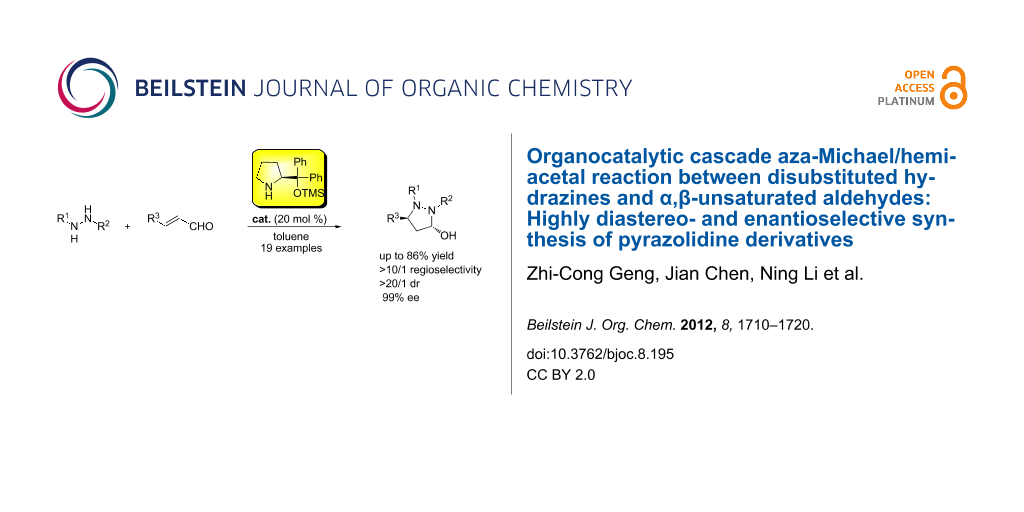
Abstract
The catalytic synthesis of nitrogen-containing heterocycles is of great importance to medicinal and synthetic chemists, and also a challenge for modern chemical methodology. In this paper, we report the synthesis of pyrazolidine derivatives through a domino aza-Michael/hemiacetal sequence with chiral or achiral secondary amines as organocatalysts. Thus, a series of achiral pyrazolidine derivatives were obtained with good yields (up to 90%) and high diastereoselectivities (>20:1) with pyrrolidine as an organocatalyst, and enantioenriched pyrazolidines are also achieved with good results (up to 86% yield, >10/1 regioselectivity, >20:1 dr, 99% ee) in the presence of (S)-diphenylprolinol trimethylsilyl ether catalyst.

Graphical Abstract
Introduction
Pyrazolidines are privileged and valuable heterocyclic compounds, which are of great importance in biological and medicinal chemistry (Figure 1) [1-5]. Besides, pyrazolidines are also important synthetic intermediates in organic chemistry. For instance, the N–N bond of pyrazolidines can be cleaved under reductive conditions to afford useful 1,3-diamines [6,7], and moreover, pyrazolidines can also be oxidized to afford pyrazolines [8-12] and pyrazoles [13-15]. The pyrazolidine structural unit is commonly constructed by [3 + 2] cycloaddition reactions using hydrazones [16-23] or azomethine amines [24-27] as dipoles. Recently, the Ma group and Toste et al. have reported efficient methods for the synthesis of pyrazolidine derivatives by metal-catalyzed aminations of allenes [28-32]. Meanwhile, Lewis acid catalyzed carboamination reactions have also been reported as efficient methods for the synthesis of pyrazolidine derivatives by Wolfe et al. [33].
![[1860-5397-8-195-1]](/bjoc/content/figures/1860-5397-8-195-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Important heterocycles containing pyrazolidine or pyrazoline structures.
Figure 1: Important heterocycles containing pyrazolidine or pyrazoline structures.
In the past decade, the research area of organocatalysis has grown rapidly and become a third brand of catalysis besides the well-established biocatalysis and metal catalysis [34-44]. Particularly, organocatalytic domino/cascade reactions have come into focus and become a powerful synthetic approach that allows the construction of structurally diverse and complex molecules, minimizes the number of manual operations, and saves time, effort, and production costs [45-47]. Thus, many nitrogen-containing heterocyclic compounds have been efficiently generated by means of organocatalytic domino reactions [48-69].
In 2009 and 2010, List et al. and the Brière group both reported, separately, the enantioselective synthesis of 2-pyrazolines starting from α,β-unsaturated ketones and phenylhydrazine or N-tert-butyloxycarbonylhydrazine in the presence of a chiral Brønsted acid or a phase-transfer catalyst [70,71]. Compared with monosubstituted hydrazines in organocatalytic asymmetric synthesis, disubstituted hydrazines were also explored by several groups [72,73]. In 2007, Jørgensen et al. reported that the organocatalyzed asymmetric aza-Michael addition of hydrazones to cyclic enones had been achieved in good yield and stereoselectivity [74]. In 2011, the Deng group developed a highly enantioselective organocatalytic synthesis of 2-pyrazolines using disubstituted hydrazines through an asymmetric conjugate addition followed by a deprotection–cyclization sequence [75]. Due to the importance of pyrazolidine derivatives in both organic and medicinal chemistry, we have become interested in developing an efficient stereoselective cascade reaction for the synthesis of the pyrazolidine compounds through organocatalysis. In this paper, we present a convenient access to racemic and enantioenriched 5-hydroxypyrazolidines through a domino aza-Michael/hemiacetal organocatalytic sequence of disubstituted hydrazines to α,β-unsaturated aldehydes. While proceeding with the submission of our results, we noticed that a related excellent work has been reported by Vicario and co-workers [76]. When comparing Vicario’s work with this manuscript, both works are complementary in scope since Vicario’s work makes use of unsaturated aldehydes containing only linear alkyl chains, whereas our work provides better results when unsaturated aldehydes bearing an aromatic moiety are employed.
Results and Discussion
First, the cascade aza-Michael/hemiacetal reactions between disubstituted hydrazines 2a–c and 4-nitrocinnamaldehyde (3a) were investigated in the presence of several common secondary amines 1a–f as organocatalysts in chloroform. Pyrrolidine (1c) turned out to be an effective catalyst and di-tert-butyl hydrazine-1,2-dicarboxylate (2c) was a potent donor (see Table S1 in the Supporting Information File 1). When di-tert-butyl hydrazine-1,2-dicarboxylate (2c) was used as the donor and 20 mol % pyrrolidine (1c) as the catalyst, 5-hydroxypyrazolidine 4a could be obtained in 68% yield with over 20:1 dr after five days (Table 1, entry 1). Thus, the tandem aza-Michael/hemiacetal reaction between di-tert-butyl hydrazine-1,2-dicarboxylate (2c) and 4-nitrocinnamaldehyde (3a) was chosen as the model reaction to further optimize the reaction conditions. The catalytic results were summarized in Table 1. In order to improve the yield, a variety of common solvents were screened (Table 1, entries 2–6). Dichloromethane was finally found to be the best medium for this reaction (74% yield with excellent diastereoselectivity was achieved, Table 1, entry 2). Subsequently, the effects of some basic/and acidic additives were examined. Inorganic bases seemed to be ineffective for the further improvement of the yields (Table 1, entries 7–10). Soluble organic bases were also tested and failed to increase the yields (Table 1, entries 11 and 12). When benzoic acid (5a) was used as an additive, the yield was slightly improved to 77% (Table 1, entry 13). With the increase of the acidity, the yield was noticeably decreased (Table 1, entry 13 versus entries 14 and 15). Finally, when increasing the amount of di-tert-butyl hydrazine-1,2-dicarboxylate (2c) to 2 equiv, the desired product 4a was obtained in 81% yield without any additive (Table 1, entry 16).
Table 1: Secondary amine catalyzed cascade aza-Michael/hemiacetal reaction.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-195-i1.svg?max-width=637&scale=1.0)
|
||||
| Entrya | Solvent | Additive | drb | Yield (%)c |
|---|---|---|---|---|
| 1 | CHCl3 | – | >20:1 | 68 |
| 2 | CH2Cl2 | – | >20:1 | 74 |
| 3 | MeOH | – | – | 48 |
| 4 | DMF | – | – | 51 |
| 5 | PhMe | – | – | 42 |
| 6 | THF | – | – | 37 |
| 7 | CH2Cl2 | NaOAc | >20:1 | 68 |
| 8 | CH2Cl2 | NaHCO3 | >20:1 | 63 |
| 9 | CH2Cl2 | Na2CO3 | – | 38 |
| 10 | CH2Cl2 | LiOAc | >20:1 | 56 |
| 11 | CH2Cl2 | Et3N | >20:1 | 42 |
| 12 | CH2Cl2 | DMAP | >20:1 | 50 |
| 13 | CH2Cl2 | benzoic acid (5a) | >20:1 | 77 |
| 14 | CH2Cl2 | 4-nitrobenzoic acid (5b) | >20:1 | 67 |
| 15 | CH2Cl2 | 3,5-dinitrobenzoic acid (5c) | – | 56 |
| 16d | CH2Cl2 | – | >20:1 | 81 |
aThe reaction was run with 2c (0.3 mmol), 3a (0.25 mmol), 1c (0.05 mmol) and the specified additive (0.05 mmol) in the given solvent (0.5 mL) at room temperature for 5 d. bDetermined by 1H NMR. cIsolated yield. dThe molar ratio of 2c/3a is 2.0:1.
Having established the optimized reaction conditions, we investigated the scope of substrates for the cascade aza-Michael/hemiacetal reaction with pyrrolidine (1c, 20 mol %) as the catalyst, without any additives, in methylene chloride (Table 2). We found that the nature of the substituents on the phenyl group of α,β-unsaturated aldehydes dramatically affected the reactivity. For example, with disubstituted hydrazine 2c as the donor, the presence of a stronger electron-deficient substituent (-NO2, -CN) on the phenyl ring of the α,β-unsaturated aldehydes 3a, 3b and 3c promoted the cascade aza-Michael/hemiacetal reaction readily to provide the desired products in good yields (75–81%, Table 2, entries 1–3). The presence of less-electron-deficient substituents (-Cl and -Br) on the phenyl ring of the α,β-unsaturated aldehydes, such as 4-chloro- or 4-bromocinnamaldehyde derivatives 3d and 3e, afforded the corresponding products in moderate yields (64 and 74%) even when 5 equiv of disubstituted hydrazine 2c was used (Table 2, entries 4 and 5). When cinnamaldehyde derivatives 3f and 3g, bearing electron-donating substituents (-Me, -OMe) on phenyl rings, were used as the substrate, the reactions became very sluggish (Table 2, entries 6 and 7). On the other hand, more symmetric and asymmetric hydrazine derivatives 2d–h were synthesized and investigated for the tandem aza-Michael/hemiacetal reaction. Generally, all the reactions between 2d–h and α,β-unsaturated aldehydes 3a and 3b proceeded smoothly with sequential catalytic actions of 1c, affording the corresponding desired products 4h–o in moderate to good yields (Table 2, entries 8–11 and 13–16). Notably, the reaction between 2e and 4-methoxycinnamaldehyde (3f) afforded the desired product in 52% yield (Table 2, entry 12). For the asymmetric disubstituted hydrazines 2g and 2h as substrates, regioselective results could be observed (Table 2, entry 14). However, with increasing catalyst loading from catalytic to stoichiometric amounts, the corresponding cascade reactions could provide the major products in 72 and 66% yields (Table 2, entries 15 and 16). In all the reactions, high diastereoselectivity could be obtained (>20:1 dr). Finally, we were able to obtain single crystals of compounds 4a and 4n, which allowed for an unambiguous assignment of the trans configuration of C3 and C5 by X-ray crystallographic analysis (Figure 2 and Figure 3).
Table 2: Substrate scope for the cascade aza-Michael/hemiacetal reactions between 2 and α,β-unsaturated aldehydes 3.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-195-i2.svg?max-width=637&scale=1.0)
|
||||
| Entrya | Donor | R3 | drb | Yield (%)c |
|---|---|---|---|---|
| 1 | 2c | 4-NO2C6H4 (3a)d | >20:1 | 4a/81 |
| 2 | 2c | 3-NO2C6H4 (3b)d | >20:1 | 4b/78 |
| 3 | 2c | 4-CNC6H4 (3c)d | >20:1 | 4c/75 |
| 4 | 2c | 4-ClC6H4 (3d)e | >20:1 | 4d/64 |
| 5 | 2c | 4-BrC6H4 (3e)e | >20:1 | 4e/74 |
| 6 | 2c | 4-MeOC6H4 (3f)d | – | 4f/<5 |
| 7 | 2c | 4-MeC6H4 (3g)d | – | 4g/<5 |
| 8 | 2d | 4-NO2C6H4 (3a)d | >20:1 | 4h/86 |
| 9 | 2d | 3-NO2C6H4 (3b)d | >20:1 | 4i/82 |
| 10 | 2e | 4-NO2C6H4 (3a)d | >20:1 | 4j/90 |
| 11 | 2e | 3-NO2C6H4 (3b)d | >20:1 | 4k/76 |
| 12 | 2e | 4-MeOC6H4 (3f)d | >20:1 | 4l/52 |
| 13 | 2f | 4-NO2C6H4 (3a)d | >20:1 | 4m/77 |
| 14 | 2g | 4-NO2C6H4 (3a)f | >20:1 | 4n/20 (43) |
| 15 | 2g | 4-NO2C6H4 (3a)f,g | >20:1 | 4n/72 (<5) |
| 16 | 2h | 3-NO2C6H4 (3b)f,g | >20:1 | 4o/66 (<5) |
aReaction was conducted on 0.25 mmol scale in solvents (0.5 mL) at room temperature for five days. bDetermined by 1H NMR. cIsolated yield (the data in parentheses is related to the isolated yield of the regioselective product). dThe ratio of 2/3 is 2.0:1. eThe ratio of 2/3 is 5.0:1. fThe ratio of 2/3 is 1.2:1. g100 mol % of pyrrolidine was used at rt for 12 h.
![[1860-5397-8-195-2]](/bjoc/content/figures/1860-5397-8-195-2.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of racemic 4a (25% thermal ellipsoids).
Figure 2: X-ray crystal structure of racemic 4a (25% thermal ellipsoids).
![[1860-5397-8-195-3]](/bjoc/content/figures/1860-5397-8-195-3.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of racemic 4n (25% thermal ellipsoids).
Figure 3: X-ray crystal structure of racemic 4n (25% thermal ellipsoids).
Then, we turned our attention to the development of the asymmetric version of the cascade aza-Michael/hemiacetal reaction. Initially, a series of readily available chiral organocatalysts 1g–o were chosen and investigated for the domino aza- Michael/hemiacetal reaction of disubstituted hydrazine 2c and 4-nitrocinnamaldehyde (3a) under catalytic loading of 20 mol % in CH2Cl2 at room temperature. The screening results are summarized in Table 3. (S)-Proline derivatives 1g–h, 1k, 1l were found to be ineffective for the reaction, because they afford only trace products after one day (Table 3, entries 1, 2, 5 and 6). Although a moderate yield was obtained with organocatalyst 1i bearing a sulfone functional group, the stereochemical induction was very poor (Table 3, entry 3). MacMillan’s catalyst 1j was proven to be inefficient for this transformation as only 13% ee was obtained (Table 3, entry 4). Subsequently, three diarylprolinol silyl ether catalysts 1m–o were investigated for this tandem reaction [77-79]. Gratifyingly, 82% ee was achieved when 1m was used as the catalyst. For catalysts 1n and 1o, slightly higher yields could be obtained, but the enantioselectivities became lower (Table 3, entry 7 versus entries 8 and 9). Relatively speaking, (S)-diphenylprolinol trimethylsilyl ether 1m turned out to be the optimal catalyst in terms of both enantioselectivity and reactivity.
Table 3: Chiral-amine-catalyzed cascade aza-Michael/hemiacetal reaction of 2c with 3a.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-195-i3.svg?max-width=637&scale=1.0)
|
||||
| Entrya | R | Time (d) | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 1g | 1 | <10 | n.d. |
| 2 | 1h | 1 | <5 | n.d. |
| 3 | 1i | 1 | 48 | 5 |
| 4 | 1j | 1 | 21 | 13 |
| 5 | 1k | 1 | <10 | n.d. |
| 6 | 1l | 1 | <5 | n.d. |
| 7 | 1m | 1 | 86 | 82 |
| 8 | 1n | 1 | 89 | (−)79d |
| 9 | 1o | 1 | 87 | 76 |
aThe reaction was run with 2c (0.5 mmol), 3a (0.25 mmol), and 1 (0.05 mmol) in CH2Cl2 (0.5 mL) at room temperature. bIsolated yield. cDetermined by HPLC analysis on a chiral stationary phase (Chiralcel OD-H), >20:1 dr. dOpposite enantiomer of the product formed. n.d. = not determined.
Having identified the readily available catalyst 1m as the optimal catalyst for the tandem aza-Michael/hemiacetal reaction of 2c and 3a, we summarize the results for the optimization of the other reaction parameters, including reaction solvents and additives, in Table 4. When the reaction was carried out in a protonic solvent, i.e., methanol, at room temperature for two days, the desired product was furnished in 56% yield with only 22% ee (Table 4, entry 1). After screening several aprotic solvents for this reaction, we were pleased to find that the enantioselectivity of the desired product was improved to 92% ee with toluene or THF as solvent after two days (Table 4, entries 4 and 5). Considering both yield and enantioselectivity, toluene was the optimal reaction medium (Table 4, entry 5). When the time was prolonged to four days, the yield of the product was increased to 80% and the enantioselectivity was retained (Table 4, entry 6). Thereafter, several Brønsted acids 5b, 5d–j were tested as additives for this transformation. Although enantioselectivity was somewhat improved from 92 to 95% ee, the reactivity dramatically decreased as evidenced by the prolonged reaction time and lower yields (Table 4, entry 6 versus entries 7–14). It seemed that the present catalytic system could be inhibited by acidic additives. Therefore, we considered whether the reaction could be accelerated by basic additives without loss of enantioselectivity and reactivity. Subsequently, several common inorganic and organic bases were investigated [80-83]. Unfortunately, the catalytic results showed that with LiOAc, DMAP, DABCO, Et3N, TMEDA as additives, the yield and enantioselectivity were only marginally influenced (Table 4, entries 15–19). When DBU was used as an additive, only 11% ee was obtained with moderate yield (Table 4, entry 20). Thus, 1m (20 mol %) as the catalyst and toluene as the reaction medium without any additive at room temperature proved to be the optimal reaction conditions for the asymmetric cascade aza-Michael/hemiacetal reaction.
Table 4: Optimization of the reaction of 2c and 3a catalyzed by chiral amine 1m.
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-195-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | Additive | Solvent | Time (d) | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | – | MeOH | 2 | 56 | 22 |
| 2 | – | CHCl3 | 1 | 84 | 75 |
| 3 | – | CHCl2 | 1 | 86 | 82 |
| 4 | – | THF | 2 | 51 | 92 |
| 5 | – | PhMe | 2 | 72 | 92 |
| 6 | – | PhMe | 4 | 80 | 92 |
| 7 | 5b | PhMe | 4 | 27 | 93 |
| 8 | 5d | PhMe | 4 | 56 | 94 |
| 9 | 5e | PhMe | 4 | 48 | 92 |
| 10 | 5f | PhMe | 4 | 45 | 92 |
| 11 | 5g | PhMe | 4 | 51 | 94 |
| 12 | 5h | PhMe | 4 | 20 | 94 |
| 13 | 5i | PhMe | 4 | 38 | 94 |
| 14 | 5j | PhMe | 4 | 53 | 95 |
| 15 | LiOAc | PhMe | 4 | 79 | 93 |
| 16 | DMAP | PhMe | 4 | 83 | 87 |
| 17 | DABCO | PhMe | 4 | 79 | 89 |
| 18 | Et3N | PhMe | 4 | 83 | 87 |
| 19 | TMEDA | PhMe | 4 | 81 | 88 |
| 20 | DBU | PhMe | 4 | 58 | 11 |
aThe reaction was run with 2c (0.5 mmol), 3a (0.25 mmol), 1m (0.05 mmol) and the specified additive (0.05 mmol) in the given solvent (0.5 mL) at room temperature. bIsolated yield. cDetermined by HPLC analysis on a chiral stationary phase (Chiralcel OD-H), >20:1 dr.
With the optimized reaction conditions in hand, the substrate scope of the organocatalyzed asymmetric domino aza-Michael/hemiacetal sequence was subsequently explored. Firstly, with symmetric di-tert-butyl hydrazine-1,2-dicarboxylate (2c) as nucleophilic reagent, aromatic α,β-unsaturated aldehydes 3a–g were examined to study the effects of electronic properties and steric hindrance on both enantioselectivity and reactivity. For the substrates 3a, 3b and 3c, bearing substituents of -NO2 and -CN at the para- or meta-position of the phenyl group, the reactions proceeded smoothly and led to the desired products 4a, 4b and 4c in 80–86% yields with 89–92% ee’s (Table 5, entries 1–3). With 3d and 3e bearing -Cl or -Br substituents at the para-position of the phenyl group as substrates, the desired products 4d and 4e were obtained in 61 and 62% yields with 74 and 77% ee, respectively (Table 5, entries 4 and 5). For substrates 3f and 3g bearing electron-donating groups (-Me, -OMe) on the phenyl rings, only a trace amount of the desired products could be observed under otherwise identical reaction conditions (Table 5, entries 6 and 7). These experimental results indicated that chemical yields and enantioselectivities were dramatically affected by the electronic properties and steric hindrance of the aryl group on the α,β-unsaturated aldehydes. High yield and good enantioselectivity could be obtained with strong electron-withdrawing substituents on the phenyl ring of cinnamaldehydes. When diisopropyl hydrazine-1,2-dicarboxylate (2d) as nucleophilic reagent reacted with 4-nitro cinnamaldehyde (3a), the product 4h was obtained in 60% yield and 72% ee (Table 5, entry 8). The result showed that the small-sized substituent on hydrazines was unfavorable on the reaction (Table 5, entry 8 versus entry 1). Subsequently, asymmetric disubstituted hydrazines 2g–j were investigated for the domino aza-Michael/hemiacetal sequence. Due to nucleophilic competition of the two nitrogens in the asymmetric disubstituted hydrazines, regioselective results were observed for these reactions. For asymmetric disubstituted hydrazines 2g, the reaction gave the 1.8:1 molar ratio of the regioselective products 4n to 4n’. The major product 4n was obtained in 58% yield and 88% ee. The enantioselectivity of the minor product 4n’ was 55% (Table 5, entry 9). For asymmetric disubstituted hydrazines 2h–i, the molar ratios of the regioselective products ranged from 3.2:1 to 6.4:1. The major products were obtained in moderate to good yields and good enantioselectivities (Table 5, entries 10–15). When disubstituted hydrazine 2j with an electron-donating group on the aromatic ring was used as the nucleophilic donor, the major, reversely regioselective product 4u’ was obtained in 75% yield, but with very low enantioselectivity (11% ee, Table 5, entry 16). To our delight, (E)-but-2-enal (3i) was a suitable substrate for this transformation. The reactions of asymmetric disubstituted hydrazines 2h and 2i with (E)-but-2-enal (3i) proceeded smoothly and provided the desired products in 78 and 83% yields with 72 and 74% ee, respectively (Table 5, entries 17 and 18). However, when the cascade aza-Michael/hemiacetal reaction of (E)-pent-2-enal (3j) and 2h was carried out, only a trace amount of the expected products could be detected (Table 5, entry 19). Fortunately, single crystals of compound 4s were obtained by recrystallization from petroleum ether/acetyl acetate, and the absolute configuration was determined by X-ray analyses (Figure 4) [84].
Table 5: Substrate scope of 2 and 3 catalyzed by chiral amine 1m.
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-195-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | Donor | R3 | Time (d) | Yield (%)b | Ratioc | ee (%)d |
|---|---|---|---|---|---|---|
| 1 | 2c | 4-NO2C6H4 (3a) | 4 | 4a/80 | – | 92 |
| 2 | 2c | 3-NO2C6H4 (3b) | 2 | 4b/83 | – | 91 |
| 3 | 2c | 4-CNC6H4 (3c) | 4 | 4c/86 | – | 89 |
| 4 | 2c | 4-ClC6H4 (3d) | 4 | 4d/61 | – | 74 |
| 5 | 2c | 4-BrC6H4 (3e) | 4 | 4e/62 | – | 77 |
| 6 | 2c | 4-MeOC6H4 (3f) | 4 | 4f/<5 | – | – |
| 7 | 2c | 4-MeC6H4 (3g) | 4 | 4g/<5 | – | – |
| 8 | 2d | 4-NO2C6H4 (3a) | 2 | 4h/60 | – | 72 |
| 9 | 2ge | 4-NO2C6H4 (3a) | 4 | 4n/58 (4n’/32) | 1.8:1 | 88/55 |
| 10 | 2he | 3-NO2C6H4 (3b) | 4 | 4o/60 | 3.2:1 | 88 |
| 11 | 2he | 4-NO2C6H4 (3a) | 4 | 4p/72 | 4.5:1 | 99 |
| 12 | 2he | 4-CNC6H4 (3c) | 4 | 4q/65 | 4.7:1 | 93 |
| 13 | 2he | 3-CF3C6H4 (3h) | 4 | 4r/53 | –g | 81 |
| 14 | 2ie | 4-NO2C6H4 (3a) | 4 | 4s/74 | 6.4:1 | 99 |
| 15 | 2ie | 4-CNC6H4 (3c) | 4 | 4t/72 | 4.0:1 | 90 |
| 16 | 2je | 4-NO2C6H4 (3a) | 4 | 4u'/75 | 1:9.0 | –/11 |
| 17 | 2hf | Me (3i) | 2 | 4v/78 | >10:1 | 72 |
| 18 | 2if | Me (3i) | 2 | 4w/83 | >10:1 | 74 |
| 19 | 2hf | Et (3j) | 3 | 4x/<10 | – | – |
aUnless noted, the reaction was run with 2 (0.5 mmol), 3 (0.25 mmol), and 1m (0.05 mmol) in toluene (0.5 mL) at room temperature. bIsolated yield of pure isomer 4 (the data in parentheses is related to the isolated yield of the 4’). cThe ratio based on isolated yield of pure 4 and 4’. dDetermined by HPLC analysis on a chiral stationary phase (Chiralcel OD-H, AD-H or AS-H), >20:1 dr. eThe ratio of 2/3 is 1.2:1. fThe reaction was run with 2 (0.25 mmol), 3 (0.38 mmol), and 1m (0.05 mmol) in toluene (0.5 mL) at room temperature. gDue to the difficulty of separation of the product 4r’ from starting material 2h.
![[1860-5397-8-195-4]](/bjoc/content/figures/1860-5397-8-195-4.png?scale=1.2&max-width=1024&background=FFFFFF)
Figure 4: The X-ray crystal structure of chiral compound 4s (40% thermal ellipsoids).
Figure 4: The X-ray crystal structure of chiral compound 4s (40% thermal ellipsoids).
Conclusion
In summary, we have developed an organocatalytic approach for the synthesis of pyrazolidine derivatives through the cascade aza-Michael/hemiacetal reaction between disubstituted hydrazines and α,β-unsaturated aldehydes. The asymmetric version of this one-pot cascade reaction has also been realized with (S)-diphenylprolinol trimethylsilyl ether 1m as a secondary amine organocatalyst, and a series of enantiomerically enriched pyrazolidine derivatives were obtained in moderate to good chemical yields with moderate to excellent enantioselectivities. The application of the products and further investigation of the reaction are ongoing in our laboratory.
Experimental
Representative experimental procedure for the cascade aza-Michael/hemiacetal reaction of disubstituted hydrazines with α,β-unsaturated aldehydes
To a stirred solution of catalyst 1c or 1m (20 mol %) in CH2Cl2 or toluene (0.5 mL) was added α,β-unsaturated aldehyde 3 (1.0 equiv, 0.25 mmol) and di-substituted hydrazine 2 (1.2 equiv or 2.0 or 5.0 equiv and 0.3 mmol or 0.5 mmol or 1.25 mmol) at rt. The reaction was vigorously stirred for 2–5 days. Then, the reaction mixture was directly subjected to flash column chromatography on silica gel (petroleum ether/ethyl acetate) to afford the corresponding products 4.
(−)-Di-tert-butyl 3-hydroxy-5-(4-nitrophenyl)pyrazolidine-1,2-dicarboxylate (4a): 80% yield, >20/1 dr, 92% ee. The enantiomeric ratio was determined by HPLC on Chiralpak OD-H column (10% 2-propanol/hexane, 1 mL/min), tmajor = 7.219 min, tminor = 6.013 min; [α]D26 −11.8 (c 0.38, acetone); 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.6 Hz, 2H), 5.92 (d, J = 4.7 Hz, 1H), 5.52–5.39 (m, 1H), 3.40 (s, 1H), 2.71 (dd, J = 13.2, 8.4 Hz, 1H), 2.13–2.03 (m, 1H), 1.53 (s, 9H), 1.43 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 154.27, 149.72, 147.21, 126.75, 124.01, 82.62, 82.56, 82.19, 61.66, 43.43, 28.33, 28.19; IR (KBr) νmax: 3354.8, 2980.4, 2934.2, 2854.2, 1728.7, 1705.9, 1600.2, 1518.9, 1456.0, 1367.7, 1345.9, 1311.6, 1243.1, 1145.0, 990.9, 853.5, 758.4 cm−1; HRMS–ESI (m/z): [Na]+ calcd for C19H27N3O7, 432.1741; found, 432.1724.
Supporting Information
| Supporting Information File 1: General experimental procedures, 1H, 13C NMR spectra and HPLC chromatograms for all new compounds, crystal data and structure refinement for enantiopure 4. | ||
| Format: PDF | Size: 2.8 MB | Download |
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21072145, 21272166), Foundation for the Author of National Excellent Doctoral Dissertation of PR China (200931), and Natural Science Foundation of Jiangsu Province of China (BK2009115). This project was also funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
-
Clark, M. P.; Laughlin, S. K.; Laufersweiler, M. J.; Bookland, R. G.; Brugel, T. A.; Golebiowski, A.; Sabat, M. P.; Townes, J. A.; VanRens, J. C.; Djung, J. F.; Natchus, M. G.; De, B.; Hsieh, L. C.; Xu, S. C.; Walter, R. L.; Mekel, M. J.; Heitmeyer, S. A.; Brown, K. K.; Juergens, K.; Taiwo, Y. O.; Janusz, M. J. J. Med. Chem. 2004, 47, 2724–2727. doi:10.1021/jm049968m
Return to citation in text: [1] -
Kornet, M. J.; Garrett, R. J. J. Pharm. Sci. 1979, 68, 377–388. doi:10.1002/jps.2600680335
Return to citation in text: [1] -
Tran, K.; Lombardi, P. J.; Leighton, J. L. Org. Lett. 2008, 10, 3165–3167. doi:10.1021/ol8011869
Return to citation in text: [1] -
Özdemir, Z.; Kandilci, H. B.; Gümüşel, B.; Çaliş, Ü.; Bilgin, A. A. Eur. J. Med. Chem. 2007, 42, 373–379. doi:10.1016/j.ejmech.2006.09.006
Return to citation in text: [1] -
Jeong, T.-S.; Kim, K. S.; An, S.-J.; Cho, K.-H.; Lee, S.; Lee, W. S. Bioorg. Med. Chem. Lett. 2004, 14, 2715–2717. doi:10.1016/j.bmcl.2004.03.079
Return to citation in text: [1] -
Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 11279–11282. doi:10.1021/ja049498l
Return to citation in text: [1] -
Shirakawa, S.; Lombardi, P. J.; Leighton, J. L. J. Am. Chem. Soc. 2005, 127, 9974–9975. doi:10.1021/ja052307
Return to citation in text: [1] -
Johnson, M.; Younglove, B.; Lee, L.; LeBlanc, R.; Holt, H., Jr.; Hills, P.; Mackay, H.; Brown, T.; Mooberry, S. L.; Lee, M. Bioorg. Med. Chem. Lett. 2007, 17, 5897–5901. doi:10.1016/j.bmcl.2007.07.105
Return to citation in text: [1] -
Ali, M. A.; Shaharyar, M. Bioorg. Med. Chem. 2007, 15, 1896–1902. doi:10.1016/j.bmc.2007.01.006
Return to citation in text: [1] -
Lange, J. H. M.; Kruse, C. G. Curr. Opin. Drug Discovery Dev. 2004, 7, 498–506.
Return to citation in text: [1] -
Alex, K.; Tillack, A.; Schwarz, N.; Beller, M. Org. Lett. 2008, 10, 2377–2379. doi:10.1021/ol800592s
Return to citation in text: [1] -
Nair, V.; Biju, A. T.; Mohanan, K.; Suresh, E. Org. Lett. 2006, 8, 2213–2216. doi:10.1021/ol0604623
Return to citation in text: [1] -
Elguero, J.; Goya, P.; Jagerovic, N.; Silva, A. M. S. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Springer: Berlin, 2003; Vol. 6, p 52.
Return to citation in text: [1] -
Martín, R.; Rodríguez Rivero, M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2006, 45, 7079–7082. doi:10.1002/anie.200602917
Return to citation in text: [1] -
Ahmed, M. S. M.; Kobayashi, K.; Mori, A. Org. Lett. 2005, 7, 4487–4489. doi:10.1021/ol051841j
Return to citation in text: [1] -
Kobayashi, S.; Hirabayashi, R.; Shimizu, H.; Ishitani, H.; Yamashita, Y. Tetrahedron Lett. 2003, 44, 3351–3354. doi:10.1016/S0040-4039(03)00606-3
Return to citation in text: [1] -
Jäger, V.; Bierer, L.; Dong, H.-Q.; Palmer, A. M.; Shaw, D.; Frey, W. J. Heterocycl. Chem. 2000, 37, 455–465. doi:10.1002/jhet.5570370304
Return to citation in text: [1] -
Gallos, J. K.; Koumbis, A. E.; Apostolakis, N. E. J. Chem. Soc., Perkin Trans. 1 1997, 2457–2460. doi:10.1039/A704207H
Return to citation in text: [1] -
Khau, V. V.; Martinelli, M. J. Tetrahedron Lett. 1996, 37, 4323–4326. doi:10.1016/0040-4039(96)00836-2
Return to citation in text: [1] -
Gergely, J.; Morgan, J. B.; Overman, L. E. J. Org. Chem. 2006, 71, 9144–9152. doi:10.1021/jo061537j
Return to citation in text: [1] -
Zamfir, A.; Schenker, S.; Bauer, W.; Clark, T.; Tsogoeva, S. B. Eur. J. Org. Chem. 2011, 3706–3709. doi:10.1002/ejoc.201100206
Return to citation in text: [1] -
Shintani, R.; Fu, G. C. J. Am. Chem. Soc. 2003, 125, 10778–10779. doi:10.1021/ja036922u
Return to citation in text: [1] -
Kobayashi, S.; Shimizu, H.; Yamashita, Y.; Ishitani, H.; Kobayashi, J. J. Am. Chem. Soc. 2002, 124, 13678–13679. doi:10.1021/ja027681d
Return to citation in text: [1] -
Chen, W.; Yuan, X.-H.; Li, R.; Du, W.; Wu, Y.; Ding, L.-S.; Chen, Y.-C. Adv. Synth. Catal. 2006, 348, 1818–1822. doi:10.1002/adsc.200606102
Return to citation in text: [1] -
Chen, W.; Du, W.; Duan, Y.-Z.; Wu, Y.; Yang, S.-Y.; Chen, Y.-C. Angew. Chem., Int. Ed. 2007, 46, 7667–7670. doi:10.1002/anie.200702618
Return to citation in text: [1] -
Hashimoto, T.; Omote, M.; Maruoka, K. Angew. Chem., Int. Ed. 2011, 50, 3489–3492. doi:10.1002/anie.201100331
Return to citation in text: [1] -
Hashimoto, T.; Maeda, Y.; Omote, M.; Nakatsu, H.; Maruoka, K. J. Am. Chem. Soc. 2010, 132, 4076–4077. doi:10.1021/ja100787a
Return to citation in text: [1] -
Ma, S.; Jiao, N.; Zheng, Z.; Ma, Z.; Lu, Z.; Ye, L.; Deng, Y.; Chen, G. Org. Lett. 2004, 6, 2193–2196. doi:10.1021/ol0493498
Return to citation in text: [1] -
Yang, Q.; Jiang, X.; Ma, S. Chem.–Eur. J. 2007, 13, 9310–9316. doi:10.1002/chem.200700620
Return to citation in text: [1] -
Shu, W.; Yang, Q.; Jia, G.; Ma, S. Tetrahedron 2008, 64, 11159–11166. doi:10.1016/j.tet.2008.09.049
Return to citation in text: [1] -
Shu, W.; Ma, S. Chem. Commun. 2009, 6198–6200. doi:10.1039/b912108k
Return to citation in text: [1] -
LaLonde, R. L.; Wang, Z. J.; Mba, M.; Lackner, A. D.; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 598–601. doi:10.1002/anie.200905000
Return to citation in text: [1] -
Giampietro, N. C.; Wolfe, J. P. J. Am. Chem. Soc. 2008, 130, 12907–12911. doi:10.1021/ja8050487
Return to citation in text: [1] -
Kotsuki, H.; Ikishima, H.; Okuyama, A. Heterocycles 2008, 75, 493–529. doi:10.3987/REV-07-620
Return to citation in text: [1] -
Kotsuki, H.; Ikishima, H.; Okuyama, A. Heterocycles 2008, 75, 757–797. doi:10.3987/REV-07-621
Return to citation in text: [1] -
Enders, D.; Narine, A. A. J. Org. Chem. 2008, 73, 7857–7870. doi:10.1021/jo801374j
Return to citation in text: [1] -
Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Angew. Chem., Int. Ed. 2008, 47, 6138–6171. doi:10.1002/anie.200705523
Return to citation in text: [1] -
Bertelsen, S.; Jørgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/b903816g
Return to citation in text: [1] -
Bella, M.; Gasperi, T. Synthesis 2009, 1583–1614. doi:10.1055/s-0029-1216796
Return to citation in text: [1] -
Enders, D.; Wang, C.; Liebich, J. X. Chem.–Eur. J. 2009, 15, 11058–11076. doi:10.1002/chem.200902236
Return to citation in text: [1] -
Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471–5569. doi:10.1021/cr0684016
Return to citation in text: [1] -
Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416–5470. doi:10.1021/cr068388p
Return to citation in text: [1] -
Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r
Return to citation in text: [1] -
Moyano, A.; Rios, R. Chem. Rev. 2011, 111, 4703–4832. doi:10.1021/cr100348t
Return to citation in text: [1] -
Enders, D.; Grondal, C.; Hüttl, M. R. M. Angew. Chem., Int. Ed. 2007, 46, 1570–1581. doi:10.1002/anie.200603129
Return to citation in text: [1] -
Yu, X.; Wang, W. Org. Biomol. Chem. 2008, 6, 2037–2046. doi:10.1039/b800245m
Return to citation in text: [1] -
Grondal, C.; Jeanty, M.; Enders, D. Nat. Chem. 2010, 2, 167–178. doi:10.1038/nchem.539
Return to citation in text: [1] -
Itoh, T.; Yokoya, M.; Miyauchi, K.; Nagata, K.; Ohsawa, A. Org. Lett. 2003, 5, 4301–4304. doi:10.1021/ol030103x
Return to citation in text: [1] -
Yamamoto, Y.; Momiyama, N.; Yamamoto, H. J. Am. Chem. Soc. 2004, 126, 5962–5963. doi:10.1021/ja049741g
Return to citation in text: [1] -
Sundén, H.; Ibrahem, I.; Eriksson, L.; Córdova, A. Angew. Chem., Int. Ed. 2005, 44, 4877–4880. doi:10.1002/anie.200500811
Return to citation in text: [1] -
Itoh, T.; Yokoya, M.; Miyauchi, K.; Nagata, K.; Ohsawa, A. Org. Lett. 2006, 8, 1533–1535. doi:10.1021/ol0530575
Return to citation in text: [1] -
Ibrahem, I.; Rios, R.; Vesely, J.; Zhao, G.-L.; Córdova, A. Chem. Commun. 2007, 849–851. doi:10.1039/b613410f
Return to citation in text: [1] -
Sundén, H.; Rios, R.; Ibrahem, I.; Zhao, G.-L.; Eriksson, L.; Córdova, A. Adv. Synth. Catal. 2007, 349, 827–832. doi:10.1002/adsc.200600513
Return to citation in text: [1] -
Vesely, J.; Ibrahem, I.; Zhao, G.-L.; Rios, R.; Córdova, A. Angew. Chem., Int. Ed. 2007, 46, 778–781. doi:10.1002/anie.200603810
Return to citation in text: [1] -
Rueping, M.; Azap, C. Angew. Chem., Int. Ed. 2006, 45, 7832–7835. doi:10.1002/anie.200603199
Return to citation in text: [1] -
Li, H.; Wang, J.; Xie, H.; Zu, L.; Jiang, W.; Duesler, E. N.; Wang, W. Org. Lett. 2007, 9, 965–968. doi:10.1021/ol062877u
Return to citation in text: [1] -
Li, H.; Zu, L.; Xie, H.; Wang, J.; Wang, W. Chem. Commun. 2008, 5636–5638. doi:10.1039/b812464g
Return to citation in text: [1] -
Pesciaioli, F.; Vincentiis, F. D.; Galzerano, F.; Bencivenni, G.; Bartoli, G.; Mazzanti, A.; Melchiorre, P. Angew. Chem., Int. Ed. 2008, 47, 8703–8706. doi:10.1002/anie.200803647
Return to citation in text: [1] -
Lu, M.; Zhu, D.; Lu, Y.; Hou, Y.; Tan, B.; Zhong, G. Angew. Chem., Int. Ed. 2008, 47, 10187–10191. doi:10.1002/anie.200803731
Return to citation in text: [1] -
Zhu, D.; Lu, M.; Chua, P. J.; Tan, B.; Wang, F.; Yang, X.; Zhong, G. Org. Lett. 2008, 10, 4585–4588. doi:10.1021/ol801864c
Return to citation in text: [1] -
Yang, H.; Carter, R. G. J. Org. Chem. 2009, 74, 5151–5156. doi:10.1021/jo9009062
Return to citation in text: [1] -
Wang, Y.-F.; Zhang, W.; Luo, S.-P.; Li, B.-L.; Xia, A.-B.; Zhong, A.-G.; Xu, D.-Q. Chem.–Asian J. 2009, 4, 1834–1838. doi:10.1002/asia.200900298
Return to citation in text: [1] -
Hong, L.; Sun, W.; Liu, C.; Wang, L.; Wang, R. Chem.–Eur. J. 2010, 16, 440–444. doi:10.1002/chem.200902638
Return to citation in text: [1] -
He, Z.-Q.; Zhou, Q.; Wu, L.; Chen, Y.-C. Adv. Synth. Catal. 2010, 352, 1904–1908. doi:10.1002/adsc.201000291
Return to citation in text: [1] -
Takizawa, S.; Inoue, N.; Hirata, S.; Sasai, H. Angew. Chem., Int. Ed. 2010, 49, 9725–9729. doi:10.1002/anie.201004547
Return to citation in text: [1] -
Enders, D.; Göddertz, D. P.; Beceño, C.; Raabe, G. Adv. Synth. Catal. 2010, 352, 2863–2868. doi:10.1002/adsc.201000658
Return to citation in text: [1] -
Deiana, L.; Zhao, G.-L.; Lin, S.; Dziedzic, P.; Zhang, Q.; Leijonmarck, H.; Córdova, A. Adv. Synth. Catal. 2010, 352, 3201–3207. doi:10.1002/adsc.201000650
Return to citation in text: [1] -
Enders, D.; Wang, C.; Raabe, G. Synthesis 2009, 4119–4124. doi:10.1055/s-0029-1217069
Return to citation in text: [1] -
Enders, D.; Wang, C.; Yang, X.; Raabe, G. Synlett 2011, 469–472. doi:10.1055/s-0030-1259326
Return to citation in text: [1] -
Müller, S.; List, B. Angew. Chem., Int. Ed. 2009, 48, 9975–9978. doi:10.1002/anie.200905035
Return to citation in text: [1] -
Mahé, O.; Dez, I.; Levacher, V.; Brière, J.-F. Angew. Chem., Int. Ed. 2010, 49, 7072–7075. doi:10.1002/anie.201002485
Return to citation in text: [1] -
Sinkkonen, J.; Ovcharenko, V.; Zelenin, K. N.; Bezhan, I. P.; Chakchir, B. A.; Al-Assar, F.; Pihlaja, K. Eur. J. Org. Chem. 2002, 3447–3454. doi:10.1002/1099-0690(200210)2002:20<3447::AID-EJOC3447>3.0.CO;2-L
Return to citation in text: [1] -
Fernández, M.; Vicario, J. L.; Reyes, E.; Carrillo, L.; Badía, D. Chem. Commun. 2012, 48, 2092–2094. doi:10.1039/C2CC17370K
Return to citation in text: [1] -
Perdicchia, D.; Jørgensen, K. A. J. Org. Chem. 2007, 72, 3565–3568. doi:10.1021/jo0626717
Return to citation in text: [1] -
Campbell, N. R.; Sun, B.; Singh, R. P.; Deng, L. Adv. Synth. Catal. 2011, 353, 3123–3128. doi:10.1002/adsc.201100447
Return to citation in text: [1] -
Fernández, M.; Reyes, E.; Vicario, J. L.; Badía, D.; Carrillo, L. Adv. Synth. Catal. 2012, 354, 371–376. doi:10.1002/adsc.201100722
Return to citation in text: [1] -
Jensen, K. L.; Dickmeiss, G.; Jiang, H.; Albrecht, Ł.; Jørgensen, K. A. Acc. Chem. Res. 2012, 45, 248–264. doi:10.1021/ar200149w
Return to citation in text: [1] -
Xu, L.-W.; Li, L.; Shi, Z.-H. Adv. Synth. Catal. 2010, 352, 243–279. doi:10.1002/adsc.200900797
Return to citation in text: [1] -
Mielgo, A.; Palomo, C. Chem.–Asian J. 2008, 3, 922–948. doi:10.1002/asia.200700417
Return to citation in text: [1] -
Brandau, S.; Maerten, E.; Jørgensen, K. A. J. Am. Chem. Soc. 2006, 128, 14986–14991. doi:10.1021/ja065507+
Return to citation in text: [1] -
Jiang, H.; Nielsen, J. B.; Nielsen, M.; Jørgensen, K. A. Chem.–Eur. J. 2007, 13, 9068–9075. doi:10.1002/chem.200700696
Return to citation in text: [1] -
Wang, Y.; Li, P.; Liang, X.; Zhang, T. Y.; Ye, J. Chem. Commun. 2008, 1232–1234. doi:10.1039/b717000a
Return to citation in text: [1] -
Marqués-López, E.; Herrera, R. P. Curr. Org. Chem. 2011, 15, 2311–2327.
Return to citation in text: [1] -
CCDC 854579 (4a), CCDC 854580 (4n) and CCDC 860007 (4s) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1]
| 84. | CCDC 854579 (4a), CCDC 854580 (4n) and CCDC 860007 (4s) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 77. | Jensen, K. L.; Dickmeiss, G.; Jiang, H.; Albrecht, Ł.; Jørgensen, K. A. Acc. Chem. Res. 2012, 45, 248–264. doi:10.1021/ar200149w |
| 78. | Xu, L.-W.; Li, L.; Shi, Z.-H. Adv. Synth. Catal. 2010, 352, 243–279. doi:10.1002/adsc.200900797 |
| 79. | Mielgo, A.; Palomo, C. Chem.–Asian J. 2008, 3, 922–948. doi:10.1002/asia.200700417 |
| 80. | Brandau, S.; Maerten, E.; Jørgensen, K. A. J. Am. Chem. Soc. 2006, 128, 14986–14991. doi:10.1021/ja065507+ |
| 81. | Jiang, H.; Nielsen, J. B.; Nielsen, M.; Jørgensen, K. A. Chem.–Eur. J. 2007, 13, 9068–9075. doi:10.1002/chem.200700696 |
| 82. | Wang, Y.; Li, P.; Liang, X.; Zhang, T. Y.; Ye, J. Chem. Commun. 2008, 1232–1234. doi:10.1039/b717000a |
| 83. | Marqués-López, E.; Herrera, R. P. Curr. Org. Chem. 2011, 15, 2311–2327. |
| 1. | Clark, M. P.; Laughlin, S. K.; Laufersweiler, M. J.; Bookland, R. G.; Brugel, T. A.; Golebiowski, A.; Sabat, M. P.; Townes, J. A.; VanRens, J. C.; Djung, J. F.; Natchus, M. G.; De, B.; Hsieh, L. C.; Xu, S. C.; Walter, R. L.; Mekel, M. J.; Heitmeyer, S. A.; Brown, K. K.; Juergens, K.; Taiwo, Y. O.; Janusz, M. J. J. Med. Chem. 2004, 47, 2724–2727. doi:10.1021/jm049968m |
| 2. | Kornet, M. J.; Garrett, R. J. J. Pharm. Sci. 1979, 68, 377–388. doi:10.1002/jps.2600680335 |
| 3. | Tran, K.; Lombardi, P. J.; Leighton, J. L. Org. Lett. 2008, 10, 3165–3167. doi:10.1021/ol8011869 |
| 4. | Özdemir, Z.; Kandilci, H. B.; Gümüşel, B.; Çaliş, Ü.; Bilgin, A. A. Eur. J. Med. Chem. 2007, 42, 373–379. doi:10.1016/j.ejmech.2006.09.006 |
| 5. | Jeong, T.-S.; Kim, K. S.; An, S.-J.; Cho, K.-H.; Lee, S.; Lee, W. S. Bioorg. Med. Chem. Lett. 2004, 14, 2715–2717. doi:10.1016/j.bmcl.2004.03.079 |
| 16. | Kobayashi, S.; Hirabayashi, R.; Shimizu, H.; Ishitani, H.; Yamashita, Y. Tetrahedron Lett. 2003, 44, 3351–3354. doi:10.1016/S0040-4039(03)00606-3 |
| 17. | Jäger, V.; Bierer, L.; Dong, H.-Q.; Palmer, A. M.; Shaw, D.; Frey, W. J. Heterocycl. Chem. 2000, 37, 455–465. doi:10.1002/jhet.5570370304 |
| 18. | Gallos, J. K.; Koumbis, A. E.; Apostolakis, N. E. J. Chem. Soc., Perkin Trans. 1 1997, 2457–2460. doi:10.1039/A704207H |
| 19. | Khau, V. V.; Martinelli, M. J. Tetrahedron Lett. 1996, 37, 4323–4326. doi:10.1016/0040-4039(96)00836-2 |
| 20. | Gergely, J.; Morgan, J. B.; Overman, L. E. J. Org. Chem. 2006, 71, 9144–9152. doi:10.1021/jo061537j |
| 21. | Zamfir, A.; Schenker, S.; Bauer, W.; Clark, T.; Tsogoeva, S. B. Eur. J. Org. Chem. 2011, 3706–3709. doi:10.1002/ejoc.201100206 |
| 22. | Shintani, R.; Fu, G. C. J. Am. Chem. Soc. 2003, 125, 10778–10779. doi:10.1021/ja036922u |
| 23. | Kobayashi, S.; Shimizu, H.; Yamashita, Y.; Ishitani, H.; Kobayashi, J. J. Am. Chem. Soc. 2002, 124, 13678–13679. doi:10.1021/ja027681d |
| 75. | Campbell, N. R.; Sun, B.; Singh, R. P.; Deng, L. Adv. Synth. Catal. 2011, 353, 3123–3128. doi:10.1002/adsc.201100447 |
| 13. | Elguero, J.; Goya, P.; Jagerovic, N.; Silva, A. M. S. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Springer: Berlin, 2003; Vol. 6, p 52. |
| 14. | Martín, R.; Rodríguez Rivero, M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2006, 45, 7079–7082. doi:10.1002/anie.200602917 |
| 15. | Ahmed, M. S. M.; Kobayashi, K.; Mori, A. Org. Lett. 2005, 7, 4487–4489. doi:10.1021/ol051841j |
| 76. | Fernández, M.; Reyes, E.; Vicario, J. L.; Badía, D.; Carrillo, L. Adv. Synth. Catal. 2012, 354, 371–376. doi:10.1002/adsc.201100722 |
| 8. | Johnson, M.; Younglove, B.; Lee, L.; LeBlanc, R.; Holt, H., Jr.; Hills, P.; Mackay, H.; Brown, T.; Mooberry, S. L.; Lee, M. Bioorg. Med. Chem. Lett. 2007, 17, 5897–5901. doi:10.1016/j.bmcl.2007.07.105 |
| 9. | Ali, M. A.; Shaharyar, M. Bioorg. Med. Chem. 2007, 15, 1896–1902. doi:10.1016/j.bmc.2007.01.006 |
| 10. | Lange, J. H. M.; Kruse, C. G. Curr. Opin. Drug Discovery Dev. 2004, 7, 498–506. |
| 11. | Alex, K.; Tillack, A.; Schwarz, N.; Beller, M. Org. Lett. 2008, 10, 2377–2379. doi:10.1021/ol800592s |
| 12. | Nair, V.; Biju, A. T.; Mohanan, K.; Suresh, E. Org. Lett. 2006, 8, 2213–2216. doi:10.1021/ol0604623 |
| 72. | Sinkkonen, J.; Ovcharenko, V.; Zelenin, K. N.; Bezhan, I. P.; Chakchir, B. A.; Al-Assar, F.; Pihlaja, K. Eur. J. Org. Chem. 2002, 3447–3454. doi:10.1002/1099-0690(200210)2002:20<3447::AID-EJOC3447>3.0.CO;2-L |
| 73. | Fernández, M.; Vicario, J. L.; Reyes, E.; Carrillo, L.; Badía, D. Chem. Commun. 2012, 48, 2092–2094. doi:10.1039/C2CC17370K |
| 6. | Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 11279–11282. doi:10.1021/ja049498l |
| 7. | Shirakawa, S.; Lombardi, P. J.; Leighton, J. L. J. Am. Chem. Soc. 2005, 127, 9974–9975. doi:10.1021/ja052307 |
| 74. | Perdicchia, D.; Jørgensen, K. A. J. Org. Chem. 2007, 72, 3565–3568. doi:10.1021/jo0626717 |
| 34. | Kotsuki, H.; Ikishima, H.; Okuyama, A. Heterocycles 2008, 75, 493–529. doi:10.3987/REV-07-620 |
| 35. | Kotsuki, H.; Ikishima, H.; Okuyama, A. Heterocycles 2008, 75, 757–797. doi:10.3987/REV-07-621 |
| 36. | Enders, D.; Narine, A. A. J. Org. Chem. 2008, 73, 7857–7870. doi:10.1021/jo801374j |
| 37. | Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Angew. Chem., Int. Ed. 2008, 47, 6138–6171. doi:10.1002/anie.200705523 |
| 38. | Bertelsen, S.; Jørgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/b903816g |
| 39. | Bella, M.; Gasperi, T. Synthesis 2009, 1583–1614. doi:10.1055/s-0029-1216796 |
| 40. | Enders, D.; Wang, C.; Liebich, J. X. Chem.–Eur. J. 2009, 15, 11058–11076. doi:10.1002/chem.200902236 |
| 41. | Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471–5569. doi:10.1021/cr0684016 |
| 42. | Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416–5470. doi:10.1021/cr068388p |
| 43. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r |
| 44. | Moyano, A.; Rios, R. Chem. Rev. 2011, 111, 4703–4832. doi:10.1021/cr100348t |
| 48. | Itoh, T.; Yokoya, M.; Miyauchi, K.; Nagata, K.; Ohsawa, A. Org. Lett. 2003, 5, 4301–4304. doi:10.1021/ol030103x |
| 49. | Yamamoto, Y.; Momiyama, N.; Yamamoto, H. J. Am. Chem. Soc. 2004, 126, 5962–5963. doi:10.1021/ja049741g |
| 50. | Sundén, H.; Ibrahem, I.; Eriksson, L.; Córdova, A. Angew. Chem., Int. Ed. 2005, 44, 4877–4880. doi:10.1002/anie.200500811 |
| 51. | Itoh, T.; Yokoya, M.; Miyauchi, K.; Nagata, K.; Ohsawa, A. Org. Lett. 2006, 8, 1533–1535. doi:10.1021/ol0530575 |
| 52. | Ibrahem, I.; Rios, R.; Vesely, J.; Zhao, G.-L.; Córdova, A. Chem. Commun. 2007, 849–851. doi:10.1039/b613410f |
| 53. | Sundén, H.; Rios, R.; Ibrahem, I.; Zhao, G.-L.; Eriksson, L.; Córdova, A. Adv. Synth. Catal. 2007, 349, 827–832. doi:10.1002/adsc.200600513 |
| 54. | Vesely, J.; Ibrahem, I.; Zhao, G.-L.; Rios, R.; Córdova, A. Angew. Chem., Int. Ed. 2007, 46, 778–781. doi:10.1002/anie.200603810 |
| 55. | Rueping, M.; Azap, C. Angew. Chem., Int. Ed. 2006, 45, 7832–7835. doi:10.1002/anie.200603199 |
| 56. | Li, H.; Wang, J.; Xie, H.; Zu, L.; Jiang, W.; Duesler, E. N.; Wang, W. Org. Lett. 2007, 9, 965–968. doi:10.1021/ol062877u |
| 57. | Li, H.; Zu, L.; Xie, H.; Wang, J.; Wang, W. Chem. Commun. 2008, 5636–5638. doi:10.1039/b812464g |
| 58. | Pesciaioli, F.; Vincentiis, F. D.; Galzerano, F.; Bencivenni, G.; Bartoli, G.; Mazzanti, A.; Melchiorre, P. Angew. Chem., Int. Ed. 2008, 47, 8703–8706. doi:10.1002/anie.200803647 |
| 59. | Lu, M.; Zhu, D.; Lu, Y.; Hou, Y.; Tan, B.; Zhong, G. Angew. Chem., Int. Ed. 2008, 47, 10187–10191. doi:10.1002/anie.200803731 |
| 60. | Zhu, D.; Lu, M.; Chua, P. J.; Tan, B.; Wang, F.; Yang, X.; Zhong, G. Org. Lett. 2008, 10, 4585–4588. doi:10.1021/ol801864c |
| 61. | Yang, H.; Carter, R. G. J. Org. Chem. 2009, 74, 5151–5156. doi:10.1021/jo9009062 |
| 62. | Wang, Y.-F.; Zhang, W.; Luo, S.-P.; Li, B.-L.; Xia, A.-B.; Zhong, A.-G.; Xu, D.-Q. Chem.–Asian J. 2009, 4, 1834–1838. doi:10.1002/asia.200900298 |
| 63. | Hong, L.; Sun, W.; Liu, C.; Wang, L.; Wang, R. Chem.–Eur. J. 2010, 16, 440–444. doi:10.1002/chem.200902638 |
| 64. | He, Z.-Q.; Zhou, Q.; Wu, L.; Chen, Y.-C. Adv. Synth. Catal. 2010, 352, 1904–1908. doi:10.1002/adsc.201000291 |
| 65. | Takizawa, S.; Inoue, N.; Hirata, S.; Sasai, H. Angew. Chem., Int. Ed. 2010, 49, 9725–9729. doi:10.1002/anie.201004547 |
| 66. | Enders, D.; Göddertz, D. P.; Beceño, C.; Raabe, G. Adv. Synth. Catal. 2010, 352, 2863–2868. doi:10.1002/adsc.201000658 |
| 67. | Deiana, L.; Zhao, G.-L.; Lin, S.; Dziedzic, P.; Zhang, Q.; Leijonmarck, H.; Córdova, A. Adv. Synth. Catal. 2010, 352, 3201–3207. doi:10.1002/adsc.201000650 |
| 68. | Enders, D.; Wang, C.; Raabe, G. Synthesis 2009, 4119–4124. doi:10.1055/s-0029-1217069 |
| 69. | Enders, D.; Wang, C.; Yang, X.; Raabe, G. Synlett 2011, 469–472. doi:10.1055/s-0030-1259326 |
| 33. | Giampietro, N. C.; Wolfe, J. P. J. Am. Chem. Soc. 2008, 130, 12907–12911. doi:10.1021/ja8050487 |
| 70. | Müller, S.; List, B. Angew. Chem., Int. Ed. 2009, 48, 9975–9978. doi:10.1002/anie.200905035 |
| 71. | Mahé, O.; Dez, I.; Levacher, V.; Brière, J.-F. Angew. Chem., Int. Ed. 2010, 49, 7072–7075. doi:10.1002/anie.201002485 |
| 28. | Ma, S.; Jiao, N.; Zheng, Z.; Ma, Z.; Lu, Z.; Ye, L.; Deng, Y.; Chen, G. Org. Lett. 2004, 6, 2193–2196. doi:10.1021/ol0493498 |
| 29. | Yang, Q.; Jiang, X.; Ma, S. Chem.–Eur. J. 2007, 13, 9310–9316. doi:10.1002/chem.200700620 |
| 30. | Shu, W.; Yang, Q.; Jia, G.; Ma, S. Tetrahedron 2008, 64, 11159–11166. doi:10.1016/j.tet.2008.09.049 |
| 31. | Shu, W.; Ma, S. Chem. Commun. 2009, 6198–6200. doi:10.1039/b912108k |
| 32. | LaLonde, R. L.; Wang, Z. J.; Mba, M.; Lackner, A. D.; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 598–601. doi:10.1002/anie.200905000 |
| 24. | Chen, W.; Yuan, X.-H.; Li, R.; Du, W.; Wu, Y.; Ding, L.-S.; Chen, Y.-C. Adv. Synth. Catal. 2006, 348, 1818–1822. doi:10.1002/adsc.200606102 |
| 25. | Chen, W.; Du, W.; Duan, Y.-Z.; Wu, Y.; Yang, S.-Y.; Chen, Y.-C. Angew. Chem., Int. Ed. 2007, 46, 7667–7670. doi:10.1002/anie.200702618 |
| 26. | Hashimoto, T.; Omote, M.; Maruoka, K. Angew. Chem., Int. Ed. 2011, 50, 3489–3492. doi:10.1002/anie.201100331 |
| 27. | Hashimoto, T.; Maeda, Y.; Omote, M.; Nakatsu, H.; Maruoka, K. J. Am. Chem. Soc. 2010, 132, 4076–4077. doi:10.1021/ja100787a |
| 45. | Enders, D.; Grondal, C.; Hüttl, M. R. M. Angew. Chem., Int. Ed. 2007, 46, 1570–1581. doi:10.1002/anie.200603129 |
| 46. | Yu, X.; Wang, W. Org. Biomol. Chem. 2008, 6, 2037–2046. doi:10.1039/b800245m |
| 47. | Grondal, C.; Jeanty, M.; Enders, D. Nat. Chem. 2010, 2, 167–178. doi:10.1038/nchem.539 |
© 2012 Geng et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)