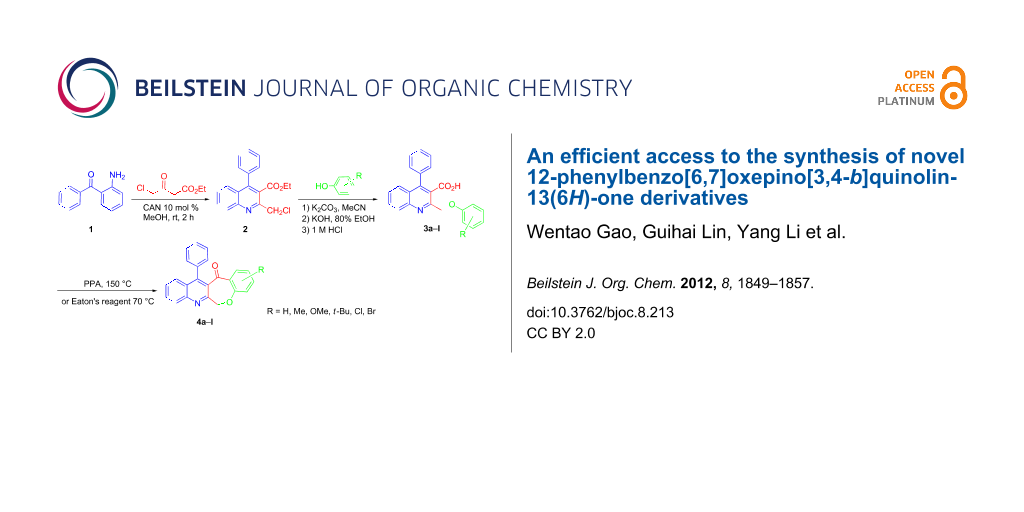
Abstract
An efficient access to the tetracyclic-fused quinoline systems, 12-phenylbenzo[6,7]oxepino[3,4-b]quinolin-13(6H)-one derivatives 4a–l, is described, involving the intramolecular Friedel–Crafts acylation reaction of 2-(phenoxymethyl)-4-phenylquinoline-3-carboxylic acid derivatives 3a–l aided by the treatment with PPA (polyphosphoric acid) or Eaton’s reagent. The required starting compound (2) was obtained by Friedländer reaction of 2-aminobenzophenone (1) with 4-chloroethylacetoacetate by using CAN (cerium ammonium nitrate, 10 mol %) as catalyst at room temperature. The substrates 3a–l were prepared through one-pot reaction of ethyl 2-(chloromethyl)-4-phenylquinoline-3-carboxylate (2) and substituted phenols. Our developed strategy, involving a three-step route, offers easy access to tetracyclic-fused quinoline systems in short reaction times, and the products are obtained in moderate to good yields.

Graphical Abstract
Introduction
Polycyclic heterocycle-fused quinoline systems as important group compounds can be found in many biologically active natural products as well as in pharmacologically significant molecules, and have wide applications in medicinal chemistry [1-4]. It has been well-established that planar heterocycle-fused tri- or tetracyclic quinoline systems on privileged templates have significant biological properties, such as antitumoral [5,6], anti-inflammatory [7], antimalarial [8], antituberculosis [9], and antiplasmodial [10] activities. Accordingly, the synthesis of new families of such quinoline systems still attracts much interest from both medicinal and synthetic organic chemists [11-14]. Most reports in the literature contain a common five- or six-membered heterocycle fused to a quinoline ring, such as pyrazolo [15], pyrano [16], indolo [12,17], benzofuro [18], benzothienoquinolines [19], and synthetic analogues thereof. However, to the best of our knowledge, there are very few reports in which a medium-sized seven-membered benzoxepin ring is fused to a quinoline unit. In this context, Bera et al. [20] described a one-pot method for the synthesis of 6,7-dihydrobenzo[2,3]oxepino[4,5-b]quinolin-12-ols. However, the report is of episodic character and no efforts have been made to develop a general synthetic approach. Furthermore, the reported approach features a major restriction in the use of the expensive and unavailable 5-chloro-2,3-dihydrobenzo[b]oxepine-4-carbaldehyde as a reactant. Thus, a facile synthesis of such compounds by using inexpensive and readily available materials represents a challenging area for exploration.
On the other hand, the seven-membered benzoxepine nucleus can be found in many medicinally relevant natural products and synthetic compounds and represents one of the most profiled chemotypes in modern drug discovery, owing to several pronounced biological activities, such as antitumor and anti-inflammatory properties, attributed to the presence of the benzoxepine unit [21-24]. Last but not least, the benzoxepine nucleus has been of increasing relevance as a synthetic building block for the synthesis of manifold biologically and pharmaceutically important compounds [25,26]. As a consequence, the remarkable bioactivity surrounding the benzoxepine moiety has elicited a significant amount of interest as demonstrated by synthetic work already published [27-30].
In light of the above findings as well as the combination principles for drug design [31], we were intrigued to explore the incorporation of a quinoline ring fused together with a benzoxepine nucleus, which would be much more attractive and valuable for medicinal chemistry and drug discovery. In recent years, our research team has been interested in the development of efficient synthesis for the quinoline-based bioactive molecules [32-37]. Thus, in connection with our continuing interest in the synthesis of highly valuable quinoline compounds, we are actively involved in diversifying our work on the synthesis of hetero-fused quinoline systems that are of interest for medical research. Thus, the aim of the present work is to present the synthesis of novel compounds combining these two bioactive components in a molecular frame work as fused forms.
Results and Discussion
The synthetic methodology developed in our laboratory for the synthesis of a new class of benzoxepino-fused quinoline compounds was achieved in a three-step procedure, commencing with the preparation of ethyl 2-(chloromethyl)-4-phenylquinoline-3-carboxylate (2) as shown in Scheme 1.
![[1860-5397-8-213-i1]](/bjoc/content/inline/1860-5397-8-213-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of ethyl 2-(chloromethyl)-4-phenylquinoline-3-carboxylate (2).
Scheme 1: Synthesis of ethyl 2-(chloromethyl)-4-phenylquinoline-3-carboxylate (2).
In fact, from the beginning, we were well aware that the preparation of ethyl 2-(chloromethyl)-4-phenylquinoline-3-carboxylate (2) is very interesting because such compounds are viewed as ideal starting materials for the flexible synthesis of a large range of quinoline derivatives [38-40]. Recently, Mizuno et al. [38] reported the preparation of ethyl 2-(chloromethyl)-4-(3,4-dimethoxyphenyl)-6,7-dimethoxy quinoline-3-carboxylate by the reaction of ethyl 4-chloroacetoacetate with 2-amino-3’,4,4’,5-tetramethoxybenzophenone hydrochloride in ethanol. However, in our case the reaction of 2-aminobenzophenone (1) with ethyl 4-chloroacetoacetate did not take place, and a gummy mass was obtained as product. In this context, Muscia et al. [39] described the synthesis of ethyl 6-chloro-2-(chloromethyl)-4-phenylquinoline-3-carboxylate by the Friedländer reaction employing microwave irradiation (MW) in the presence of a catalytic amount of hydrochloric acid. Subsequently, we reported a similar reaction under ultrasound irradiation conditions by using KHSO4 as catalyst [36]. Although the two methodologies are elegant and impressive, our attempts to follow both routes to synthesize 2 were frustrated by very low yields. In this regard, Bose et al. [40] reported the preparation of ethyl 6-chloro-2-(chloromethyl)-4-phenylquinoline-3-carboxylate by the treatment of 2-amino-5-chlorobenzophenone with 4-(chloroethyl)acetoacetate in MeOH by using 10 mol % CAN as catalyst at room temperature. To our delight, under similar reaction conditions, we were able to obtain 2 from 2-aminobenzophenone (1) and 4-(chloroethyl)acetoacetate in a good yield of 84% (Scheme 1). Moreover, the obtained product was very pure, and a chromatographic purification was unnecessary.
Next, the resulting 2-(chloromethyl)quinoline 2 was subjected to the Williamson reaction with a variety of phenols with varying substituents in the presence of K2CO3 as base in MeCN under reflux as shown in Scheme 2.
![[1860-5397-8-213-i2]](/bjoc/content/inline/1860-5397-8-213-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2-(phenoxymethyl)-4-phenylquinoline-3-carboxylic acid derivatives 3a–l.
Scheme 2: Synthesis of 2-(phenoxymethyl)-4-phenylquinoline-3-carboxylic acid derivatives 3a–l.
In this reaction, we adopted MeCN as the solvent of choice simply because of its low boiling point, bringing much convenience to the workup procedure. Thus, upon the completion of the Williamson reaction as observed on TLC, MeCN was simply evaporated to dryness, 80% ethanolic potassium hydroxide solution (15 mL) was directly added to the residue, and the resulting reaction mixture was stirred under reflux. When the reactions were completed (usually within four hours) the quinoline-3-carboxylic acid ether compounds were obtained in high yields (80–93%) after simple recrystallization from ethanol. The structures assigned to these compounds are confirmed by spectral data and elemental analysis, which were fully consistent with the assigned molecular structure as depicted in Supporting Information File 1. The beauty of this reaction is that two chemical transformations, i.e., Williamson ether synthesis and subsequent ester hydrolysis take place in one-pot, thereby providing the acids in good yields of 66–93% with operational and experimental simplicity. Moreover, the presence of sterically hindered tert-butyl groups is not problematic although slightly lower yields were obtained when the aryl-ring was substituted in o-position by a tert-butyl group. The scope and generality of the newly synthesized compounds 3a–l during the present investigation are listed in Table 1 together with yields and melting points.
Table 1: Yields and melting points of compounds 3a–l.
| Entry | Product | Yield (%)a | Mp (°C) |
|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-213-i5.svg?max-width=637&scale=1.0)
3a |
83 | 191–192 |
| 2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-213-i6.svg?max-width=637&scale=1.0)
3b |
91 | 209–210 |
| 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-213-i7.svg?max-width=637&scale=1.0)
3c |
88 | 212–213 |
| 4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-213-i8.svg?max-width=637&scale=1.0)
3d |
93 | 207–208 |
| 5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-213-i9.svg?max-width=637&scale=1.0)
3e |
86 | 197–198 |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-213-i10.svg?max-width=637&scale=1.0)
3f |
83 | 167–168 |
| 7 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-213-i11.svg?max-width=637&scale=1.0)
3g |
88 | 165–166 |
| 8 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-213-i12.svg?max-width=637&scale=1.0)
3h |
82 | 196–197 |
| 9 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-213-i13.svg?max-width=637&scale=1.0)
3i |
71 | 192–194 |
| 10 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-213-i14.svg?max-width=637&scale=1.0)
3j |
85 | 188–190 |
| 11 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-213-i15.svg?max-width=637&scale=1.0)
3k |
66 | 230–232 |
| 12 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-213-i16.svg?max-width=637&scale=1.0)
3l |
82 | 239–241 |
aIsolated yield.
As shown in Table 1, high yields of 3 were achieved irrespective of the electronic nature or positions of the substituents, except for in the cases of 3i and 3k (Table 1, entries 9 and 11). The relatively lower yields of 3i and 3k may be ascribed to the sterically hindered nature of the bulky tert-butyl group at the o-position of aryl.
Thus, the resulting substrates, quinoline-3-carboxylic acids 3a–l, further served as active synthons for the intramolecular Friedel–Crafts acylation reaction to construct the desired tetracyclic benzoxepino-fused quinoline systems. Of the commonly available cyclization agents screened for the intramolecular Friedel–Crafts acylation reaction (e.g., AlCl3, H2SO4, p-TsOH, TiCl4, P2O5), the use of inexpensive and readily available polyphosphoric acid (PPA), requiring no additional solvent, was found to be very suitable for such a reaction in terms of good yield, short reaction time and simple workup [37,41]. Upon use of PPA as the cyclization agent, we found that the cyclization reaction of 3a–h could be performed smoothly at 150 °C as shown in Scheme 3.
![[1860-5397-8-213-i3]](/bjoc/content/inline/1860-5397-8-213-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 12-phenylbenzo[6,7]oxepino[3,4-b]quinolin-13(6H)-ones 4a–h.
Scheme 3: Synthesis of 12-phenylbenzo[6,7]oxepino[3,4-b]quinolin-13(6H)-ones 4a–h.
After the reaction was completed (monitored by TLC), the reaction mixture was poured into cold water to induce precipitation, followed by neutralization with NaHCO3 solution. Thus, the cyclized products 4a–h were obtained in good yields, ranging from 69–85% after recrystallization from ethanol, and their identities were unequivocally ascertained from their satisfactory elemental and spectral data. The compounds 4a–h, newly synthesized in the present investigation, are listed in Table 2.
Table 2: Structures and yields of the cyclized products 4a–h.
| Entry | Product | Time (h) | Yield (%)a | Mp (°C) |
|---|---|---|---|---|
| 1 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-213-i17.svg?max-width=637&scale=1.0)
4a |
6 | 77 | 197–198 |
| 2 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-213-i18.svg?max-width=637&scale=1.0)
4b |
5 | 80 | 215–216 |
| 3 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-213-i19.svg?max-width=637&scale=1.0)
4c |
5 | 83 | 199–200 |
| 4 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-213-i20.svg?max-width=637&scale=1.0)
4d |
5 | 82 | 195–197 |
| 5 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-213-i21.svg?max-width=637&scale=1.0)
4e |
5 | 85 | 197–199 |
| 6 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-213-i22.svg?max-width=637&scale=1.0)
4f |
7 | 74 | 175–177 |
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-213-i23.svg?max-width=637&scale=1.0)
4g |
7 | 69 | 171–172 |
| 8 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-213-i24.svg?max-width=637&scale=1.0)
4h |
7 | 72 | 179–180 |
aIsolated yield.
As shown in Table 2, the cyclization reaction appears to be generally applicable, as most of the substrates 3 were consumed within 5–7 hours to give the corresponding cyclized products with reasonable yields. However, the method was limited due to difficulties in tolerating the tert-butyl groups. For example, when substrate 3l was treated by PPA under the reaction conditions, the resulting product was assigned not to the expected 4l, but characterized as the de-tert-butylation product 4l′ as shown in Scheme 4. The product was easily characterized from its 1H NMR spectrum, which showed no signals attributable to the carboxylic acid proton and tert-butyl protons of its precursor 3l, along with the presence of a total count of twelve aromatic protons between 7.15–8.20 ppm, perfectly matched with their structures with additional support from its 13C NMR spectrum and other analytical data.
![[1860-5397-8-213-i4]](/bjoc/content/inline/1860-5397-8-213-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cyclization and de-tert-butylation reaction of 3l by using PPA.
Scheme 4: Cyclization and de-tert-butylation reaction of 3l by using PPA.
Similarly, in the present investigation the substrates 3i–k also showed the same reaction patterns and the obtained product was identified as being the same structure as 4a. Obviously, the problem with this is the loss of the tert-butyl group during the cyclization reaction course. In addition, slow and gradual warming to the reaction temperature was also tried, but this had no effect on the outcome of the reaction. Thus, according to our previous experience [33], coupled with the fact that the tert-butyl substituent could be easily removed from aromatic nuclei by Friedel–Crafts reaction at a high reaction temperature using a high-boiling-point solvent [42], we presumed the occurrence of debutylation reaction during the cyclization course probably due to the high reaction temperature.
Considering these results, an alternative route would be desirable. Recently, the Eaton’s reagent, a mixture of P2O5 and MeSO3H [43], has gained wide application as an advantageous medium for cyclization reactions [44], and we have also reported the serendipitous discovery of its excellent performance in these types of transformations [32,33,45]. Thus, we resorted to the use of our reliable approach to conduct the cyclization reaction. Under the same experimental conditions as in our previous methods, the ring closure of compounds 3i–l was achieved and the corresponding cyclized products retaining the tert-butyl moiety were afforded in satisfactory yields of 59–70%. The reaction results are summarized in Table 3.
Table 3: Synthesis of tert-butyl-substituted compounds 4i–l.
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-213-i25.svg?max-width=637&scale=1.0)
|
||||
| Entry | Product | Time (h) | Yield (%)a | Mp (°C) |
|---|---|---|---|---|
| 1 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-213-i26.svg?max-width=637&scale=1.0)
4i |
6 | 68 | 193–195 |
| 2 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-213-i27.svg?max-width=637&scale=1.0)
4j |
5 | 70 | 210–212 |
| 3 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-213-i28.svg?max-width=637&scale=1.0)
4k |
5 | 62 | 166–168 |
| 4 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-8-213-i29.svg?max-width=637&scale=1.0)
4l |
5 | 59 | 209–210 |
aIsolated yield.
As shown in Table 3, by the treatment with Eaton’s reagent, the tert-butyl-substituted quinoline-3-carboxylic acids 3i–l underwent the conversion smoothly and gave the corresponding cyclized products 4i–l as expected. Compounds 4i–l containing tert-butyl groups are interesting candidates for medicinal applications since it was reported that the introduction of tert-butyl groups into organic molecules could increase the lipophilicity of the molecule, which is very important for allowing passage through the extraordinarily thick and tight cell wall [46]. Altogether, the strategy using Eaton’s reagent instead of PPA showed a satisfactory conversion and gave us easy access to mono- and di-tert-butyl-substituted tetracyclic-fused quinoline systems. In addition, it is noteworthy that we also attempted additional experiments by conducting the cyclization reaction of 3a–l under the given conditions (Eaton’s reagent, 70 °C). Although the cyclization reaction proceeded, the yields of the desired products were not as good as with PPA.
Conclusion
In conclusion, we have described the synthesis of a series of structurally new 12-phenylbenzo[6,7]oxepino[3,4-b]quinolin-13(6H)-ones 4a–l. The advantages of the current protocol include the ready availability of starting materials, ease of experimental operation, and satisfactory yields, which contribute to the usefulness of this method. These compounds belong to a new class of linearly fused tetracyclic heterocyclic quinoline systems, which could be potentially applied for the development of biologically and pharmaceutically important compounds. Access to such biologically intriguing structures should allow us to study their biological activities, and currently we are exploring this possibility.
Experimental
All chemicals (AR graded) were commercially available and used without further purification. The melting points were determined by using a WRS-1B melting-points apparatus and were uncorrected. The IR spectra were obtained as KBr pellets in the range of 400–4000 cm−1 on a Shimadzu FTIR-8400S spectrophotometer (Shimadzu, Japan). 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE NMR spectrometer with CDCl3 or DMSO-d6 as the solvent. The reported chemical shifts (δ values) are given in parts per million (ppm) downfield from tetramethylsilane (TMS) as the internal standard. Mass spectra were determined on a MSD VL ESI1 spectrometer. Elemental analysis was recorded on an Elementar vario EL-III element analyzer.
Procedure for the preparation of 2-(chloromethyl)-4-phenylquinoline-3-carboxylate (2) [40]. To a stirred solution of 2-aminobenzophenone (1, 1.97 g, 10 mmol) and ethyl 4-chloroacetoacetate (1.65 g, 10 mmol) in methanol (15 mL), was added CAN (0.55 g, 1 mmol, 10 mol %). The resulting reaction mixture was stirred at room temperature for 2 h. After the reaction was completed (monitored by TLC), the mixture was washed with water (15 mL), extracted with ethyl acetate (30 × 2 mL), dried over Na2SO4, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with petroleum ether/EtOAc (5:1) as eluent to afford 2.74 g of the product 2 as yellow crystals in 84% yield. Mp 103–105 °C (lit. [47] mp 109–111 °C).
General procedure for the synthesis of 2-(phenoxymethyl)-4-phenylquinoline-3-carboxylic acid derivatives 3a–l. A mixture of ethyl 2-(chloromethyl)-4-phenylquinoline-3-carboxylate (0.325 g, 1 mmol), substituted phenol (1 mmol) and K2CO3 (0.414 g, 3 mmol) was stirred in CH3CN (10 mL) under reflux. After completion of the reaction (monitored by TLC), CH3CN was evaporated to dryness. Then, a solution of KOH (2.8 g, 20 mmol) in 80% ethanol (15 mL) was added to the residue, and the mixture was heated under reflux for 4 h, cooled, and acidified with 1 M hydrochloric acid solution. The resulting crude product was recrystallized from ethanol to afford 3a–l. The yields and melting points of all compounds are summarized in Table 1 and the spectral and analytical data are given in Supporting Information File 1.
General procedure for the synthesis of 12-phenylbenzo[6,7]oxepino[3,4-b]quinolin-13(6H)-one derivatives 4a–h. The precursors 2-(phenoxymethyl)-4-phenylquinoline-3-carboxylic acid derivatives (3a–h, 0.5 mmol) and PPA (10 g) were added to a round flask (25 mL) and stirred at 150 °C for 5–7 h. The conversion was monitored by TLC. After the reaction was completed, the reaction mixture was poured slowly into cold water under stirring to induce precipitation, followed by neutralization with NaHCO3 solution. The obtained crude products were recrystallized from ethanol to afford products 4a–h. The yields are summarized in Table 2 and the spectra and analytical data are given in Supporting Information File 1.
General procedure for the synthesis of tert-butyl-substituted 12-phenylbenzo[6,7]oxepino[3,4-b]quinolin-13(6H)-one derivatives 4i–l. The tert-butyl-substituted precursors 2-(phenoxymethyl)-4-phenylquinoline-3-carboxylic acid derivatives (3i–l, 0.5 mmol) and Eaton’s reagent (5 mL) were added to a round flask (10 mL) and stirred at 70 °C for 5–6 h. The conversion was monitored by TLC. After the reaction was completed, the reaction mixture was poured slowly into cold water under stirring to induce precipitation, followed by neutralization with NaHCO3 solution. The crude products were obtained after filtration and washing with water. The pure products 4i–l were obtained by recrystallization from ethanol. The yields are summarized in Table 3 and the spectra and analytical data are given in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Characterization data of the title compounds and NMR and HRMS spectra. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Moyer, M. P.; Weber, F. H.; Gross, J. L. J. Med. Chem. 1992, 35, 4595–4601. doi:10.1021/jm00102a013
Return to citation in text: [1] -
Gerster, J. F.; Lindstrom, K. J.; Miller, R. L.; Tomai, M. A.; Birmachu, W.; Bomersine, S. N.; Gibson, S. J.; Imbertson, L. M.; Jacobson, J. R.; Knafla, R. T.; Maye, P. V.; Nikolaides, N.; Oneyemi, F. Y.; Parkhurst, G. J.; Pecore, S. E.; Reiter, M. J.; Scribner, L. S.; Testerman, T. L.; Thompson, N. J.; Wagner, T. L.; Weeks, C. E.; Andre, J.-D.; Lagain, D.; Bastard, Y.; Lupu, M. J. Med. Chem. 2005, 48, 3481–3491. doi:10.1021/jm049211v
Return to citation in text: [1] -
Miert, S. V.; Miert, S. V.; Hostyn, S.; Maes, B. U. W.; Cimanga, K.; Brun, R.; Kaiser, M.; Mátyus, P.; Dommisse, R.; Lemière, G.; Vlietinck, A.; Pieters, L. J. Nat. Prod. 2005, 68, 674–677. doi:10.1021/np0496284
Return to citation in text: [1] -
Chilin, A.; Marzaro, G.; Marzano, C.; Via, L. D.; Ferlin, M. G.; Pastorini, G.; Guiotto, A. Bioorg. Med. Chem. 2009, 17, 523–529. doi:10.1016/j.bmc.2008.11.072
Return to citation in text: [1] -
Yamato, M.; Takeuchi, Y.; Hashigaki, K.; Ikeda, Y.; Chang, M. R.; Takeuchi, K.; Matsushima, M.; Tsuruo, T.; Tashiro, T. J. Med. Chem. 1989, 32, 1295–1300. doi:10.1021/jm00126a025
Return to citation in text: [1] -
Sánchez, I.; Reches, R.; Caignard, D. H.; Renard, P.; Pujol, M. D. Eur. J. Med. Chem. 2006, 41, 340–342. doi:10.1016/j.ejmech.2005.11.006
Return to citation in text: [1] -
Chen, Y.-L.; Chen, I.-L.; Lu, C.-M.; Tzeng, C.-C.; Tsao, L.-T.; Wang, J.-P. Bioorg. Med. Chem. 2004, 12, 387–392. doi:10.1016/j.bmc.2003.10.051
Return to citation in text: [1] -
Wright, C. W.; Addae-Kyereme, J.; Breen, A. G.; Brown, J. E.; Cox, M. F.; Croft, S. L.; Gökçek, Y.; Kendrick, H.; Phillips, R. M.; Pollet, P. L. J. Med. Chem. 2001, 44, 3187–3194. doi:10.1021/jm010929+
Return to citation in text: [1] -
Eswaran, S.; Adhikari, A. V.; Kumar, R. A. Eur. J. Med. Chem. 2010, 45, 957–966. doi:10.1016/j.ejmech.2009.11.036
Return to citation in text: [1] -
Jonckers, T. H. M.; van Miert, S.; Cimanga, K.; Bailly, C.; Colson, P.; De Pauw-Gillet, M. C.; van den Heuvel, H.; Claeys, M.; Lemière, F.; Esmans, E. L.; Rozenski, J.; Quirijnen, L.; Maes, L.; Dommisse, R.; Lemière, G. L. F.; Vlietinck, A.; Pieters, L. J. Med. Chem. 2002, 45, 3497–3508. doi:10.1021/jm011102i
Return to citation in text: [1] -
Zhong, W.; Ma, W.; Liu, Y. Tetrahedron 2011, 67, 3509–3518. doi:10.1016/j.tet.2011.03.039
Return to citation in text: [1] -
Parvatkar, P. T.; Parameswaran, P. S.; Tilve, S. G. J. Org. Chem. 2009, 74, 8369–8372. doi:10.1021/jo901361x
Return to citation in text: [1] [2] -
Yu, F.; Yan, S.; Hu, L.; Wang, Y.; Lin, J. Org. Lett. 2011, 13, 4782–4785. doi:10.1021/ol201783d
Return to citation in text: [1] -
Singh, V.; Hutait, S.; Batra, S. Eur. J. Org. Chem. 2009, 3454–3466. doi:10.1002/ejoc.200900336
Return to citation in text: [1] -
Mali, J. R.; Pratap, U. R.; Jawale, D. V.; Mane, R. A. Tetrahedron Lett. 2010, 51, 3980–3982. doi:10.1016/j.tetlet.2010.05.117
Return to citation in text: [1] -
Kalita, P. K.; Baruah, B.; Bhuyan, P. J. Tetrahedron Lett. 2006, 47, 7779–7782. doi:10.1016/j.tetlet.2006.08.086
Return to citation in text: [1] -
Chen, Y. L.; Hung, H. M.; Lu, C. M.; Li, K. C.; Tzeng, C. C. Bioorg. Med. Chem. 2004, 12, 6539–6546. doi:10.1016/j.bmc.2004.09.025
Return to citation in text: [1] -
Yang, C.-L.; Tseng, C.-H.; Chen, Y.-L.; Lu, C.-M.; Kao, C.-L.; Wu, M.-H.; Tzeng, C.-C. Eur. J. Med. Chem. 2010, 45, 602–607. doi:10.1016/j.ejmech.2009.10.050
Return to citation in text: [1] -
David, E.; Pellet-Rostaing, S.; Lemaire, M. Tetrahedron 2007, 63, 8999–9006. doi:10.1016/j.tet.2007.05.110
Return to citation in text: [1] -
Bera, R.; Dhananjaya, G.; Singh, S. N.; Ramu, B.; Kiran, S. U.; Kumar, P. R.; Mukkanti, K.; Pal, M. Tetrahedron 2008, 64, 582–589. doi:10.1016/j.tet.2007.10.101
Return to citation in text: [1] -
Gruijters, B. W. T.; van Veldhuizen, A.; Weijers, C. A. G. M.; Wijnberg, J. B. P. A. J. Nat. Prod. 2002, 65, 558–561. doi:10.1021/np010510m
Return to citation in text: [1] -
Kahnberg, P.; Sterner, O. Tetrahedron 2001, 57, 7181–7184. doi:10.1016/S0040-4020(01)00640-8
Return to citation in text: [1] -
Staben, S. T.; Siu, M.; Goldsmith, R.; Olivero, A. G.; Do, S.; Burdick, D. J.; Heffron, T. P.; Dotson, J.; Sutherlin, D. P.; Zhu, B. Y.; Tsui, V.; Le, H.; Lee, L.; Lesnick, J.; Lewis, C.; Murray, J. M.; Nonomiya, J.; Pang, J.; Prior, W. W.; Salphati, L.; Rouge, L.; Sampath, D.; Sideris, S.; Wiesmann, C.; Wu, P. Bioorg. Med. Chem. Lett. 2011, 21, 4054–4058. doi:10.1016/j.bmcl.2011.04.124
Return to citation in text: [1] -
Liu, J.-h.; Steigel, A.; Reininger, E.; Bauer, R. J. Nat. Prod. 2000, 63, 403–405. doi:10.1021/np990362o
Return to citation in text: [1] -
Sugaya, T.; Kato, N.; Sakaguchi, A.; Tomioka, S. Synthesis 1995, 1257–1262. doi:10.1055/s-1995-4088
Return to citation in text: [1] -
Kumar, S.; Ila, H.; Junjappa, H. Tetrahedron 2007, 63, 10067–10076. doi:10.1016/j.tet.2007.07.045
Return to citation in text: [1] -
Lautens, M.; Paquin, J.-F.; Piguel, S. J. Org. Chem. 2002, 67, 3972–3974. doi:10.1021/jo025730z
Return to citation in text: [1] -
Arnold, L. A.; Luo, W.; Guy, R. K. Org. Lett. 2004, 6, 3005–3007. doi:10.1021/ol0487884
Return to citation in text: [1] -
Rotzoll, S.; Appel, B.; Langer, P. Tetrahedron Lett. 2005, 46, 4057–4059. doi:10.1016/j.tetlet.2005.04.009
Return to citation in text: [1] -
Barrett, I.; Meegan, M. J.; Hughes, R. B.; Carr, M.; Knox, A. J. S.; Artemenko, N.; Golfis, G.; Zisterer, D. M.; Lloyd, D. G. Bioorg. Med. Chem. 2008, 16, 9554–9573. doi:10.1016/j.bmc.2008.09.035
Return to citation in text: [1] -
Dolle, R. E.; Nelson, K. H., Jr. J. Comb. Chem. 1999, 1, 235–282. doi:10.1021/cc9900192
Return to citation in text: [1] -
Li, Y.; Chang, M. Q.; Gao, F.; Gao, W. T. J. Chem. Res. 2008, 640–641. doi:10.3184/030823408X375070
Return to citation in text: [1] [2] -
Li, Y.; Zhang, C. H.; Sun, M. C.; Gao, W. T. J. Heterocycl. Chem. 2009, 46, 1190–1194. doi:10.1002/jhet.203
Return to citation in text: [1] [2] [3] -
Gao, W. T.; Zhang, C. H.; Li, Y. J. Braz. Chem. Soc. 2010, 21, 806–812.
Return to citation in text: [1] -
Li, Y.; Gao, W. T. Beilstein J. Org. Chem. 2010, 6, 966–972. doi:10.3762/bjoc.6.108
Return to citation in text: [1] -
Gao, W. T.; Liu, J.; Jiang, Y.; Li, Y. Beilstein J. Org. Chem. 2011, 7, 210–217. doi:10.3762/bjoc.7.28
Return to citation in text: [1] [2] -
Gao, W. T.; Zhao, Z. G.; Li, Y.; Yan, Y.; Li, F. Chin. J. Chem. 2012, 30, 1127–1132. doi:10.1002/cjoc.201100420
Return to citation in text: [1] [2] -
Mizuno, M.; Inagaki, A.; Yamashita, M.; Soma, N.; Maeda, Y.; Nakatani, H. Tetrahedron 2006, 62, 4065–4070. doi:10.1016/j.tet.2006.02.021
Return to citation in text: [1] [2] -
Muscia, G. C.; Cazorla, S. I.; Frank, F. M.; Borosky, G. L.; Buldain, G. Y.; Asís, S. E.; Malchiodi, E. L. Eur. J. Med. Chem. 2011, 46, 3696–3703. doi:10.1016/j.ejmech.2011.05.035
Return to citation in text: [1] [2] -
Bose, D. S.; Idrees, M.; Jakka, N. M.; Rao, J. V. J. Comb. Chem. 2010, 12, 100–110. doi:10.1021/cc900129t
Return to citation in text: [1] [2] [3] -
Popp, F. D.; McEwen, W. E. Chem. Rev. 1958, 58, 321–401. doi:10.1021/cr50020a004
Return to citation in text: [1] -
Irlapati, N. R.; Baldwin, J. E.; Adlington, R. M.; Pritchard, G. J.; Cowley, A. R. Tetrahedron 2006, 62, 4603–4614. doi:10.1016/j.tet.2006.01.090
Return to citation in text: [1] -
Eaton, P. E.; Carlson, G. R.; Lee, J. T. J. Org. Chem. 1973, 38, 4071–4073. doi:10.1021/jo00987a028
Return to citation in text: [1] -
Zewge, D.; Chen, C. Y.; Deer, C.; Dormer, P. G.; Hughes, D. L. J. Org. Chem. 2007, 72, 4276–4279. doi:10.1021/jo070181o
Return to citation in text: [1] -
Gao, W.-T.; Hou, W.-D.; Zheng, M.-R.; Tang, L.-J. Synth. Commun. 2010, 40, 732–738. doi:10.1080/00397910903013713
Return to citation in text: [1] -
Vinsova, J.; Cermakova, K.; Tomeckova, A.; Ceckova, M.; Jampilek, J.; Cermak, P.; Kunes, J.; Dolezal, M.; Staud, F. Bioorg. Med. Chem. 2006, 14, 5850–5865. doi:10.1016/j.bmc.2006.05.030
Return to citation in text: [1] -
Stevenson, L.; Tavares, A. A. S.; Brunet, A.; McGonagle, F. I.; Dewar, D.; Pimlott, S. L.; Sutherland, A. Bioorg. Med. Chem. Lett. 2010, 20, 954–957. doi:10.1016/j.bmcl.2009.12.061
Return to citation in text: [1]
| 44. | Zewge, D.; Chen, C. Y.; Deer, C.; Dormer, P. G.; Hughes, D. L. J. Org. Chem. 2007, 72, 4276–4279. doi:10.1021/jo070181o |
| 32. | Li, Y.; Chang, M. Q.; Gao, F.; Gao, W. T. J. Chem. Res. 2008, 640–641. doi:10.3184/030823408X375070 |
| 33. | Li, Y.; Zhang, C. H.; Sun, M. C.; Gao, W. T. J. Heterocycl. Chem. 2009, 46, 1190–1194. doi:10.1002/jhet.203 |
| 45. | Gao, W.-T.; Hou, W.-D.; Zheng, M.-R.; Tang, L.-J. Synth. Commun. 2010, 40, 732–738. doi:10.1080/00397910903013713 |
| 46. | Vinsova, J.; Cermakova, K.; Tomeckova, A.; Ceckova, M.; Jampilek, J.; Cermak, P.; Kunes, J.; Dolezal, M.; Staud, F. Bioorg. Med. Chem. 2006, 14, 5850–5865. doi:10.1016/j.bmc.2006.05.030 |
| 1. | Moyer, M. P.; Weber, F. H.; Gross, J. L. J. Med. Chem. 1992, 35, 4595–4601. doi:10.1021/jm00102a013 |
| 2. | Gerster, J. F.; Lindstrom, K. J.; Miller, R. L.; Tomai, M. A.; Birmachu, W.; Bomersine, S. N.; Gibson, S. J.; Imbertson, L. M.; Jacobson, J. R.; Knafla, R. T.; Maye, P. V.; Nikolaides, N.; Oneyemi, F. Y.; Parkhurst, G. J.; Pecore, S. E.; Reiter, M. J.; Scribner, L. S.; Testerman, T. L.; Thompson, N. J.; Wagner, T. L.; Weeks, C. E.; Andre, J.-D.; Lagain, D.; Bastard, Y.; Lupu, M. J. Med. Chem. 2005, 48, 3481–3491. doi:10.1021/jm049211v |
| 3. | Miert, S. V.; Miert, S. V.; Hostyn, S.; Maes, B. U. W.; Cimanga, K.; Brun, R.; Kaiser, M.; Mátyus, P.; Dommisse, R.; Lemière, G.; Vlietinck, A.; Pieters, L. J. Nat. Prod. 2005, 68, 674–677. doi:10.1021/np0496284 |
| 4. | Chilin, A.; Marzaro, G.; Marzano, C.; Via, L. D.; Ferlin, M. G.; Pastorini, G.; Guiotto, A. Bioorg. Med. Chem. 2009, 17, 523–529. doi:10.1016/j.bmc.2008.11.072 |
| 9. | Eswaran, S.; Adhikari, A. V.; Kumar, R. A. Eur. J. Med. Chem. 2010, 45, 957–966. doi:10.1016/j.ejmech.2009.11.036 |
| 25. | Sugaya, T.; Kato, N.; Sakaguchi, A.; Tomioka, S. Synthesis 1995, 1257–1262. doi:10.1055/s-1995-4088 |
| 26. | Kumar, S.; Ila, H.; Junjappa, H. Tetrahedron 2007, 63, 10067–10076. doi:10.1016/j.tet.2007.07.045 |
| 8. | Wright, C. W.; Addae-Kyereme, J.; Breen, A. G.; Brown, J. E.; Cox, M. F.; Croft, S. L.; Gökçek, Y.; Kendrick, H.; Phillips, R. M.; Pollet, P. L. J. Med. Chem. 2001, 44, 3187–3194. doi:10.1021/jm010929+ |
| 27. | Lautens, M.; Paquin, J.-F.; Piguel, S. J. Org. Chem. 2002, 67, 3972–3974. doi:10.1021/jo025730z |
| 28. | Arnold, L. A.; Luo, W.; Guy, R. K. Org. Lett. 2004, 6, 3005–3007. doi:10.1021/ol0487884 |
| 29. | Rotzoll, S.; Appel, B.; Langer, P. Tetrahedron Lett. 2005, 46, 4057–4059. doi:10.1016/j.tetlet.2005.04.009 |
| 30. | Barrett, I.; Meegan, M. J.; Hughes, R. B.; Carr, M.; Knox, A. J. S.; Artemenko, N.; Golfis, G.; Zisterer, D. M.; Lloyd, D. G. Bioorg. Med. Chem. 2008, 16, 9554–9573. doi:10.1016/j.bmc.2008.09.035 |
| 7. | Chen, Y.-L.; Chen, I.-L.; Lu, C.-M.; Tzeng, C.-C.; Tsao, L.-T.; Wang, J.-P. Bioorg. Med. Chem. 2004, 12, 387–392. doi:10.1016/j.bmc.2003.10.051 |
| 20. | Bera, R.; Dhananjaya, G.; Singh, S. N.; Ramu, B.; Kiran, S. U.; Kumar, P. R.; Mukkanti, K.; Pal, M. Tetrahedron 2008, 64, 582–589. doi:10.1016/j.tet.2007.10.101 |
| 5. | Yamato, M.; Takeuchi, Y.; Hashigaki, K.; Ikeda, Y.; Chang, M. R.; Takeuchi, K.; Matsushima, M.; Tsuruo, T.; Tashiro, T. J. Med. Chem. 1989, 32, 1295–1300. doi:10.1021/jm00126a025 |
| 6. | Sánchez, I.; Reches, R.; Caignard, D. H.; Renard, P.; Pujol, M. D. Eur. J. Med. Chem. 2006, 41, 340–342. doi:10.1016/j.ejmech.2005.11.006 |
| 21. | Gruijters, B. W. T.; van Veldhuizen, A.; Weijers, C. A. G. M.; Wijnberg, J. B. P. A. J. Nat. Prod. 2002, 65, 558–561. doi:10.1021/np010510m |
| 22. | Kahnberg, P.; Sterner, O. Tetrahedron 2001, 57, 7181–7184. doi:10.1016/S0040-4020(01)00640-8 |
| 23. | Staben, S. T.; Siu, M.; Goldsmith, R.; Olivero, A. G.; Do, S.; Burdick, D. J.; Heffron, T. P.; Dotson, J.; Sutherlin, D. P.; Zhu, B. Y.; Tsui, V.; Le, H.; Lee, L.; Lesnick, J.; Lewis, C.; Murray, J. M.; Nonomiya, J.; Pang, J.; Prior, W. W.; Salphati, L.; Rouge, L.; Sampath, D.; Sideris, S.; Wiesmann, C.; Wu, P. Bioorg. Med. Chem. Lett. 2011, 21, 4054–4058. doi:10.1016/j.bmcl.2011.04.124 |
| 24. | Liu, J.-h.; Steigel, A.; Reininger, E.; Bauer, R. J. Nat. Prod. 2000, 63, 403–405. doi:10.1021/np990362o |
| 16. | Kalita, P. K.; Baruah, B.; Bhuyan, P. J. Tetrahedron Lett. 2006, 47, 7779–7782. doi:10.1016/j.tetlet.2006.08.086 |
| 18. | Yang, C.-L.; Tseng, C.-H.; Chen, Y.-L.; Lu, C.-M.; Kao, C.-L.; Wu, M.-H.; Tzeng, C.-C. Eur. J. Med. Chem. 2010, 45, 602–607. doi:10.1016/j.ejmech.2009.10.050 |
| 15. | Mali, J. R.; Pratap, U. R.; Jawale, D. V.; Mane, R. A. Tetrahedron Lett. 2010, 51, 3980–3982. doi:10.1016/j.tetlet.2010.05.117 |
| 19. | David, E.; Pellet-Rostaing, S.; Lemaire, M. Tetrahedron 2007, 63, 8999–9006. doi:10.1016/j.tet.2007.05.110 |
| 11. | Zhong, W.; Ma, W.; Liu, Y. Tetrahedron 2011, 67, 3509–3518. doi:10.1016/j.tet.2011.03.039 |
| 12. | Parvatkar, P. T.; Parameswaran, P. S.; Tilve, S. G. J. Org. Chem. 2009, 74, 8369–8372. doi:10.1021/jo901361x |
| 13. | Yu, F.; Yan, S.; Hu, L.; Wang, Y.; Lin, J. Org. Lett. 2011, 13, 4782–4785. doi:10.1021/ol201783d |
| 14. | Singh, V.; Hutait, S.; Batra, S. Eur. J. Org. Chem. 2009, 3454–3466. doi:10.1002/ejoc.200900336 |
| 40. | Bose, D. S.; Idrees, M.; Jakka, N. M.; Rao, J. V. J. Comb. Chem. 2010, 12, 100–110. doi:10.1021/cc900129t |
| 10. | Jonckers, T. H. M.; van Miert, S.; Cimanga, K.; Bailly, C.; Colson, P.; De Pauw-Gillet, M. C.; van den Heuvel, H.; Claeys, M.; Lemière, F.; Esmans, E. L.; Rozenski, J.; Quirijnen, L.; Maes, L.; Dommisse, R.; Lemière, G. L. F.; Vlietinck, A.; Pieters, L. J. Med. Chem. 2002, 45, 3497–3508. doi:10.1021/jm011102i |
| 12. | Parvatkar, P. T.; Parameswaran, P. S.; Tilve, S. G. J. Org. Chem. 2009, 74, 8369–8372. doi:10.1021/jo901361x |
| 17. | Chen, Y. L.; Hung, H. M.; Lu, C. M.; Li, K. C.; Tzeng, C. C. Bioorg. Med. Chem. 2004, 12, 6539–6546. doi:10.1016/j.bmc.2004.09.025 |
| 47. | Stevenson, L.; Tavares, A. A. S.; Brunet, A.; McGonagle, F. I.; Dewar, D.; Pimlott, S. L.; Sutherland, A. Bioorg. Med. Chem. Lett. 2010, 20, 954–957. doi:10.1016/j.bmcl.2009.12.061 |
| 38. | Mizuno, M.; Inagaki, A.; Yamashita, M.; Soma, N.; Maeda, Y.; Nakatani, H. Tetrahedron 2006, 62, 4065–4070. doi:10.1016/j.tet.2006.02.021 |
| 39. | Muscia, G. C.; Cazorla, S. I.; Frank, F. M.; Borosky, G. L.; Buldain, G. Y.; Asís, S. E.; Malchiodi, E. L. Eur. J. Med. Chem. 2011, 46, 3696–3703. doi:10.1016/j.ejmech.2011.05.035 |
| 40. | Bose, D. S.; Idrees, M.; Jakka, N. M.; Rao, J. V. J. Comb. Chem. 2010, 12, 100–110. doi:10.1021/cc900129t |
| 31. | Dolle, R. E.; Nelson, K. H., Jr. J. Comb. Chem. 1999, 1, 235–282. doi:10.1021/cc9900192 |
| 32. | Li, Y.; Chang, M. Q.; Gao, F.; Gao, W. T. J. Chem. Res. 2008, 640–641. doi:10.3184/030823408X375070 |
| 33. | Li, Y.; Zhang, C. H.; Sun, M. C.; Gao, W. T. J. Heterocycl. Chem. 2009, 46, 1190–1194. doi:10.1002/jhet.203 |
| 34. | Gao, W. T.; Zhang, C. H.; Li, Y. J. Braz. Chem. Soc. 2010, 21, 806–812. |
| 35. | Li, Y.; Gao, W. T. Beilstein J. Org. Chem. 2010, 6, 966–972. doi:10.3762/bjoc.6.108 |
| 36. | Gao, W. T.; Liu, J.; Jiang, Y.; Li, Y. Beilstein J. Org. Chem. 2011, 7, 210–217. doi:10.3762/bjoc.7.28 |
| 37. | Gao, W. T.; Zhao, Z. G.; Li, Y.; Yan, Y.; Li, F. Chin. J. Chem. 2012, 30, 1127–1132. doi:10.1002/cjoc.201100420 |
| 42. | Irlapati, N. R.; Baldwin, J. E.; Adlington, R. M.; Pritchard, G. J.; Cowley, A. R. Tetrahedron 2006, 62, 4603–4614. doi:10.1016/j.tet.2006.01.090 |
| 43. | Eaton, P. E.; Carlson, G. R.; Lee, J. T. J. Org. Chem. 1973, 38, 4071–4073. doi:10.1021/jo00987a028 |
| 37. | Gao, W. T.; Zhao, Z. G.; Li, Y.; Yan, Y.; Li, F. Chin. J. Chem. 2012, 30, 1127–1132. doi:10.1002/cjoc.201100420 |
| 41. | Popp, F. D.; McEwen, W. E. Chem. Rev. 1958, 58, 321–401. doi:10.1021/cr50020a004 |
| 33. | Li, Y.; Zhang, C. H.; Sun, M. C.; Gao, W. T. J. Heterocycl. Chem. 2009, 46, 1190–1194. doi:10.1002/jhet.203 |
| 36. | Gao, W. T.; Liu, J.; Jiang, Y.; Li, Y. Beilstein J. Org. Chem. 2011, 7, 210–217. doi:10.3762/bjoc.7.28 |
| 40. | Bose, D. S.; Idrees, M.; Jakka, N. M.; Rao, J. V. J. Comb. Chem. 2010, 12, 100–110. doi:10.1021/cc900129t |
| 38. | Mizuno, M.; Inagaki, A.; Yamashita, M.; Soma, N.; Maeda, Y.; Nakatani, H. Tetrahedron 2006, 62, 4065–4070. doi:10.1016/j.tet.2006.02.021 |
| 39. | Muscia, G. C.; Cazorla, S. I.; Frank, F. M.; Borosky, G. L.; Buldain, G. Y.; Asís, S. E.; Malchiodi, E. L. Eur. J. Med. Chem. 2011, 46, 3696–3703. doi:10.1016/j.ejmech.2011.05.035 |
© 2012 Gao et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)