further transformation of 3c was performed in the presence of RuCl3 and NaIO4 under mild conditions (Scheme 3) [22,60]. Upon dihydroxylation of 3c, the corresponding product 4c containing five stereocenters was produced in 69% yield and good diastereoselectivity on the basis of NMR spectroscopic data (dr = 20:1, see Supporting Information File 1).
![[1860-5397-8-121-i3]](/bjoc/content/inline/1860-5397-8-121-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have developed a novel multifunctional thiourea-phosphine catalyzed asymmetric [3 + 2] annulation of MBH carbonates with maleimides, which can efficiently construct functionalized cyclopentenes bearing three contiguous stereocenters in moderate to excellent yields and excellent diastereo- and enantioselectivities, and the product can be efficiently transformed into a cyclopentane containing five stereocenters under mild conditions, which was difficult to construct by other synthetic methodologies. Current efforts are in progress to apply this new methodology to synthesize biologically active products.
Experimental
General procedure for asymmetric [3 + 2] annulation
Under an argon atmosphere, a mixture of maleimide 1 (0.2 mmol), MBH carbonate 2 (0.2 mmol) and catalyst TP (0.02 mmol, 11 mg) in toluene (1.0 mL) was stirred at room temperature for 24–48 h. Then the solvent was removed under reduced pressure, and the residue was chromatographed on silica gel (elution with petroleum ether/EtOAc 10:1–4:1) to provide compound 3.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data of compounds given in this article. | ||
| Format: PDF | Size: 2.0 MB | Download |
| Supporting Information File 2: Crystal structure data of compound 3r. | ||
| Format: PDF | Size: 255.1 KB | Download |
| Supporting Information File 3: cif data of 3r. | ||
| Format: CIF | Size: 15.4 KB | Download |
Acknowledgements
We thank the Shanghai Municipal Committee of Science and Technology (11JC1402600), National Basic Research Program of China (973)-2010CB833302, and the National Natural Science Foundation of China for financial support (21072206, 20472096, 20872162, 20672127, 21121062 and 20732008) and Mr. Jie Sun for performing X-ray diffraction.
References
-
Hartley, R. C.; Caldwell, S. T. J. Chem. Soc., Perkin Trans. 1 2000, 477–501. doi:10.1039/A804421J
Return to citation in text: [1] -
Wu, H.; Zhang, H.; Zhao, G. Tetrahedron 2007, 63, 6454–6461. doi:10.1016/j.tet.2007.03.031
Return to citation in text: [1] -
Lu, X.; Zhang, C.; Xu, Z. Acc. Chem. Res. 2001, 34, 535–544. doi:10.1021/ar000253x
Return to citation in text: [1] -
Methot, J. L.; Roush, W. R. Adv. Synth. Catal. 2004, 346, 1035–1050. doi:10.1002/adsc.200404087
Return to citation in text: [1] -
Ye, L.-W.; Zhou, J.; Tang, Y. Chem. Soc. Rev. 2008, 37, 1140–1152. doi:10.1039/b717758e
Return to citation in text: [1] -
Cowen, B. J.; Miller, S. J. Chem. Soc. Rev. 2009, 38, 3102–3116. doi:10.1039/b816700c
Return to citation in text: [1] -
Marinetti, A.; Voituriez, A. Synlett 2010, 174–194. doi:10.1055/s-0029-1219157
Return to citation in text: [1] -
Wei, Y.; Shi, M. Acc. Chem. Res. 2010, 43, 1005–1018. doi:10.1021/ar900271g
Return to citation in text: [1] -
López, F.; Mascareñas, J. L. Chem.–Eur. J. 2011, 17, 418–428. doi:10.1002/chem.201002366
Return to citation in text: [1] -
Zhao, Q.-Y.; Lian, Z.; Wei, Y.; Shi, M. Chem. Commun. 2012, 48, 1724–1732. doi:10.1039/c1cc15793k
Return to citation in text: [1] -
Zhang, C.; Lu, X. J. Org. Chem. 1995, 60, 2906–2908. doi:10.1021/jo00114a048
Return to citation in text: [1] -
Xu, Z.; Lu, X. J. Org. Chem. 1998, 63, 5031–5041. doi:10.1021/jo9723063
Return to citation in text: [1] -
Xu, Z.; Lu, X. Tetrahedron Lett. 1999, 40, 549–552. doi:10.1016/S0040-4039(98)02405-8
Return to citation in text: [1] -
Du, Y.; Lu, X. J. Org. Chem. 2003, 68, 6463–6465. doi:10.1021/jo034281f
Return to citation in text: [1] -
Lu, Z.; Zheng, S.; Zhang, X.; Lu, X. Org. Lett. 2008, 10, 3267–3270. doi:10.1021/ol8011452
Return to citation in text: [1] [2] -
Zheng, S.; Lu, X. Org. Lett. 2008, 10, 4481–4484. doi:10.1021/ol801661y
Return to citation in text: [1] -
Du, Y.; Lu, X.; Zhang, C. Angew. Chem., Int. Ed. 2003, 42, 1035–1037. doi:10.1002/anie.200390266
Return to citation in text: [1] -
Zhu, X.-F.; Lan, J.; Kwon, O. J. Am. Chem. Soc. 2003, 125, 4716–4717. doi:10.1021/ja0344009
Return to citation in text: [1] -
Tran, Y. S.; Kwon, O. J. Am. Chem. Soc. 2007, 129, 12632–12633. doi:10.1021/ja0752181
Return to citation in text: [1] -
Na, R.; Jing, C.; Xu, Q.; Shi, J.; Jiang, H.; Wu, X.; Shi, J.; Zhong, J.; Wang, M.; Benitez, D.; Tkatchouk, E.; Goddard, W. A., III; Guo, H.; Kwon, O. J. Am. Chem. Soc. 2011, 133, 13337–13348. doi:10.1021/ja200231v
Return to citation in text: [1] -
Zhu, G.; Chen, Z.; Jiang, Q.; Xiao, D.; Cao, P.; Zhang, X. J. Am. Chem. Soc. 1997, 119, 3836–3837. doi:10.1021/ja9644687
Return to citation in text: [1] -
Wurz, R. P.; Fu, G. C. J. Am. Chem. Soc. 2005, 127, 12234–12235. doi:10.1021/ja053277d
Return to citation in text: [1] [2] -
Wilson, J. E.; Fu, G. C. Angew. Chem., Int. Ed. 2006, 45, 1426–1429. doi:10.1002/anie.200503312
Return to citation in text: [1] -
Fujiwara, Y.; Fu, G. C. J. Am. Chem. Soc. 2011, 133, 12293–12297. doi:10.1021/ja2049012
Return to citation in text: [1] -
Mercier, E.; Fonovic, B.; Henry, C.; Kwon, O.; Dudding, T. Tetrahedron Lett. 2007, 48, 3617–3620. doi:10.1016/j.tetlet.2007.03.030
Return to citation in text: [1] -
Voituriez, A.; Panossian, A.; Fleury-Brégeot, N.; Retailleau, P.; Marinetti, A. J. Am. Chem. Soc. 2008, 130, 14030–14031. doi:10.1021/ja806060a
Return to citation in text: [1] -
Voituriez, A.; Panossian, A.; Fleury-Brégeot, N.; Retailleau, P.; Marinetti, A. Adv. Synth. Catal. 2009, 351, 1968–1976. doi:10.1002/adsc.200900193
Return to citation in text: [1] -
Voituriez, A.; Pinto, N.; Neel, M.; Retailleau, P.; Marinetti, A. Chem.–Eur. J. 2010, 16, 12541–12544. doi:10.1002/chem.201001791
Return to citation in text: [1] -
Han, X.; Wang, Y.; Zhong, F.; Lu, Y. J. Am. Chem. Soc. 2011, 133, 1726–1729. doi:10.1021/ja1106282
Return to citation in text: [1] -
Han, X.; Wang, S.-X.; Zhong, F.; Lu, Y. Synthesis 2011, 1859–1964. doi:10.1055/s-0030-1260460
Return to citation in text: [1] -
Han, X.; Zhong, F.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2012, 51, 767–770. doi:10.1002/anie.201106672
Return to citation in text: [1] -
Cowen, B. J.; Miller, S. J. J. Am. Chem. Soc. 2007, 129, 10988–10989. doi:10.1021/ja0734243
Return to citation in text: [1] -
Fang, Y.-Q.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 5660–5661. doi:10.1021/ja801344w
Return to citation in text: [1] -
Xiao, H.; Chai, Z.; Zheng, C.-W.; Yang, Y.-Q.; Liu, W.; Zhang, J.-K.; Zhao, G. Angew. Chem., Int. Ed. 2010, 49, 4467–4470. doi:10.1002/anie.201000446
Return to citation in text: [1] -
Sampath, M.; Loh, T.-P. Chem. Sci. 2010, 1, 739–742. doi:10.1039/c0sc00123f
Return to citation in text: [1] -
Steurer, M.; Jensen, K. L.; Worgull, D.; Jørgensen, K. A. Chem.–Eur. J. 2012, 18, 76–79. doi:10.1002/chem.201103502
Return to citation in text: [1] -
Xiao, H.; Chai, Z.; Wang, H.-F.; Wang, X.-W.; Cao, D.-D.; Liu, W.; Lu, Y.-P.; Yang, Y.-Q.; Zhao, G. Chem.–Eur. J. 2011, 17, 10562–10565. doi:10.1002/chem.201100850
Return to citation in text: [1] -
Du, Y.; Feng, J.; Lu, X. Org. Lett. 2005, 7, 1987–1989. doi:10.1021/ol050443d
Return to citation in text: [1] -
Feng, J.; Lu, X.; Kong, A.; Han, X. Tetrahedron 2007, 63, 6035–6041. doi:10.1016/j.tet.2007.02.115
Return to citation in text: [1] -
Zheng, S.; Lu, X. Tetrahedron Lett. 2009, 50, 4532–4535. doi:10.1016/j.tetlet.2009.05.085
Return to citation in text: [1] -
Zheng, S.; Lu, X. Org. Lett. 2009, 11, 3978–3981. doi:10.1021/ol901618h
Return to citation in text: [1] -
Ye, L.-W.; Sun, X.-L.; Wang, Q.-G.; Tang, Y. Angew. Chem., Int. Ed. 2007, 46, 5951–5954. doi:10.1002/anie.200701460
Return to citation in text: [1] -
Han, X.; Ye, L.-W.; Sun, X.-L.; Tang, Y. J. Org. Chem. 2009, 74, 3394–3397. doi:10.1021/jo9001917
Return to citation in text: [1] -
Zhou, R.; Wang, J.; Song, H.; He, Z. Org. Lett. 2011, 13, 580–583. doi:10.1021/ol102738b
Return to citation in text: [1] -
Chen, Z.; Zhang, J. Chem.–Asian J. 2010, 5, 1542–1545. doi:10.1002/asia.201000193
Return to citation in text: [1] -
Xie, P.; Huang, Y.; Chen, R. Org. Lett. 2010, 12, 3768–3771. doi:10.1021/ol101611v
Return to citation in text: [1] -
Tian, J.; Zhou, R.; Sun, H.; Song, H.; He, Z. J. Org. Chem. 2011, 76, 2374–2378. doi:10.1021/jo200164v
Return to citation in text: [1] -
Wang, Q.-G.; Zhu, S.-F.; Ye, L.-W.; Zhou, C.-Y.; Sun, X.-L.; Tang, Y.; Zhou, Q.-L. Adv. Synth. Catal. 2010, 352, 1914–1919. doi:10.1002/adsc.201000129
Return to citation in text: [1] -
Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w
Return to citation in text: [1] -
Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2012, 354, 783–789. doi:10.1002/adsc.201101012
Return to citation in text: [1] [2] -
Shi, Y.-L.; Shi, M. Adv. Synth. Catal. 2007, 349, 2129–2135. doi:10.1002/adsc.200700155
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Eur. J. Org. Chem. 2011, 1956–1960. doi:10.1002/ejoc.201001660
Return to citation in text: [1] -
Yang, Y.-L.; Pei, C.-K.; Shi, M. Org. Biomol. Chem. 2011, 9, 3349–3358. doi:10.1039/c1ob00017a
Return to citation in text: [1] -
Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f
Return to citation in text: [1] -
Ito, M.; Osaku, A.; Kobayashi, C.; Shiibashi, A.; Ikariya, T. Organometallics 2009, 28, 390–393. doi:10.1021/om801042b
Return to citation in text: [1] -
The crystal data of 3r have been deposited in the CCDC with number 863228 (Supporting Information File 2).
Return to citation in text: [1] -
Jiang, Y.-Q.; Shi, Y.-L.; Shi, M. J. Am. Chem. Soc. 2008, 130, 7202–7203. doi:10.1021/ja802422d
Return to citation in text: [1] -
Plietker, B.; Niggemann, M. Org. Lett. 2003, 5, 3353–3356. doi:10.1021/ol035335a
Return to citation in text: [1]
| 15. | Lu, Z.; Zheng, S.; Zhang, X.; Lu, X. Org. Lett. 2008, 10, 3267–3270. doi:10.1021/ol8011452 |
| 51. | Deng, H.-P.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2012, 354, 783–789. doi:10.1002/adsc.201101012 |
| 58. | The crystal data of 3r have been deposited in the CCDC with number 863228 (Supporting Information File 2). |
| 59. | Jiang, Y.-Q.; Shi, Y.-L.; Shi, M. J. Am. Chem. Soc. 2008, 130, 7202–7203. doi:10.1021/ja802422d |
| 1. | Hartley, R. C.; Caldwell, S. T. J. Chem. Soc., Perkin Trans. 1 2000, 477–501. doi:10.1039/A804421J |
| 2. | Wu, H.; Zhang, H.; Zhao, G. Tetrahedron 2007, 63, 6454–6461. doi:10.1016/j.tet.2007.03.031 |
| 22. | Wurz, R. P.; Fu, G. C. J. Am. Chem. Soc. 2005, 127, 12234–12235. doi:10.1021/ja053277d |
| 23. | Wilson, J. E.; Fu, G. C. Angew. Chem., Int. Ed. 2006, 45, 1426–1429. doi:10.1002/anie.200503312 |
| 24. | Fujiwara, Y.; Fu, G. C. J. Am. Chem. Soc. 2011, 133, 12293–12297. doi:10.1021/ja2049012 |
| 52. | Shi, Y.-L.; Shi, M. Adv. Synth. Catal. 2007, 349, 2129–2135. doi:10.1002/adsc.200700155 |
| 53. | Deng, H.-P.; Wei, Y.; Shi, M. Eur. J. Org. Chem. 2011, 1956–1960. doi:10.1002/ejoc.201001660 |
| 54. | Yang, Y.-L.; Pei, C.-K.; Shi, M. Org. Biomol. Chem. 2011, 9, 3349–3358. doi:10.1039/c1ob00017a |
| 55. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 56. | Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f |
| 21. | Zhu, G.; Chen, Z.; Jiang, Q.; Xiao, D.; Cao, P.; Zhang, X. J. Am. Chem. Soc. 1997, 119, 3836–3837. doi:10.1021/ja9644687 |
| 57. | Ito, M.; Osaku, A.; Kobayashi, C.; Shiibashi, A.; Ikariya, T. Organometallics 2009, 28, 390–393. doi:10.1021/om801042b |
| 11. | Zhang, C.; Lu, X. J. Org. Chem. 1995, 60, 2906–2908. doi:10.1021/jo00114a048 |
| 12. | Xu, Z.; Lu, X. J. Org. Chem. 1998, 63, 5031–5041. doi:10.1021/jo9723063 |
| 13. | Xu, Z.; Lu, X. Tetrahedron Lett. 1999, 40, 549–552. doi:10.1016/S0040-4039(98)02405-8 |
| 14. | Du, Y.; Lu, X. J. Org. Chem. 2003, 68, 6463–6465. doi:10.1021/jo034281f |
| 15. | Lu, Z.; Zheng, S.; Zhang, X.; Lu, X. Org. Lett. 2008, 10, 3267–3270. doi:10.1021/ol8011452 |
| 16. | Zheng, S.; Lu, X. Org. Lett. 2008, 10, 4481–4484. doi:10.1021/ol801661y |
| 17. | Du, Y.; Lu, X.; Zhang, C. Angew. Chem., Int. Ed. 2003, 42, 1035–1037. doi:10.1002/anie.200390266 |
| 18. | Zhu, X.-F.; Lan, J.; Kwon, O. J. Am. Chem. Soc. 2003, 125, 4716–4717. doi:10.1021/ja0344009 |
| 19. | Tran, Y. S.; Kwon, O. J. Am. Chem. Soc. 2007, 129, 12632–12633. doi:10.1021/ja0752181 |
| 20. | Na, R.; Jing, C.; Xu, Q.; Shi, J.; Jiang, H.; Wu, X.; Shi, J.; Zhong, J.; Wang, M.; Benitez, D.; Tkatchouk, E.; Goddard, W. A., III; Guo, H.; Kwon, O. J. Am. Chem. Soc. 2011, 133, 13337–13348. doi:10.1021/ja200231v |
| 50. | Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094 |
| 3. | Lu, X.; Zhang, C.; Xu, Z. Acc. Chem. Res. 2001, 34, 535–544. doi:10.1021/ar000253x |
| 4. | Methot, J. L.; Roush, W. R. Adv. Synth. Catal. 2004, 346, 1035–1050. doi:10.1002/adsc.200404087 |
| 5. | Ye, L.-W.; Zhou, J.; Tang, Y. Chem. Soc. Rev. 2008, 37, 1140–1152. doi:10.1039/b717758e |
| 6. | Cowen, B. J.; Miller, S. J. Chem. Soc. Rev. 2009, 38, 3102–3116. doi:10.1039/b816700c |
| 7. | Marinetti, A.; Voituriez, A. Synlett 2010, 174–194. doi:10.1055/s-0029-1219157 |
| 8. | Wei, Y.; Shi, M. Acc. Chem. Res. 2010, 43, 1005–1018. doi:10.1021/ar900271g |
| 9. | López, F.; Mascareñas, J. L. Chem.–Eur. J. 2011, 17, 418–428. doi:10.1002/chem.201002366 |
| 10. | Zhao, Q.-Y.; Lian, Z.; Wei, Y.; Shi, M. Chem. Commun. 2012, 48, 1724–1732. doi:10.1039/c1cc15793k |
| 51. | Deng, H.-P.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2012, 354, 783–789. doi:10.1002/adsc.201101012 |
| 37. | Xiao, H.; Chai, Z.; Wang, H.-F.; Wang, X.-W.; Cao, D.-D.; Liu, W.; Lu, Y.-P.; Yang, Y.-Q.; Zhao, G. Chem.–Eur. J. 2011, 17, 10562–10565. doi:10.1002/chem.201100850 |
| 48. | Wang, Q.-G.; Zhu, S.-F.; Ye, L.-W.; Zhou, C.-Y.; Sun, X.-L.; Tang, Y.; Zhou, Q.-L. Adv. Synth. Catal. 2010, 352, 1914–1919. doi:10.1002/adsc.201000129 |
| 32. | Cowen, B. J.; Miller, S. J. J. Am. Chem. Soc. 2007, 129, 10988–10989. doi:10.1021/ja0734243 |
| 33. | Fang, Y.-Q.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 5660–5661. doi:10.1021/ja801344w |
| 34. | Xiao, H.; Chai, Z.; Zheng, C.-W.; Yang, Y.-Q.; Liu, W.; Zhang, J.-K.; Zhao, G. Angew. Chem., Int. Ed. 2010, 49, 4467–4470. doi:10.1002/anie.201000446 |
| 35. | Sampath, M.; Loh, T.-P. Chem. Sci. 2010, 1, 739–742. doi:10.1039/c0sc00123f |
| 36. | Steurer, M.; Jensen, K. L.; Worgull, D.; Jørgensen, K. A. Chem.–Eur. J. 2012, 18, 76–79. doi:10.1002/chem.201103502 |
| 49. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w |
| 29. | Han, X.; Wang, Y.; Zhong, F.; Lu, Y. J. Am. Chem. Soc. 2011, 133, 1726–1729. doi:10.1021/ja1106282 |
| 30. | Han, X.; Wang, S.-X.; Zhong, F.; Lu, Y. Synthesis 2011, 1859–1964. doi:10.1055/s-0030-1260460 |
| 31. | Han, X.; Zhong, F.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2012, 51, 767–770. doi:10.1002/anie.201106672 |
| 22. | Wurz, R. P.; Fu, G. C. J. Am. Chem. Soc. 2005, 127, 12234–12235. doi:10.1021/ja053277d |
| 60. | Plietker, B.; Niggemann, M. Org. Lett. 2003, 5, 3353–3356. doi:10.1021/ol035335a |
| 25. | Mercier, E.; Fonovic, B.; Henry, C.; Kwon, O.; Dudding, T. Tetrahedron Lett. 2007, 48, 3617–3620. doi:10.1016/j.tetlet.2007.03.030 |
| 26. | Voituriez, A.; Panossian, A.; Fleury-Brégeot, N.; Retailleau, P.; Marinetti, A. J. Am. Chem. Soc. 2008, 130, 14030–14031. doi:10.1021/ja806060a |
| 27. | Voituriez, A.; Panossian, A.; Fleury-Brégeot, N.; Retailleau, P.; Marinetti, A. Adv. Synth. Catal. 2009, 351, 1968–1976. doi:10.1002/adsc.200900193 |
| 28. | Voituriez, A.; Pinto, N.; Neel, M.; Retailleau, P.; Marinetti, A. Chem.–Eur. J. 2010, 16, 12541–12544. doi:10.1002/chem.201001791 |
| 38. | Du, Y.; Feng, J.; Lu, X. Org. Lett. 2005, 7, 1987–1989. doi:10.1021/ol050443d |
| 39. | Feng, J.; Lu, X.; Kong, A.; Han, X. Tetrahedron 2007, 63, 6035–6041. doi:10.1016/j.tet.2007.02.115 |
| 40. | Zheng, S.; Lu, X. Tetrahedron Lett. 2009, 50, 4532–4535. doi:10.1016/j.tetlet.2009.05.085 |
| 41. | Zheng, S.; Lu, X. Org. Lett. 2009, 11, 3978–3981. doi:10.1021/ol901618h |
| 42. | Ye, L.-W.; Sun, X.-L.; Wang, Q.-G.; Tang, Y. Angew. Chem., Int. Ed. 2007, 46, 5951–5954. doi:10.1002/anie.200701460 |
| 43. | Han, X.; Ye, L.-W.; Sun, X.-L.; Tang, Y. J. Org. Chem. 2009, 74, 3394–3397. doi:10.1021/jo9001917 |
| 44. | Zhou, R.; Wang, J.; Song, H.; He, Z. Org. Lett. 2011, 13, 580–583. doi:10.1021/ol102738b |
| 45. | Chen, Z.; Zhang, J. Chem.–Asian J. 2010, 5, 1542–1545. doi:10.1002/asia.201000193 |
| 46. | Xie, P.; Huang, Y.; Chen, R. Org. Lett. 2010, 12, 3768–3771. doi:10.1021/ol101611v |
| 47. | Tian, J.; Zhou, R.; Sun, H.; Song, H.; He, Z. J. Org. Chem. 2011, 76, 2374–2378. doi:10.1021/jo200164v |
© 2012 Deng et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)