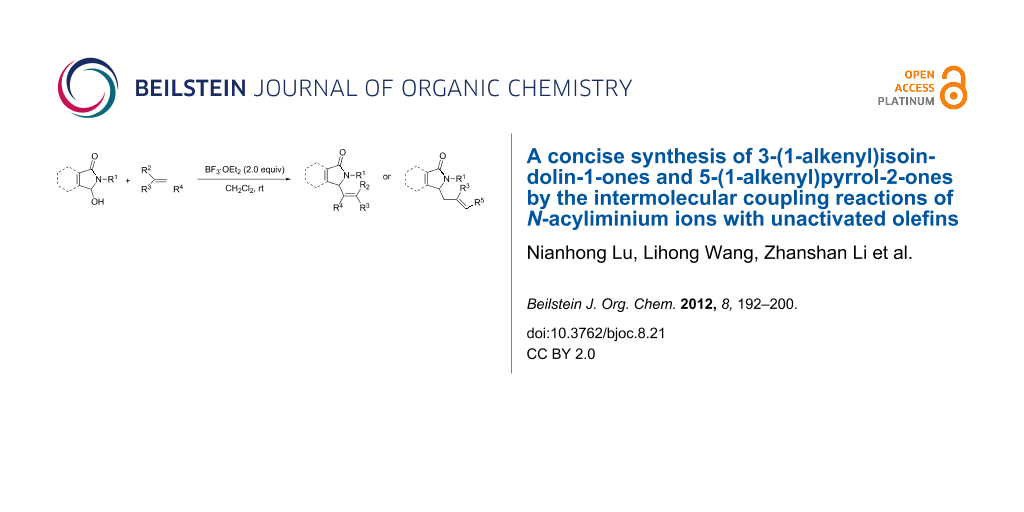
Abstract
A concise synthesis of 3-(1-alkenyl)isoindolin-1-ones and 5-(1-alkenyl)pyrrol-2-ones has been achieved by the coupling reactions of N-acyliminium ions produced from 3-hydroxyisoindol-1-ones or 5-hydroxy-1-pyrrol-2-ones with unactivated olefins in the presence of BF3·OEt2 at room temperature. For most of the olefins, the reactions afforded the Csp3–Csp2 cross-coupling products, but for the α-methylstyrene and 1-hexene, the Csp3–Csp3 cross-coupling products were obtained.

Graphical Abstract
Introduction
The coupling of alcohols with alkynes, aromatics and active methylene compounds has attracted great attention in recent years as an effective and environmentally benign strategy for the construction of carbon–carbon bonds with the concomitant loss of water. For example, the metal-catalyzed coupling of allyl, benzyl, and propargyl alcohols with terminal alkynes to give the doubly alkyl-substituted acetylenes [1-3]; the Brønsted acid and Lewis acid catalyzed coupling of alcohols with indoles to give the 3-alkyl-substituted indoles [4-6]; and the Brønsted acid and Lewis acid-catalyzed coupling of alcohols with 1,3-dicarbonyls to give the 2-alkyl-substituted 1,3-dicarbonyls [7-9]. All these reactions generally proceed by the addition of carbon cations to multiple bonds and subsequent deprotonation. In comparison, the reports for the coupling of alcohols with unactivated olefins to give the corresponding alkyl-substituted alkenes are rare. Lee recently reported a coupling of alcohols with olefins catalyzed by a ruthenium complex to give alkyl-substituted alkenes through the formation of Csp3–Csp2 bonds [10]; Liu reported the FeCl3/TsOH catalyzed coupling of diarylmethanol with styrenes to afford the alkyl-substituted styrenes [11]. We have long been interested in the reactions of N-acyliminium ions produced easily by the Brønsted acid and Lewis acid catalyzed dehydroxylation of α-hydroxyamides [12-14]. The high electrophilicity of these species is very suitable for electrophilic addition to carbon–carbon multiple bonds. The coupling reactions of N-acyliminium ions with various carbon nucleophiles, such as allylsilanes, alkylmetals, TMSCN, 1,3-dicarbonyls, isonitriles, enol derivatives and aromatics has been studied extensively [15-17]. Few reports are found to deal with the intermolecular coupling reactions of N-acyliminium ions with unactivated olefins, although the intramolecular addition of acyliminium ions to olefins has been reported [18]. The reported olefins that coupled with N-acyliminium ions were generally activated alkenes, such as 1-alkenylsilanes [19], 1-alkenylcoppers [20,21], 1-alkenylalanes [22] and 1-alkenylboronic acid, or esters [23,24] besides allylsilane. For example, Angst reported the coupling of styrylsilanes with N-acyl-2-chloroglycine esters catalyzed by SnCl4 to give the 3-styryl glycine derivatives in 1987 [19]; Wistrand reported the coupling of methyl 1-acyl-5-methoxy-L-proline with 1-alkenylcoppers catalyzed by BF3·OEt2 to give methyl 1-acyl-5-(1-alkenyl)-L-proline in 1992 [20]; Menicagli reported the coupling of N-acylisoquinolium chloride with di-isobutyl 1-hexenylalanes to give 1,2-dihydro-2-acyl-1-hexenylisoquinolines in 2008 [22]; Schaus reported the coupling of 1-alkenylboronates with 2-ethoxy-N-acylquinolines catalyzed by tartaric acid to produce 2-(1-alkenyl)-N-acylquinolines in 2011 [23]. We report here a concise synthesis of 3-(1-alkenyl)isoindolin-1-ones and 5-(1-alkenyl)pyrrol-2-ones by the cross-coupling reactions of N-acyliminium ions derived from 3-hydroxyisoindol-1-ones or 5-hydroxypyrrol-2-ones with unactivated olefins such as styrene (2a) (Scheme 1 and Scheme 2).
![[1860-5397-8-21-i1]](/bjoc/content/inline/1860-5397-8-21-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of 3-hydroxyisoindol-1-one with styrene.
Scheme 1: Reaction of 3-hydroxyisoindol-1-one with styrene.
![[1860-5397-8-21-i2]](/bjoc/content/inline/1860-5397-8-21-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of 5-hydroxypyrrol-1-one with styrene.
Scheme 2: Reaction of 5-hydroxypyrrol-1-one with styrene.
Isoindolinones and pyrrolones are the core structures of numerous natural alkaloids [25-27] as well as many drug candidates [28-30]. Isoindolinones demonstrate a remarkably wide range of biological activities, including anti-inflammatory, antihypertensive, antipsychotic and antileukemic and antiviral effects [31-33]. Thus, many methods have been developed to synthesize 2- or 3-functionalized isoindolinones. Among them, only two reports dealt with the synthesis of 3-(1-alkenyl)isoindolin-1-one derivatives. One was, as mentioned above, by the Cp2ZrCl2 catalyzed coupling of N-acyliminium ions with in situ generated dimethyl 1-alkenylalanes [24], another was performed by the palladium-catalyzed coupling of 2-iodobenzoyl chloride with aldimines and subsequent cyclization [34]. The results of our investigation have furnished another route to the synthesis of 3-(1-alkenyl)isoindolin-1-ones and 5-(1-alkenyl)pyrrol-2-ones.
Results and Discussion
Two kinds of N-acyliminium ion precursors, 3-hydroxyisoindol-1-ones (1a–c) and 5-hydroxypyrrol-2-ones (5a,b) were easily prepared by the reduction of the parent phthalimide [35] and maleimide [36] derivatives. In order to explore the effects of the experimental conditions on the coupling reactions, the reaction of 1a with styrene (2a) was selected as a representative and carried out at room temperature under different conditions (Table 1). The use of a larger amount of catalyst led to an increase in the yield of the coupling product 3a (Table 1, entries 1–3). This observation is general for most of the intermolecular coupling reactions of N-acyliminium ions with the weakest nucleophiles [14-16]. Of the catalysts examined, BF3·OEt2 was very efficient for the formation of 3a compared to other catalysts such as CF3SO3H, CH3CO2H, TiCl4, SnCl4 and InCl3. Among various solvents tested, anhydrous dichloromethane (DCM) appeared to be the best choice, providing the desired adduct in the highest yield (>80%). Thus, the reaction employing 2.0 equiv BF3·OEt2 as catalyst and anhydrous DCM as solvent at room temperature was selected as the model for the general conditions for all of the other reactions.
Table 1: Optimization of the intermolecular coupling reaction of 1a with 2a.a
| Entry | Solvent | Catalyst |
t
(h) |
T
(°C) |
Yieldb
(%) |
|---|---|---|---|---|---|
| 1 | CH2Cl2 | 1.0 equiv BF3·OEt2 | 1.0 | 25 | 65 |
| 2 | CH2Cl2 | 1.5 equiv BF3·OEt2 | 1.0 | 25 | 80 |
| 3 | CH2Cl2 | 2.0 equiv BF3·OEt2 | 1.0 | 25 | 83 |
| 4 | CH2Cl2 | 2.0 equiv CF3SO3H | 1.0 | 25 | 50 |
| 5 | CH2Cl2 | 2.0 equiv CF3CO2H | 1.0 | 25 | 37 |
| 6 | CH2Cl2 | 2.0 equiv TiCl4 | 1.0 | 25 | 30 |
| 7 | CH2Cl2 | 2.0 equiv SnCl4 | 1.0 | 25 | 25 |
| 8 | CH2Cl2 | 2.0 equiv InCl3 | 1.0 | 25 | 21 |
| 9 | CH3CN | 2.0 equiv BF3·OEt2 | 1.0 | 25 | 66 |
| 10 | Et2O | 2.0 equiv BF3·OEt2 | 1.0 | 25 | 64 |
aReactions were carried out on 1.0 mmol scale in 15.0 mL of solvent for 1.0 h with 1a (0.1 mmol), 2a (2.0 mmol) and catalyst (2.0 mmol); bisolated yields based on 1a.
Under the selected conditions, the reactions of substrates 1a–c with different olefins, such as styrene (2a), α-methylstyrene (2b), 1,1-diphenylethene (2c), indene (2d), cyclohexene (2e), 3,4-dihydropyran (2f), 2,3-dihydofuran (2g) and 1-hexene (2h), were examined (Scheme 3). All reactions proceeded quickly to afford the corresponding coupling products 3a–o or 4a–d in moderate to high yields (Table 2 and Table 3). The products were fully characterized by 1H, 13C NMR and HRMS, and the structure of 3h was further confirmed by X-ray crystallography (Figure 1).
![[1860-5397-8-21-i3]](/bjoc/content/inline/1860-5397-8-21-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of 5-hydroxyisoindol-1-ones with olefins in the presence of BF3·OEt2.
Scheme 3: Reactions of 5-hydroxyisoindol-1-ones with olefins in the presence of BF3·OEt2.
![[1860-5397-8-21-1]](/bjoc/content/figures/1860-5397-8-21-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-Ray structure (ORTEP drawing) of 3h.
Figure 1: X-Ray structure (ORTEP drawing) of 3h.
Table 2: The reactions of 3-hydroxyisoindol-1-one 1a with olefins 2 in the presence of BF3·OEt2.a
| Entry | Reactants | t (h) | T (°C) | Product | Yieldb (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | |||||||
| 1 | 1a | PhCH2 | 2a | H | Ph | H | 0.5 | 25 | — |
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-21-i5.svg?max-width=637&scale=1.0)
3a |
83 |
| 2 | 1a | PhCH2 | 2b | CH3 | Ph | H | 0.5 | 25 | H |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-21-i6.svg?max-width=637&scale=1.0)
4a |
90 |
| 3 | 1a | PhCH2 | 2c | Ph | Ph | H | 0.25 | 25 | — |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-21-i7.svg?max-width=637&scale=1.0)
3b |
93 |
| 4 | 1a | PhCH2 | 2d | H | –CH2C6H4– | 1.0 | 25 | — |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-21-i8.svg?max-width=637&scale=1.0)
3c |
78 | |
| 5 | 1a | PhCH2 | 2e | H | –(CH2)4– | 1.0 | 25 | — |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-21-i9.svg?max-width=637&scale=1.0)
3d |
77 | |
| 6 | 1a | PhCH2 | 2f | H | –(CH2)3O– | 1.0 | 25 | — |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-21-i10.svg?max-width=637&scale=1.0)
3e |
59 | |
| 7 | 1a | PhCH2 | 2g | H | –(CH2)2O– | 1.0 | 25 | — |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-21-i11.svg?max-width=637&scale=1.0)
3f |
54 | |
| 8 | 1a | PhCH2 | 2h | H | H | n-Bu | 2.0 | 25 | n-Pr |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-21-i12.svg?max-width=637&scale=1.0)
4b |
47 |
aAll reactions were performed under the optimal conditions; bisolated yields based on 1a.
Table 3: The reactions of 3-hydroxyisoindol-1-one (1b,c) with olefins 2 in the presence of BF3·OEt2.a
| Entry | Reactants | t (h) | T (°C) | Product | Yieldb (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | |||||||
| 1 | 1b | CH3 | 2a | H | Ph | H | 0.5 | 25 | — |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-21-i13.svg?max-width=637&scale=1.0)
3g |
70 |
| 2 | 1b | CH3 | 2b | CH3 | Ph | H | 0.5 | 25 | H |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-21-i14.svg?max-width=637&scale=1.0)
4c |
93 |
| 3 | 1b | CH3 | 2c | Ph | Ph | H | 0.25 | 25 | — |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-21-i15.svg?max-width=637&scale=1.0)
3h |
94 |
| 4 | 1b | CH3 | 2d | H | –CH2C6H4– | 0.5 | 25 | — |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-21-i16.svg?max-width=637&scale=1.0)
3i |
65 | |
| 5 | 1b | CH3 | 2e | H | –(CH2)4– | 1.0 | 25 | — |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-21-i17.svg?max-width=637&scale=1.0)
3j |
53 | |
| 6 | 1b | CH3 | 2f | H | –(CH2)3O– | 1.0 | 25 | — |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-21-i18.svg?max-width=637&scale=1.0)
3k |
48 | |
| 7 | 1b | CH3 | 2g | H | –(CH2)2O– | 1.0 | 25 | — |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-21-i19.svg?max-width=637&scale=1.0)
3l |
45 | |
| 8 | 1c | H | 2a | H | Ph | H | 1.0 | 25 | — |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-21-i20.svg?max-width=637&scale=1.0)
3m |
58 |
| 9 | 1c | H | 2b | CH3 | Ph | H | 1.0 | 25 | H |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-21-i21.svg?max-width=637&scale=1.0)
4d |
66 |
| 10 | 1c | H | 2c | Ph | Ph | H | 1.0 | 25 | — |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-21-i22.svg?max-width=637&scale=1.0)
3n |
73 |
| 11 | 1c | H | 2d | H | –CH2C6H4– | 1.0 | 25 | — |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-21-i23.svg?max-width=637&scale=1.0)
3o |
50 | |
aAll reactions were performed under the optimal conditions; bisolated yields based on 1b,c.
It can be seen from Table 2 and Table 3 that substituents such as the benzyl and methyl group at the N-atom in 1a,b favored the formation of the coupling products and, thus, higher yields of product were produced from 1a,b. Moreover, both the reaction efficiency and selectivity appeared to be strongly dependent upon variation of the structure of the alkene component. The yields of the coupling adducts are seen to gradually decrease as the nucleophilicity of the alkene diminishes, as is exemplified by the yields recorded for the reactions between 1a and 1b and diphenyl ethylene, α-methylstyrene and styrene (case of 1a: Table 2, entries 1–3 and case of 1b: Table 3, entries 1–3). The same trend is also observed in the less favorable case of 1c (Table 3, entries 8–10). Consistent with this reactivity profile, hexene gave only a moderate yield of adduct 4b when reacted with 1a (Table 2, entry 8). Likewise, alkenes bearing allylic protons prone to β-elimination, such as α-methylstyrene and hexane, did not afford the “normal” Csp3–Csp2 vinylic adducts of type 3, but instead the Csp3–Csp3 coupling products 4 were isolated (Table 2, entries 2 and 8 and Table 3, entries 2 and 9) much like the ene-type adducts of oxonium ion with olefins [37,38]. This means that these alkenes may be envisioned as surrogates of their corresponding, more expensive and less atom-economical, allylsilane derivatives, which are typically used in N-acyliminium ion chemistry to produce amide compounds substituted with an α-allyl group. The reactions of cyclic alkenes (2d–g) with 1a,b all gave the normal Csp3–Csp2 coupling products in moderate yields.
The coupling reactions were examined under the same conditions with alternate substrates (5a,b), and olefins (2a–c) (Scheme 4). All these reactions gave the cross-coupling products (Table 4). As compared with 1a–c, the rates of the coupling reactions of 5a,b with 2a–c were somewhat slower and the yields of the corresponding products were also decreased, probably as a result of both the limited nucleophilicity parameter of the alkenes [39] and the lower stability of the transient N-acyliminium intermediate derived from 5a,b. Similarly to the reactions of 1a–c with α-methylstyrene (2b), the reactions of 5a,b with 2b also gave the Csp3–Csp3 coupling products 7a,b instead of the Csp3–Csp2 coupling products.
![[1860-5397-8-21-i4]](/bjoc/content/inline/1860-5397-8-21-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactions of 5-hydroxypyrrol-1-ones with olefins in the presence of BF3·OEt2.
Scheme 4: Reactions of 5-hydroxypyrrol-1-ones with olefins in the presence of BF3·OEt2.
Table 4: The reactions of 5-hydroxypyrrol-2-ones 5 with olefins 2 in the presence of BF3·OEt2.a
| Entry | Reactants | t (h) | T (°C) | Product | Yieldb (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | |||||||
| 1 | 5a | PhCH2 | 2a | H | Ph | 2.0 | 25 | 6a | 55 |
| 2 | 5a | PhCH2 | 2b | CH3 | Ph | 2.0 | 25 | 7a | 82 |
| 3 | 5a | PhCH2 | 2c | Ph | Ph | 2.0 | 25 | 6b | 72 |
| 4 | 5b | CH3 | 2a | CH3 | Ph | 2.0 | 25 | 7b | 72 |
| 5 | 5b | CH3 | 2b | Ph | Ph | 2.0 | 25 | 6c | 65 |
aAll reactions were performed under the optimal conditions; bisolated yields based on 5a,b.
Conclusion
In summary, we have developed a concise route for the synthesis of 3-(1-alkenyl)isoindolin-1-ones and 5-(1-alkenyl)pyrrol-2-ones by the coupling reactions of N-acyliminium ions derived from 3-hydroxyisoindol-1-ones or 5-hydroxypyrrol-2-ones with unactivated olefins in the presence of BF3·OEt2 at room temperature. For most of the olefins, the reactions afforded the Csp3–Csp2 cross-coupling products, but for α-methylstyrene and 1-hexene, the Csp3–Csp3 cross-coupling products were produced.
Experimental
General information
All reagents were purchased from commercial suppliers and used without further purification. All solvents were dried and redistilled before use. Flash chromatography was carried out with silica gel (200–300 mesh). Analytical TLC was performed with silica gel GF254 plates, and the products were visualized by UV detection. Melting points were determined on a Yanagimoto melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker AM-400 NMR or a Bruker DRX-300 NMR spectrometer in CDCl3 with TMS as an internal standard. EIMS were recorded with a HP 5988 A mass spectrometer. HRMS (ESI) were measured on a Bruker Dattonics APEX 47e mass spectrometer.
General procedure for the coupling reactions
To a solution of 1a (1.0 mmol) and olefin 2a (2.0 mmol) in 15 mL of anhydrous methylene dichloride, BF3·OEt2 (2.0 mmol) was added at 25 °C in one portion under stirring. After continued stirring at 25 °C until 1a disappeared (monitored by TLC), the reaction was quenched with water. The mixture was separated and the aqueous phase was extracted with methylene dichloride (10 mL). The combined organic layers were washed with water (20 mL), dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was separated by silica-gel column chromatography, eluted by hexane/acetone (10:1 v/v), to give the corresponding product 3a.
(E)-2-Benzyl-3-(2-phenylethenyl)isoindolin-1-one (3a): Colorless syrup; 1H NMR (400 MHz, CDCl3) δ 4.22 (d, J = 14.8 Hz, 1H), 4.90 (d, J = 9.2 Hz, 1H), 5.33 (d, J = 14.8 Hz, 1H), 5.82 (dd, J = 9.2 Hz, 15.6 Hz, 1H), 6.77 (d, J = 15.6 Hz, 1H), 7.28–7.37 (m, 11H), 7.50–7.55 (m, 2H), 7.92 (dd, J = 1.6 Hz, 6.4 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 44.1, 62.7, 123.2, 123.8, 125.6, 126.7 (2C), 127.5, 128.4 (2C), 128.5 (2C), 128.6 (2C), 128.7, 128.7, 131.7, 131.8, 135.7, 135.9, 137.4, 144.5, 168.0 (CO) ppm; EIMS m/z (% relative intensity): 325 (56), 310 (29), 234 (89), 220 (31), 149 (46), 91 (45), 57 (53), 44 (100); HRMS–ESI (m/z): [M + H]+ calculated for C23H20NO, 326.1540; found, 326.1536.
2-Benzyl-3-(2-phenyl-2-propenyl)isoindolin-1-one (4a): Colorless solid, mp 69–72 °C; 1H NMR (400 MHz, CDCl3) δ 2.54 (dd, J = 9.2 Hz, 14.0 Hz, 1H), 3.40 (dd, J = 4.0 Hz, 14.0 Hz, 1H), 4.24 (d, J = 15.6 Hz, 1H), 4.39 (dd, J = 4.0 Hz, 9.2 Hz, 1H), 5.00 (s, 1H), 5.38 (s, 1H), 5.40 (d, J = 15.6 Hz, 1H), 7.20–7.31 (m, 11H), 7.41 (t, J = 4.0 Hz, 2H), 7.86 (t, J = 4.0 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 38.1, 44.1, 56.9, 116.9, 123.2, 123.7, 126.1 (2C), 127.6, 127.8, 128.1, 128.1 (2C), 128.5 (2C), 128.8 (2C), 130.9, 131.8, 137.0, 139.8, 143.6, 145.2, 168.4 (CO) ppm; MS m/z (% relative intensity): 339 (1), 253 (4), 237 (6), 222 (100), 197 (5), 149 (13), 91 (71); HRMS–ESI (m/z): [M + H]+ calcd for C24H22NO, 340.1696; found, 340.1699.
2-Benzyl-3-cyclohexenylisoindolin-1-one (3d): Colorless solid, mp 109–112 °C; 1H NMR (400 MHz, CDCl3) δ 1.15–1.19 (m, 1H), 1.38–1.43 (m, 3H), 1.50–1.59 (m, 2H), 2.13 (t, J = 2.4 Hz, 2H), 4.06 (d, J = 14.8 Hz, 1H), 4.71 (s, 1H), 5.19 (d, J = 14.8 Hz, 1H), 5.93 (s, 1H), 7.26–7.30 (m, 5H), 7.41–7.50 (m, 3H), 7.87 (d, J = 7.2 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 21.8, 22.0, 22.2, 25.4, 43.9, 66.7, 122.4, 123.4, 127.3, 128.1, 128.4 (4C), 130.2, 131.4, 132.3, 133.4, 137.4, 144.4, 168.3 (CO) ppm; MS m/z (% relative intensity): 303 (64), 222 (27), 199 (70), 183 (6), 170 (12), 157 (15), 129 (27), 91 (100), 40 (37); HRMS–ESI (m/z): [M + H]+ calcd for C21H22NO, 304.1696; found, 304.1691.
(E)-2-Benzyl-3-(hex-2-enyl)isoindolin-1-one (4b): Colorless syrup; 1H NMR (400 MHz, CDCl3) δ 0.74 (t, J = 7.2 Hz, 3H), 1.25–1.87 (m, 2H), 1.79–1.86 (m, 2H), 2.55–2.70 (m, 2H), 4.17 (d, J = 15.2 Hz, 1H), 4.39 (dd, J = 4.0 Hz, 5.6 Hz, 1H), 4.91–4.98 (m, 1H), 5.36–5.42 (m, 1H), 5.42 (d, J = 15.2 Hz, 1H), 7.28–7.32 (m, 5H), 7.37 (d, J = 7.2 Hz, 1H), 7.43–7.53 (m, 2H), 7.88 (d, J = 7.6 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.4, 22.3, 34.1, 34.5, 43.9, 58.4, 122.4 (2C), 123.7, 127.5, 128.0, 128.1 (2C), 128.7 (2C), 131.2, 132.4, 135.4, 137.2, 145.1, 168.5 (CO) ppm; MS m/z (% relative intensity): 305 (4), 223 (18), 222 (100), 186 (6), 172 (6), 132 (8), 104 (5), 91 (89); HRMS–ESI (m/z): [M + H]+ calcd for C21H24NO, 306.1853; found, 306.1851.
3-(2,2-Diphenylethenyl)-2-methylisoindolin-1-one (3h): Colorless solid, mp 146–148 °C; 1H NMR (400 MHz, CDCl3) δ 3.08 (s, 3H), 5.01 (d, J = 10.0 Hz, 1H), 5.71 (d, J = 10.0 Hz, 1H), 7.25–7.27 (m, 5H), 7.38–7.46 (m, 5H), 7.47–7.52 (m, 3H), 7.83 (d, J = 7.6 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 27.5, 61.3, 122.8, 123.4, 124.3, 127.2 (2C), 127.9, 128.1, 128.3 (3C), 128.8 (2C), 129.5 (2C), 131.3, 132.3, 138.5, 140.4, 144.5, 148.1, 168.0 (CO) ppm; MS m/z (% relative intensity): 325 (28), 310 (15), 294 (9), 265 (5), 248 (11), 220 (18), 188 (10), 178 (11), 165 (13), 149 (37), 91 (30), 57 (63), 43 (100); HRMS–ESI (m/z): [M + H]+ calcd for C23H20NO, 326.1540; found, 326.1545.
Crystal data for 3h (recrystallized from ethanol): C23H19NO, Mr = 325.39. Monoclinic, a = 17.373(11) Å, b = 17.241(11) Å, c = 24.421(16) Å, β = 91.219(9), V = 7313(8) Å3, colorless plates, ρ = 1.182 g cm−3, T = 296(2) K, space group P2(1)/c, Z = 4, μ (Mo Kα) = 0.084 mm−1, 2θmax = 51°, 9126 reflections measured, 3995 unique (Rint = 0.0696), which were used in all calculations. The final wR(F2) was 0.1427 (for all data), R1 = 0.0764. CCDC file No. 835330.
3-(3,4-Dihydro-2H-pyran-5-yl)-2-methylisoindolin-1-one (3k): Colorless solid, mp 94–97 °C; 1H NMR (400 MHz, CDCl3) δ 1.20–1.27 (m, 1H), 1.40–1.47 (m, 1H), 1.71–1.79 (m, 2H), 3.00 (s, 3H), 3.92–4.04 (m, 2H), 4.65 (s, 1H), 6.79 (s, 1H), 7.36 (d, J = 7.2 Hz, 1H), 7.44 (t, J = 7.2 Hz, 1H), 7.53 (t, J = 7.2 Hz, 1H), 7.81 (d, J = 7.2 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 17.0, 21.5, 26.6, 65.2, 66.0, 108.2, 122.2, 123.0, 128.1, 131.3, 132.7, 144.2, 144.6, 168.2 (CO) ppm; MS m/z (% relative intensity): 229 (100), 200 (47), 186 (35), 172 (54), 146 (51), 128 (20), 115 (17), 91 (24); HRMS–ESI (m/z): [M + H]+ calcd for C14H16NO2, 230.1176; found, 230.1175.
(E)-1-Benzyl-5-(2-phenylethenyl)-1H-pyrrol-2(5H)-one (6a): Colorless syrup; 1H NMR (400 MHz, CDCl3) δ 4.08 (d, J = 14.8 Hz, 1H), 4.54 (d, J = 9.2 Hz, 1H), 5.12 (d, J = 14.8 Hz, 1H), 5.69 (dd, J = 9.2 Hz, 15.6 Hz, 1H), 6.26 (dd, J = 1.6 Hz, 5.6 Hz, 1H), 6.59 (d, J = 15.6 Hz, 1H), 6.96 (dd, J = 1.6 Hz, 6.0 Hz, 1H), 7.23–7.35 (m, 8H), 7.40 (dd, J = 1.6 Hz, 8.0 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 42.4, 64.8, 126.1, 126.6, 127.4, 128.0, 128.2, 128.6 (2C), 128.7 (2C), 128.7 (2C), 128.9 (2C), 135.7, 137.6, 146.6, 170.9 (CO) ppm; MS m/z (% relative intensity): 275 (22), 190 (11), 189 (100), 184 (30), 161 (29), 160 (39), 132 (37), 119 (22), 104 (48), 91 (21); HRMS–ESI (m/z): [M + H]+ calcd for C19H18NO, 276.1383; found, 276.1385.
References
-
Ren, K.; Li, P.; Wang, L.; Zhang, X. Tetrahedron 2011, 67, 2753–2759. doi:10.1016/j.tet.2011.02.050
Return to citation in text: [1] -
Wang, T.; Chen, X.-L.; Chen, L.; Zhan, Z.-P. Org. Lett. 2011, 13, 3324–3327. doi:10.1021/ol201054z
Return to citation in text: [1] -
Xiang, S.-K.; Zhang, L.-H.; Jiao, N. Chem. Commun. 2009, 6487–6489. doi:10.1039/B911905A
Return to citation in text: [1] -
Wu, Y.-C.; Li, H.-J.; Liu, L.; Demoulin, N.; Liu, Z.; Wang, D.; Chen, Y.-J. Adv. Synth. Catal. 2011, 353, 907–912. doi:10.1002/adsc.201000930
Return to citation in text: [1] -
Sanz, R.; Miguel, D.; Martínez, A.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; Álvarez, E.; Rodríguez, F. Eur. J. Org. Chem. 2010, 7027–7039. doi:10.1002/ejoc.201001055
Return to citation in text: [1] -
Davoust, M.; Kitching, J. A.; Fleming, M. J.; Lautens, M. Chem.–Eur. J. 2010, 16, 50–54. doi:10.1002/chem.200902694
Return to citation in text: [1] -
Liu, P. N.; Dang, L.; Wang, Q. W.; Zhao, S. L.; Xia, F.; Ren, Y. J.; Gong, X. Q.; Chen, J. Q. J. Org. Chem. 2010, 75, 5017–5030. doi:10.1021/jo100517k
Return to citation in text: [1] -
Reddy, C. R.; Vijaykumar, J.; Grée, R. Synthesis 2010, 3715–3723. doi:10.1055/s-0030-1258214
Return to citation in text: [1] -
Theerthagiri, P.; Lalitha, A. Tetrahedron Lett. 2010, 51, 5454–5458. doi:10.1016/j.tetlet.2010.08.019
Return to citation in text: [1] -
Lee, D.-H.; Kwon, K.-H.; Yi, C. S. Science 2011, 333, 1613–1616. doi:10.1126/science.1208839
Return to citation in text: [1] -
Liu, Z.-Q.; Zhang, Y.; Zhao, L.; Li, Z.; Wang, J.; Li, H.; Wu, L.-M. Org. Lett. 2011, 13, 2208–2211. doi:10.1021/ol200372y
Return to citation in text: [1] -
Zhang, W.; Zheng, A.; Liu, Z.; Yang, L.; Liu, Z. Tetrahedron Lett. 2005, 46. doi:10.1016/j.tetlet.2005.06.097
Return to citation in text: [1] -
Zhang, W.; Huang, L.; Wang, J. Synthesis 2006, 2053–2063. doi:10.1055/s-2006-942372
Return to citation in text: [1] -
Zhou, Y.; Qian, L.; Zhang, W. Synlett 2009, 843–847. doi:10.1055/s-0028-1087955
Return to citation in text: [1] [2] -
Yazici, A.; Pyne, S. G. Synthesis 2009, 339–368. doi:10.1055/s-0028-1083325
Return to citation in text: [1] [2] -
Yazici, A.; Pyne, S. G. Synthesis 2009, 513–541. doi:10.1055/s-0028-1083346
Return to citation in text: [1] [2] -
Speckamp, W. N.; Moolenaar, M. J. Tetrahedron 2000, 56, 3817–3856. doi:10.1016/S0040-4020(00)00159-9
Return to citation in text: [1] -
Maryanoff, B. E.; Zhang, H.-C.; Cohen, J. H.; Turchi, I. J.; Maryanoff, C. A. Chem. Rev. 2004, 104, 1431–1628. doi:10.1021/cr0306182
Return to citation in text: [1] -
Angst, C. Pure Appl. Chem. 1987, 59, 373–380. doi:10.1351/pac198759030373
Return to citation in text: [1] [2] -
Thaning, M.; Wistrand, L.-G. Acta Chem. Scand. 1992, 46, 194–199. doi:10.3891/acta.chem.scand.46-0194
Return to citation in text: [1] [2] -
McClure, K. F.; Renold, P.; Kemp, D. S. J. Org. Chem. 1995, 60, 454–457. doi:10.1021/jo00107a028
Return to citation in text: [1] -
Signore, G.; Malanga, C.; Menicagli, R. Tetrahedron 2008, 64, 197–203. doi:10.1016/j.tet.2007.10.077
Return to citation in text: [1] [2] -
Kodama, T.; Moquist, P. N.; Schaus, S. E. Org. Lett. 2011, 13, 6316–6319. doi:10.1021/ol2028702
Return to citation in text: [1] [2] -
Morgan, I. R.; Yazici, A.; Pyne, S. G. Tetrahedron 2008, 64, 1409–1419. doi:10.1016/j.tet.2007.11.046
Return to citation in text: [1] [2] -
Scherlach, K.; Schuemann, J.; Dahse, H.-M.; Hertweck, C. J. Antibiot. 2010, 63, 375–377. doi:10.1038/ja.2010.46
Return to citation in text: [1] -
Chen, J.; Huang, P.-Q.; Queneau, Y. J. Org. Chem. 2009, 74, 7457–7463. doi:10.1021/jo901557h
Return to citation in text: [1] -
Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Org. Biomol. Chem. 2007, 5, 1466–1471. doi:10.1039/B701661A
Return to citation in text: [1] -
Sorbera, L. A.; Leeson, P. A.; Silvestre, J.; Castaner, J. Drugs Future 2001, 26, 651–657. doi:10.1358/dof.2001.026.07.630003
Return to citation in text: [1] -
Wada, T.; Fukuda, N. Psychopharmacology (Berlin) 1991, 103, 314–322. doi:10.1007/BF02244284
Return to citation in text: [1] -
Kawasuji, T.; Fuji, M.; Yoshinaga, T.; Sato, A.; Fujiwara, T.; Kiyama, R. Bioorg. Med. Chem. 2007, 15, 5487–5492. doi:10.1016/j.bmc.2007.05.052
Return to citation in text: [1] -
Zhuang, Z.-P.; Kung, M.-P.; Mu, M.; Kung, H. F. J. Med. Chem. 1998, 41, 157–166. doi:10.1021/jm970296s
Return to citation in text: [1] -
Norman, M. H.; Minick, D. J.; Rigdon, G. C. J. Med. Chem. 1996, 39, 149–157. doi:10.1021/jm9502201
Return to citation in text: [1] -
De Clercq, E. J. Med. Chem. 1995, 38, 2491–2517. doi:10.1021/jm00014a001
Return to citation in text: [1] -
Cho, C. S.; Wu, X.; Jiang, L. H.; Shim, S. C.; Choi, H.-J.; Kim, T. J. J. Heterocycl. Chem. 1998, 35, 265–268. doi:10.1002/jhet.5570350147
Return to citation in text: [1] -
Horii, Z.-I.; Iwata, C.; Tamura, Y. J. Org. Chem. 1961, 26, 2273–2276. doi:10.1021/jo01351a031
Return to citation in text: [1] -
Mase, N.; Nishi, T.; Hiyoshi, M.; Ichihara, K.; Bessho, J.; Yoda, H.; Takabe, K. J. Chem. Soc., Perkin Trans. 1 2002, 707–709. doi:10.1039/B200729K
Return to citation in text: [1] -
Mikami, K.; Kishino, H. J. Chem. Soc., Chem. Commun. 1993, 1843–1844. doi:10.1039/C39930001843
Return to citation in text: [1] -
Mikami, K.; Shimizu, M. Chem. Rev. 1992, 92, 1021–1050. doi:10.1021/cr00013a014
Return to citation in text: [1] -
Mayr, H.; Kempf, B.; Ofial, A. R. Acc. Chem. Res. 2003, 36, 66–77. doi:10.1021/ar020094c
Return to citation in text: [1]
| 1. | Ren, K.; Li, P.; Wang, L.; Zhang, X. Tetrahedron 2011, 67, 2753–2759. doi:10.1016/j.tet.2011.02.050 |
| 2. | Wang, T.; Chen, X.-L.; Chen, L.; Zhan, Z.-P. Org. Lett. 2011, 13, 3324–3327. doi:10.1021/ol201054z |
| 3. | Xiang, S.-K.; Zhang, L.-H.; Jiao, N. Chem. Commun. 2009, 6487–6489. doi:10.1039/B911905A |
| 11. | Liu, Z.-Q.; Zhang, Y.; Zhao, L.; Li, Z.; Wang, J.; Li, H.; Wu, L.-M. Org. Lett. 2011, 13, 2208–2211. doi:10.1021/ol200372y |
| 22. | Signore, G.; Malanga, C.; Menicagli, R. Tetrahedron 2008, 64, 197–203. doi:10.1016/j.tet.2007.10.077 |
| 10. | Lee, D.-H.; Kwon, K.-H.; Yi, C. S. Science 2011, 333, 1613–1616. doi:10.1126/science.1208839 |
| 23. | Kodama, T.; Moquist, P. N.; Schaus, S. E. Org. Lett. 2011, 13, 6316–6319. doi:10.1021/ol2028702 |
| 7. | Liu, P. N.; Dang, L.; Wang, Q. W.; Zhao, S. L.; Xia, F.; Ren, Y. J.; Gong, X. Q.; Chen, J. Q. J. Org. Chem. 2010, 75, 5017–5030. doi:10.1021/jo100517k |
| 8. | Reddy, C. R.; Vijaykumar, J.; Grée, R. Synthesis 2010, 3715–3723. doi:10.1055/s-0030-1258214 |
| 9. | Theerthagiri, P.; Lalitha, A. Tetrahedron Lett. 2010, 51, 5454–5458. doi:10.1016/j.tetlet.2010.08.019 |
| 4. | Wu, Y.-C.; Li, H.-J.; Liu, L.; Demoulin, N.; Liu, Z.; Wang, D.; Chen, Y.-J. Adv. Synth. Catal. 2011, 353, 907–912. doi:10.1002/adsc.201000930 |
| 5. | Sanz, R.; Miguel, D.; Martínez, A.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; Álvarez, E.; Rodríguez, F. Eur. J. Org. Chem. 2010, 7027–7039. doi:10.1002/ejoc.201001055 |
| 6. | Davoust, M.; Kitching, J. A.; Fleming, M. J.; Lautens, M. Chem.–Eur. J. 2010, 16, 50–54. doi:10.1002/chem.200902694 |
| 20. | Thaning, M.; Wistrand, L.-G. Acta Chem. Scand. 1992, 46, 194–199. doi:10.3891/acta.chem.scand.46-0194 |
| 22. | Signore, G.; Malanga, C.; Menicagli, R. Tetrahedron 2008, 64, 197–203. doi:10.1016/j.tet.2007.10.077 |
| 18. | Maryanoff, B. E.; Zhang, H.-C.; Cohen, J. H.; Turchi, I. J.; Maryanoff, C. A. Chem. Rev. 2004, 104, 1431–1628. doi:10.1021/cr0306182 |
| 23. | Kodama, T.; Moquist, P. N.; Schaus, S. E. Org. Lett. 2011, 13, 6316–6319. doi:10.1021/ol2028702 |
| 24. | Morgan, I. R.; Yazici, A.; Pyne, S. G. Tetrahedron 2008, 64, 1409–1419. doi:10.1016/j.tet.2007.11.046 |
| 15. | Yazici, A.; Pyne, S. G. Synthesis 2009, 339–368. doi:10.1055/s-0028-1083325 |
| 16. | Yazici, A.; Pyne, S. G. Synthesis 2009, 513–541. doi:10.1055/s-0028-1083346 |
| 17. | Speckamp, W. N.; Moolenaar, M. J. Tetrahedron 2000, 56, 3817–3856. doi:10.1016/S0040-4020(00)00159-9 |
| 12. | Zhang, W.; Zheng, A.; Liu, Z.; Yang, L.; Liu, Z. Tetrahedron Lett. 2005, 46. doi:10.1016/j.tetlet.2005.06.097 |
| 13. | Zhang, W.; Huang, L.; Wang, J. Synthesis 2006, 2053–2063. doi:10.1055/s-2006-942372 |
| 14. | Zhou, Y.; Qian, L.; Zhang, W. Synlett 2009, 843–847. doi:10.1055/s-0028-1087955 |
| 20. | Thaning, M.; Wistrand, L.-G. Acta Chem. Scand. 1992, 46, 194–199. doi:10.3891/acta.chem.scand.46-0194 |
| 21. | McClure, K. F.; Renold, P.; Kemp, D. S. J. Org. Chem. 1995, 60, 454–457. doi:10.1021/jo00107a028 |
| 31. | Zhuang, Z.-P.; Kung, M.-P.; Mu, M.; Kung, H. F. J. Med. Chem. 1998, 41, 157–166. doi:10.1021/jm970296s |
| 32. | Norman, M. H.; Minick, D. J.; Rigdon, G. C. J. Med. Chem. 1996, 39, 149–157. doi:10.1021/jm9502201 |
| 33. | De Clercq, E. J. Med. Chem. 1995, 38, 2491–2517. doi:10.1021/jm00014a001 |
| 25. | Scherlach, K.; Schuemann, J.; Dahse, H.-M.; Hertweck, C. J. Antibiot. 2010, 63, 375–377. doi:10.1038/ja.2010.46 |
| 26. | Chen, J.; Huang, P.-Q.; Queneau, Y. J. Org. Chem. 2009, 74, 7457–7463. doi:10.1021/jo901557h |
| 27. | Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Org. Biomol. Chem. 2007, 5, 1466–1471. doi:10.1039/B701661A |
| 28. | Sorbera, L. A.; Leeson, P. A.; Silvestre, J.; Castaner, J. Drugs Future 2001, 26, 651–657. doi:10.1358/dof.2001.026.07.630003 |
| 29. | Wada, T.; Fukuda, N. Psychopharmacology (Berlin) 1991, 103, 314–322. doi:10.1007/BF02244284 |
| 30. | Kawasuji, T.; Fuji, M.; Yoshinaga, T.; Sato, A.; Fujiwara, T.; Kiyama, R. Bioorg. Med. Chem. 2007, 15, 5487–5492. doi:10.1016/j.bmc.2007.05.052 |
| 39. | Mayr, H.; Kempf, B.; Ofial, A. R. Acc. Chem. Res. 2003, 36, 66–77. doi:10.1021/ar020094c |
| 14. | Zhou, Y.; Qian, L.; Zhang, W. Synlett 2009, 843–847. doi:10.1055/s-0028-1087955 |
| 15. | Yazici, A.; Pyne, S. G. Synthesis 2009, 339–368. doi:10.1055/s-0028-1083325 |
| 16. | Yazici, A.; Pyne, S. G. Synthesis 2009, 513–541. doi:10.1055/s-0028-1083346 |
| 37. | Mikami, K.; Kishino, H. J. Chem. Soc., Chem. Commun. 1993, 1843–1844. doi:10.1039/C39930001843 |
| 38. | Mikami, K.; Shimizu, M. Chem. Rev. 1992, 92, 1021–1050. doi:10.1021/cr00013a014 |
| 35. | Horii, Z.-I.; Iwata, C.; Tamura, Y. J. Org. Chem. 1961, 26, 2273–2276. doi:10.1021/jo01351a031 |
| 36. | Mase, N.; Nishi, T.; Hiyoshi, M.; Ichihara, K.; Bessho, J.; Yoda, H.; Takabe, K. J. Chem. Soc., Perkin Trans. 1 2002, 707–709. doi:10.1039/B200729K |
| 24. | Morgan, I. R.; Yazici, A.; Pyne, S. G. Tetrahedron 2008, 64, 1409–1419. doi:10.1016/j.tet.2007.11.046 |
| 34. | Cho, C. S.; Wu, X.; Jiang, L. H.; Shim, S. C.; Choi, H.-J.; Kim, T. J. J. Heterocycl. Chem. 1998, 35, 265–268. doi:10.1002/jhet.5570350147 |
© 2012 Lu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)