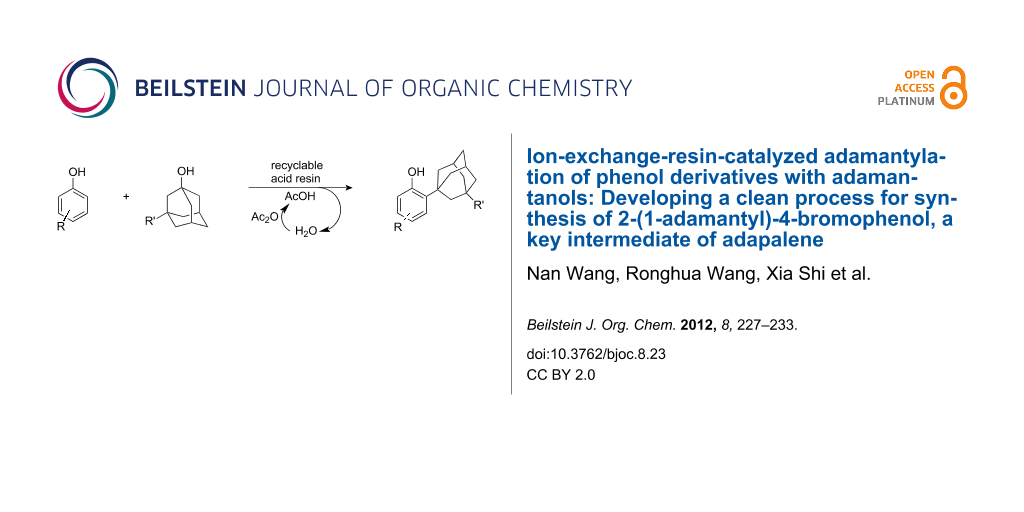
Abstract
A clean process has been developed for the synthesis of 2-adamantylphenol derivatives through adamantylation of substituted phenols with adamantanols catalyzed by commercially available and recyclable ion-exchange sulfonic acid resin in acetic acid. The sole byproduct of the adamantylation reaction, namely water, could be converted into the solvent acetic acid by addition of a slight excess of acetic anhydride during the work-up procedure, making the process waste-free except for regeneration of the ion-exchange resin, and facilitating the recycling of the resin catalyst. The ion-exchange sulfonic acid resin catalyst could be readily recycled by filtration and directly reused at least ten times without a significant loss of activity. The key intermediate of adapalene, 2-(1-adamantyl)-4-bromophenol, could be produced by means of this waste-free process.

Graphical Abstract
Introduction
o-Adamantylphenols and their derivatives are the key skeletons of synthetic retinoid analogues [1-9] and supporting ligands of many homogeneous transition-metal catalysts [10-14]. Introduction of an adamantyl group to the phenol ring has largely relied on acid-catalyzed Friedel–Crafts alkylation with haloadamantanes or adamantanols [1-9,15-19]. Compared with adamantylation by haloadamantanes, the alkylation employing adamantanols combined with mineral acids, such as concentrated H2SO4, ensures that the process affords adamantylphenols under milder conditions, and thus often with higher selectivities. However, the work-up procedure of mineral-acid-promoted reactions necessitates aqueous quenching and neutralization steps to remove the acids, resulting in enormous quantities of waste. For example, although the state-of-the-art process promoted by combination of concentrated H2SO4 and glacial acetic acid in CH2Cl2 was adapted [20-22], it was still found that at least 15 kg of waste water, besides solid waste such as Na2SO4 and NaOAc, was generated per kilogram of 2-(1-adamantyl)-4-bromophenol, simply in an effort to remove the acids from the reaction mixture by means of aqueous quenching and washing. Therefore, the development of alternative procedures that can reduce or eliminate the waste from the work-up procedure is highly needed. It was reported that adamantylation of aromatics promoted by strong organic acids, such as trifluoroacetic acid [18] and triflic acid [19], could circumvent the drawbacks associated with using concentrated H2SO4. However, these strong organic protic acids are expensive and volatile, and thus their recovery poses many difficulties.
Strongly acidic ion-exchange sulfonic acid resins have been extensively exploited as recyclable acid catalysts for organic syntheses in both laboratory and industry since they became commercially available [23-26]. The success of recyclable ion-exchange resins as substitutes for mineral acids in organic syntheses prompted us to study the possibility of using acidic sulfonic acid resin as a recyclable catalyst in the adamantylation reaction of 4-bromophenol (1a) with 1-adamantanol (2a) to develop a clean process for the production of the key intermediate of adapalene, namely 2-(1-adamantyl)-4-bromophenol (3aa). Olah et al. [15] reported that amberlyst and nafion-H could effectively catalyze adamantylation of aromatics with 1-bromoadamantane similarly to organic sulfonic acids or their fluorinated analogues. However, due to the detrimental effects of water on the catalytic activity of the cation-exchange resin in Friedel–Crafts alkylation, there is, to the best of our knowledge, no report on cation-exchange-resin-catalyzed adamantylation of phenols with adamantanols, although alkylation with alcohols is greener than that with alkyl halides. Herein, we report a process catalyzed by a recyclable acidic resin for an efficient and clean adamantylation of phenol derivatives by using adamantanols [27].
Results and Discussion
Treatment of 4-bromophenol (1a) with 1.05 equiv of 1-adamantanol (2a) in the presence of a macroporous sulfonic acid cation-exchange resin (Amberlite 200, H+ form) in ethyl acetate or diethyl carbonate under reflux for 10–12 h gave the desired product 2-(1-adamantyl)-4-bromophenol (3aa) in good yields; however, no reaction occurred in acetonitrile under similar conditions (Table 1, entries 1–3). The reaction proceeded faster in 1,2-dichloroethane but with lower selectivity, generating some (9%) doubly adamantylated product, 2,6-bis(1-adamantyl)-4-bromophenol, (Table 1, entry 4).
Table 1: Ion-exchange-resin-catalyzed adamantylation of 4-bromophenol (1a) with 1-adamantanol (2a).a
| entry | resin loading (g)b | solvent | T (°C) | t (h) | yield (%)c |
|---|---|---|---|---|---|
| 1 | 1.0 | CH3CO2Et | reflux | 12 | 94 |
| 2 | 1.0 | CO(OEt)2 | reflux | 10 | 96 |
| 3 | 1.0 | CH3CN | reflux | 12 | trace |
| 4 | 1.0 | (CH2Cl)2 | reflux | 5 | 83 |
| 5 | 1.0, 1st recycled | CO(OEt)2 | reflux | 24 | 76e |
| 6 | 1.0, 2nd recyclef | CO(OEt)2 | reflux | 15 | 93 |
| 7 | 1.0, 3rd recyclef | CO(OEt)2 | reflux | 15 | 89 |
| 8 | 1.0 | EtOH | reflux | 12 | trace |
| 9 | 1.0 | CH3CO2H | 100 | 2 | 98 |
| 10 | 1.0 | CH3CO2H | 60 | 12 | 75 |
| 11 | 1.0 | CH3CO2H | 80 | 7 | 95 |
| 12 | 0 | CH3CO2H | 100 | 6 | trace |
| 13 | 0.30 | CH3CO2H | 100 | 6 | 92 |
| 14 | 0.75 | CH3CO2H | 100 | 4 | 98 |
| 15 | 0.75g | CH3CO2H | 100 | 18 | 85 |
aThe reaction was run at 3 mmol scale with respect to 4-bromophenol in air. bAmberlite 200, H+ form used. cIsolated yields. dThe recovered resin catalyst was dried in vacuum at 50 °C for 1 h. e14% 4-bromophenol recovered. f The recovered resin catalyst was dried in vacuum at 90 °C for 3 h. gAmberlite IR120 used instead of Amberlite 200.
The cation-exchange-resin catalyst could be easily recovered by filtration and washing with diethyl carbonate. However, the catalytic activity of the recovered resin catalyst strongly depended on the dryness of the resin. That is, the water byproduct had to be thoroughly removed from the recovered resin by drying the resin under vacuum (<0.5 mmHg) at 90 °C for 3 h before reuse, otherwise the reaction could not be carried to completion even after heating under reflux for 24 h (Table 1, entries 5–7). To facilitate recycling of the resin catalyst, we tested the reaction in ethanol, which was supposed to help in removing the byproduct water from the resin, but no reaction was observed after 12 h under reflux. However, when the reaction was conducted in acetic acid at 100 °C (bath temperature), almost a quantitative yield of the target product 2-(1-adamantyl)-4-bromophenol (3aa) was obtained (98% after purification by chromatography) in 2 h (Table 1, entry 9).
The control experiment showed that no reaction occurred in the absence of the acidic ion-exchange resin, clearly excluding the possibility of acetic acid catalysis in the adamantylation (Table 1, entry 12). When the loading of the resin catalyst was reduced to 0.3 g and 0.75 g from 1.0 g, for 3 mmol 4-bromophenol (1a), the adamantylation reaction still gave 92% and 98% yields of the target product, respectively, within 4–6 h (Table 1, entries 13 and 14). However, the microporous sulfonic acid cation-exchange resin (Amberlite IR120, H+ form) was found to be less effective in promoting the adamantylation compared to the macroporous analogue. For example, the reaction took a longer time and still gave a lower yield with Amberlite IR120 (Table 1, entry 15).
Acetic acid appeared to be the best choice of solvent for the resin-catalyzed adamantylation with 1-adamantanol (2a), as it not only gave the desired 2-(1-adamantyl)-4-bromophenol (3aa) in high yields within a shorter reaction time, but it also made it possible to directly reuse the recovered resin catalyst since the byproduct, water, which is detrimental to the catalytic activity of such resins, could be easily converted into acetic acid by the addition of an equivalent amount of acetic anhydride during the work-up procedure. Further, after the sole byproduct, water, is converted into the solvent acetic acid, the resulting process is waste-free except for regeneration of the resin at its first use. Therefore, both the resin catalyst and the acetic acid solvent can be easily recovered by simple filtration and readily reused.
The recyclability of the acidic ion-exchange-resin catalyst was then investigated (Figure 1). To facilitate the operation, the reaction was run at 15 mmol scale with respect to 1a, with 5.0 g of the resin catalyst. When the reaction was completed, the mixture was cooled to 50–60 °C, and an acetic acid solution containing 1.05 equiv acetic anhydride was added to the mixture in order to convert the byproduct, water, to the solvent, acetic acid, thereby realizing a waste-free process and removal of water from the resin in order to facilitate recycling of the resin catalyst. After being stirred for a while (1–2 h) to ensure complete consumption of water in the mixture, the resin catalyst was filtered, washed with acetic acid and used directly in the subsequent run. In fact, the resin catalyst, Amberlite 200, H+ form, was reused ten times without significant loss of activity in the reaction of 4-bromophenol (1a) with 1-adamantanol (2a) in acetic acid. Solvent acetic acid was recovered from the filtrate by distillation, and the residue, crude product 2-(1-adamantyl)-4-bromophenol (3aa), was further purified by recrystallization in CH2Cl2/petroleum ether.
![[1860-5397-8-23-1]](/bjoc/content/figures/1860-5397-8-23-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Recycling of the ion-exchange-resin catalyst in the adamantylation reaction of 4-bromophenol (1a) with 1-adamantanol (2a).
Figure 1: Recycling of the ion-exchange-resin catalyst in the adamantylation reaction of 4-bromophenol (1a) w...
The scope of the sulfonic acid resin catalyzed adamantylation of phenol derivatives with adamantanols was further explored (Table 2).
Table 2: Scope of the ion-exchange-resin catalyzed adamantylation.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-23-i1.svg?max-width=637&scale=1.0)
|
|||||
| entry | R/R1 | R2 | t (h) | R3 | yield (%)a |
|---|---|---|---|---|---|
| 1 | H/4-Cl (1b) | H (2a) | 2 | H (3ba) | 90 |
| 2 | H/4-CH3 (1c) | H (2a) | 2 | H (3ca) | 87 |
| 3 | H/3-CH3 (1d) | H (2a) | 2 | H (3da) | 94 |
| 4 | H/4-OMe (1e) | H (2a) | 1 | H (3ea) | 80b |
| 5 | H/4-CHO (1f) | H (2a) | 4 | H (3fa) | 84 |
| 6 | H/4-CO2Et (1g) | H (2a) | 4 | H (3ga) | 75 |
| 7 | H/4-CH3CO (1h) | H (2a) | 10 | H (3ha) | 82 |
| 8 | H/4-NO2 (1i) | H (2a) | 12 | H (3ia) | 66 |
| 9 | Me/4-Br (1j) | H (2a) | 7 | H (3ja) | 80 |
| 10 | H/H (1k) | H (2a) | 4 | H (3ka) | –c |
| 11 | H/4-Br (1a) | COOH (2b) | 9 | COOH (3ab) | 75 |
| 12 | H/4-Br (1a) | CH2OH (2c) | 7 | CH2OAc (3ac) | 90 |
| 13 | H/4-CH3 (1c) | CH2OH (2c) | 4 | CH2OAc (3cc) | 94 |
| 14 | Me/4-Br (1j) | CH2OH (2c) | 7 | CH2OAc (3jc) | 94 |
aThe reaction was run at 1 mmol scale with Amberlite 200, H+ form (0.35 g/mmol with respect to phenols) in air. bThe lower yield was partly due to crystallization to completely remove 3-(1-adamantyl)-4-methoxyphenol after chromatography. cA mixture of isomers was formed.
The reaction rate was slightly decreased by electron-withdrawing groups compared to 4-bromophenol (1a). For example, cresols (1c, 1d) and 4-chlorophenol (1b) reacted similarly to the model reaction of 4-bromophenol (1a), giving the mono-adamantylation products 3ca, 3da and 3ba in good to excellent yields and selectivities (Table 2, entries 1–3). No isomers of 3da, e.g., 2-(1-adamantyl)-3-methylphenol or 3-methyl-4-(1-adamantyl)phenol, were observed for m-cresol (1d). The reaction of phenols bearing an electron-withdrawing group, e.g., –CHO (1f), –COCH3 (1h), –CO2Et (1g) and –NO2 (1i), needed a longer time to afford the 2-adamantylation products in good yields (Table 2, entries 5–8). When 4-methoxyphenol (1e) was subjected to 2a under similar conditions, both 2-(1-adamantyl)-4-methoxyphenol (3ea) and its isomer 3-(1-adamantyl)-4-methoxyphenol (3ea’) formed in 9:1 molar ratio as determined by 1H NMR analysis of the crude products, indicating that the hydroxy group (–OH) favourably directed the adamantylation compared to the methoxy group (–OMe), although the reaction of 4-bromoanisole (1j) provided 2-(1-adamantyl)-4-bromoanisole (3ja) as the solely isolated adamantylation product (Table 2, entries 4 and 9). However, phenol itself reacted to give a mixture of bi- and mono-adamantylphenol isomers, whereas 1,4-hydroquinone formed an insoluble solid material immediately upon being heated with sulfonic acid resin in acetic acid.
On the 1-adamantanol counterpart, the presence of the carboxyl acid group and the carbinol group appeared not to affect the adamantylation significantly. The reaction of 3-hydroxy-1-adamantanecarboxylic acid (2b) with 4-bromophenol (1a) gave the desired product 3ab in 75% yield (Table 2, entry 11). Interestingly, reactions of 3-hydroxymethyl-1-adamantanol (2c) with 4-bromophenol (1a), p-cresol (1c), or 4-bromoanisole (1j), afforded solely acetylated adamantylation products, 2-(3-acetoxymethyl-1-adamantyl)-4-bromophenol (3ac), 2-(3-acetoxymethyl-1-adamantyl)-p-cresol (3cc), or 2-(3-acetoxymethyl-1-adamantyl)-4-bromoanisole (3jc) in 90%, 94% and 94% yields, respectively (Table 2, entries 12–14). That is, the carbinol group was completely acetylated under the reaction conditions. Moreover, no unacetylated product was detected during the monitoring of the reaction progress, implying that acetylation of carbinol proceeded faster than the adamantylation under these acidic conditions.
Conclusion
In summary, we have developed a clean process for the synthesis of 2-adamantylphenol derivatives in acetic acid using a commercially available, strongly acidic cross-linked polystyrene-type sulfonic acid resin as a recyclable catalyst. The features of this process include (a) an almost waste-free procedure, except for the regeneration of the resin catalyst at its first use, after conversion of the sole byproduct, water, to the solvent acetic acid by anhydride; (b) the acidic ion-exchange-resin catalyst could be readily separated from the products by simple filtration and directly reused at least ten times without any reactivation, whilst showing no significant loss of activity; (c) the technique is effective with a wide range of substituted phenols giving o-adamantylation products with respect to the OH group, in good to excellent yields with high selectivities. The key intermediate of adapalene, 2-(1-adamantyl)-4-bromophenol, was produced by this procedure in excellent yields under the optimized conditions.
Experimental
General
Unless stated otherwise, all reagents and chemicals obtained commercially were used without further purification. Melting points are reported uncorrected and were recorded by using an electrothermal melting-point apparatus. 1H and 13C NMR spectra were recorded on a Bruker Avance Spectrometer. Mass-spectrum analysis was performed at the Center for Analysis, ECUST. 3-Hydroxyadamantane-1-carboxylic acid and 3-(hydroxymethyl)-1-adamantol were purchased from Alfa Aesar and Sigma-Aldrich, respectively.
Regeneration of the sulfonic acid resins (H+ form)
Sulfonic acid resins (Amberlite 200 and Amberlite-IR 120, Polysciences Inc.) were regenerated following a procedure reported in the literature [28]: 100 g of resin was stirred with 20% sulfuric acid (500 mL) overnight, then filtered and washed with deionized water until the washings reached a pH value of 5–6. The resin was then washed with THF (2 × 50 mL), dried in vacuum at 90 °C for 2 h, and kept over P2O5 in a vacuum desiccator.
Representative procedure for the acidic-ion-exchange-resin-catalyzed adamantylation with resin recycling
2-(1-Adamantyl)-4-bromophenol (3aa) [29]: To a suspension of dry sulfonic acid resin (Amberlite 200, H+ form, 5.0 g) in acetic acid (15 mL), adamantan-1-ol (2a; 2.43 g, 16 mmol) and p-bromophenol (1a; 2.60 g, 15 mmol) were added. The mixture was heated to 100 °C (bath temperature) and stirred for 2 h. After being cooled to about 60 °C, acetic anhydride (1.61 g, 16 mmol) in acetic acid (5 mL) was added to the reaction mixture and was stirred for 30 min followed by filtration in order to separate the resin catalyst from the solution. The resin was washed with acetic acid until it was free of product and was directly reused in the next run. The filtrate and the washings were combined, from which acetic acid was recovered by distillation, and the crude residue was purified by recrystallization from petroleum ether/CH2Cl2 to afford 2-(1-adamantanyl)-4-bromophenol (3aa) as a colourless fine crystals (4.48 g, 98%); mp 146–148 °C; 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.29 (d, J = 2.8 Hz, 1H), 7.14 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 1H), 6.52 (d, J = 8.4 Hz, 1H), 4.80 (s, 1H, OH), 2.08 (s, 9H), 1.77 (s, 6H); 13C NMR δ 153.54, 138.78, 130.24, 129.34, 118.40, 113.26, 40.30, 36.94, 28.95.
Representative procedure for ion-exchange-resin-catalyzed adamantylation on a small scale
2-(1-Adamantyl)-4-chlorophenol (3ba): A mixture of adamantan-1-ol (2a; 0.160 g, 1.05 mmol), 4-chlorophenol (1c; 0.128 g, 1.0 mmol) and resin (0.35 g) was stirred at 90 °C for 2 h in acetic acid (2 mL). After the reaction was complete, the resin catalyst was filtered and washed with ethyl acetate. The solvents were removed from the combined filtrate to give the crude product, which was further purified by column chromatography (petroleum ether/ethyl acetate as eluent) to afford a white powder (0.236 g, 90%); mp 139–141 °C; 1H NMR (500 MHz, CDCl3) δ 7.15 (d, J = 3.5 Hz, 1H), 6.99 (dd, J1 = 3.0 Hz, J2 = 10.5 Hz, 1H), 6.53 (d, J = 10.5 Hz, 1H), 4.86 (s, 1H), 2.07 (s, 9H), 1.76 (s, 6H); 13C NMR δ 153.05, 138.30, 127.36, 126.32, 125.67, 117.87, 40.30, 36.95, 28.96; ESI–HRMS: [M − 1]+ calcd for C16H18OCl, 261.1052; found 261.1046.
Characterization data for the adamantylation products
2-(1-Adamantyl)-4-methylphenol (3ca) [30]: mp 129–131 °C; 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.06 (s, 1H), 6.90 (d, J = 7.6 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H), 4.68 (s, 1H, OH), 2.32 (s, 3H), 2.17 (s, 6H), 2.12 (s, 3H), 1.83 (s, 6H); 13C NMR δ 152.18, 136.17, 129.71, 127.72, 127.03, 116.68, 40.63, 37.14, 36.61, 29.12, 20.88.
2-(1-Adamantyl)-5-methylphenol (3da) [30]: mp 84–86 °C; 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 8.0 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 6.51 (s, 1H), 4.85 (s, 1H), 2.30 (s, 3H), 2.16 (s, 6H), 2.12 (s, 3H), 1.83 (s, 6H); 13C NMR δ 154.25, 136.71, 133.57, 126.89, 121.48, 117.66, 40.78, 37.15, 36.39, 29.13, 20.60.
3-(1-Adamantyl)-4-hydroxyanisole (3ea): White powder; mp 207–209 °C; 1H NMR (500 MHz, CDCl3) δ 6.81 (s, 1H), 6.59 (br s, 2H), 4.62 (s, 1H), 3.76 (s, 3H), 2.11 (s, 6H), 2.07 (s, 3H), 1.77 (s, 6H); 13C NMR δ 153.57, 148.62, 137.86, 117.05, 113.95, 110.43, 55.71, 40.42, 37.04, 36.82, 29.04; ESI–HRMS: [M − 1]+ calcd for C17H21O2, 257.1542; found, 257.1538.
3-Adamantyl-4-hydroxybenzaldehyde (3fa) [31]: mp 217–219 °C; 1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H), 7.79 (s, 1H), 7.62 (d, J = 8.4 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.09 (s, 1H), 2.14–2.10 (m, 9H), 1.79 (s, 6H); 13C NMR δ (125 MHz, C5D5N) δ 191.71, 164.37, 138.12, 130.79, 129.92, 129.80, 117.89, 40.87, 37.70, 29.82.
2-(1-Adamantyl)-4-hydroxybenzoic acid ethyl ester (3ga): White powder; mp 250–252 °C; 1H NMR (500 MHz, C6D6N) δ 13.85 (s, 1H), 9.80 (d, J = 3.0 Hz, 1H), 9.53 (dd, J1 = 2.5 Hz, J2 = 10.5 Hz, 1H), 8.66 (d, J = 10.0 Hz, 1H), 5.88 (q, J = 9.0 Hz, 2H), 3.85 (d, J = 2.5, 6H), 3.54 (s, 3H), 3.25 (m, 6H), 2.78 (t, J = 9.0 Hz, 3H); 13C NMR δ 167.59, 162.83, 137.37, 129.95, 129.89, 122.07, 117.36, 61.00, 40.98, 37.71, 29.84, 15.08; ESI–HRMS: [M − 1]+ calcd for C19H23O3, 299.1647; found, 299.1648.
3-(1-Adamantyl)-4-hydroxyacetophenone (3ha): White powder; mp 183–185 °C; 1H NMR (400 MHz, CDCl3, 25 °C), δ 7.92 (d, J = 2.0 Hz, 1H), 7.74 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 7.01 (s, 1H), 6.80 (d, J = 8.4 Hz, 1H), 2.60 (s, 3H), 2.16, 2.10 (s + s overlapped, 9H), 1.80 (s, 6H); 13C NMR δ (100 MHz, C5D5N, 25 °C) δ 197.09, 163.02, 137.39, 129.90, 129.35, 128.59, 117.18, 41.01, 37.76, 29.89, 26.78; ESI–HRMS: [M − 1]+ calcd for C18H21O2, 269.1542; found, 269.1540.
2-(1-Adamantyl)-4-nitrophenol (3ia): White powder; mp 227–229 °C; 1H NMR (500 MHz, C6D6N) δ 8.29 (d, J = 3.5 Hz, 1H), 8.10 (dd, J1 = 3.5 Hz, J2 = 11.0 Hz, 1H), 7.06 (d, J = 11.5 Hz, 1H), 2.27 (s, 6H), 2.06 (s, 3H), 1.78 (m, 6H); 13C NMR δ 164.76, 141.08, 138.14, 124.20, 124.16 (overlapped with C6D6N), 117.31, 40.60, 37.81, 37.57, 29.74; ESI–HRMS: [M − 1]+ calcd for C16H18NO3, 272.1287; found, 272.1291.
2-(1-Adamantyl)-4-bromoanisole (3ja) [29]: mp 136–138 °C; 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.31 (s, 1H), 7.28 (d, J = 8.4 Hz, 1H), 6.75 (d, J = 8.8 Hz, 1H), 3.83 (s, 3H), 2.08 (s, 9H), 1.79 (s, 6H); 13C NMR δ 157.95, 140.83, 129.79, 129.33, 113.36, 113.32, 55.23, 40.39, 37.22, 37.06, 29.07.
2-(3-Acetoxymethyl-1-adamantyl)-4-bromophenol (3ac): White powder; mp 184–186 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 7.16 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 1H), 7.11 (d, J = 2.4 Hz, 1H), 6.73 (d, J = 8.4 Hz, 1H), 3.69 (s, 2H), 2.10 (s, 2H), 2.01–1.94 (m, 7H), 1.80 (s, 2H), 1.60–1.50 (m, 2H), 1.49 (m, 4H); 13C NMR δ 170.38, 155.38, 137.47, 129.14, 128.80, 118.32, 110.38, 72.89, 40.99, 39.04 (overlapped with DMSO-d6), 38.04, 36.62, 35.77, 33.69, 28.09, 20.57; ESI–HRMS: [M − 1]+ calcd for C19H22BrO3, 377.0752; found, 377.0756.
2-(3-Acetoxymethyl-1-adamantyl)-4-methylphenol (3cc): White powder; mp 217–219 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 6.85 (s, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.63 (d, J = 8.0 Hz, 1H), 3.69 (s, 2H), 2.17 (s, 3H), 2.10 (s, 2H), 2.01–1.95 (m, 7H), 1.67 (s, 2H), 1.65–1.55 (m, 2H), 1.50 (br s, 4H); 13C NMR δ 170.46, 153.58, 134.47, 126.84, 126.75, 126.69, 116.14, 73.02, 41.38, 39.30 (overlapped with DMSO-d6), 38.20, 36.26, 35.95, 33.71, 28.19, 20.60; ESI–HRMS: [M − 1]+ calcd for C20H25O3, 313.1804; found, 313.1798.
2-(3-Acetoxymethyl-1-adamantyl)-4-bromoanisole (3jc): White powder; mp 110–112 °C; 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 9.6 Hz, 1H), 7.26 (s, 1H), 6.73 (d, J = 9.2 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 2H), 2.16 (s, 2H), 2.06 (s, 3H), 2.00–1.95 (m, 4H), 1.83 (s, 2H), 1.75–1.65 (m, 2H), 1.55 (br s, 4H); 13C NMR δ 171.32, 157.82, 139.86, 129.68, 129.56, 113.40, 113.28, 73.92, 55.21, 41.78, 39.78, 38.61, 37.44, 36.31, 34.09, 28.75, 20.91; ESI–HRMS: [M]+ calcd for C20H25BrO3, 392.0987; found, 392.0984.
2-(3-Carboxy-1-adamantyl)-4-bromophenol (3ab): White powder; mp 209–211 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.02 (br s, 1H), 9.68 (br s, 1H), 7.17 (d, J = 8.0 Hz, 1H), 7.11 (s, 1H), 6.74 (d, J = 8.4 Hz, 1H), 2.11–2.01 (m, 4H), 1.95–1.92 (m, 4H), 1.80 (s, 4H), 1.66 (s, 2H); 13C NMR δ 178.36, 155.37, 137.28, 129.25, 128.78, 118.37, 110.42, 40.56, 40.51 (overlapped with DMSO-d6), 38.62, 37.90, 36.57, 35.41, 28.11; ESI–HRMS: [M − 1]+ calcd for C17H18BrO3, 349.0439; found, 349.0440.
References
-
Shroot, B.; Eustache, J.; Bernardon, J.-M. Benzonaphthalene derivatives and compositions. U.S. Patent 4,717,720, Jan 5, 1988.
Return to citation in text: [1] [2] -
Dawson, M. I.; Harris, D. L.; Liu, G.; Hobbs, P. D.; Lange, C. W.; Jong, L.; Bruey-Sedano, N.; James, S. Y.; Zhang, X.-k.; Peterson, V. J.; Leid, M.; Farhana, L.; Rishi, A. K.; Fontana, J. A. J. Med. Chem. 2004, 47, 3518–3536. doi:10.1021/jm030524k
Return to citation in text: [1] [2] -
Cincinelli, R.; Dallavalle, S.; Nannei, R.; Carella, S.; de Zani, D.; Merlini, L.; Penco, S.; Garattini, E.; Giannini, G.; Pisano, C.; Vesci, L.; Carminati, P.; Zuco, V.; Zanchi, C.; Zunino, F. J. Med. Chem. 2005, 48, 4931–4946. doi:10.1021/jm049440h
Return to citation in text: [1] [2] -
Lorenzo, P.; Alvarez, R.; Ortiz, M. A.; Alvarez, S.; Piedrafita, F. J.; de Lera, Á. R. J. Med. Chem. 2008, 51, 5431–5440. doi:10.1021/jm800285f
Return to citation in text: [1] [2] -
Pérez-Rodríguez, S.; Ortiz, M. A.; Pereira, R.; Rodríguez-Barrios, F.; de Lera, Á. R.; Piedrafita, F. J. Eur. J. Med. Chem. 2009, 44, 2434–2446. doi:10.1016/j.ejmech.2009.01.011
Return to citation in text: [1] [2] -
Dawson, M. I.; Xia, Z.; Jiang, T.; Ye, M.; Fontana, J. A.; Farhana, L.; Patel, B.; Xue, L. P.; Bhuiyan, M.; Pellicciari, R.; Macchiarulo, A.; Nuti, R.; Zhang, X.-K.; Han, Y.-H.; Tautz, L.; Hobbs, P. D.; Jong, L.; Waleh, N.; Chao, W.; Feng, G.-S.; Pang, Y.; Su, Y. J. Med. Chem. 2008, 51, 5650–5662. doi:10.1021/jm800456k
Return to citation in text: [1] [2] -
Cincinelli, R.; Dallavalle, S.; Nannei, R.; Merlini, L.; Penco, S.; Giannini, G.; Pisano, C.; Vesci, L.; Ferrara, F. F.; Zuco, V.; Zanchi, C.; Zunino, F. Bioorg. Med. Chem. 2007, 15, 4863–4875. doi:10.1016/j.bmc.2007.04.057
Return to citation in text: [1] [2] -
Dawson, M. I.; Xia, Z.; Liu, G.; Fontana, J. A.; Farhana, L.; Patel, B. B.; Arumugarajah, S.; Bhuiyan, M.; Zhang, X.-K.; Han, Y.-H.; Stallcup, W. B.; Fukushi, J.-i.; Mustelin, T.; Tautz, L.; Su, Y.; Harris, D. L.; Waleh, N.; Hobbs, P. D.; Jong, L.; Chao, W.-r.; Schiff, L. J.; Sani, B. P. J. Med. Chem. 2007, 50, 2622–2639. doi:10.1021/jm0613323
Return to citation in text: [1] [2] -
Tribulovich, V. G.; Garabadzhiu, A. V.; Kalvin’sh, I. Pharm. Chem. J. 2011, 45, 241–244. doi:10.1007/s11094-011-0605-z
Return to citation in text: [1] [2] -
Alexander, J. B.; Schrock, R. R.; Davis, W. M.; Hultzsch, K. C.; Hoveyda, A. H.; Houser, J. H. Organometallics 2000, 19, 3700–3715. doi:10.1021/om000336h
Return to citation in text: [1] -
Gademann, K.; Chavez, D. E.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2002, 41, 3059–3061. doi:10.1002/1521-3773(20020816)41:16<3059::AID-ANIE3059>3.0.CO;2-I
Return to citation in text: [1] -
Golisz, S. R.; Bercaw, J. E. Macromolecules 2009, 42, 8751–8762. doi:10.1021/ma901659q
Return to citation in text: [1] -
Waltman, A. W.; Grubbs, R. H. Organometallics 2004, 23, 3105–3107. doi:10.1021/om049727c
Return to citation in text: [1] -
Watanabe, T.; Ishida, Y.; Matsuo, T.; Kawaguchi, H. Dalton Trans. 2010, 39, 484–491. doi:10.1039/b911082h
Return to citation in text: [1] -
Olah, G. A.; Török, B.; Shamma, T.; Török, M.; Prakash, G. K. S. Catal. Lett. 1996, 42, 5–13. doi:10.1007/BF00814460
Return to citation in text: [1] [2] -
Prakash, G. K. S.; Yan, P.; Tórók, B.; Bucsi, I.; Tanaka, M.; Olah, G. A. Catal. Lett. 2003, 85, 1–6. doi:10.1023/A:1022133227407
Return to citation in text: [1] -
Shokova, E. A.; Khomich, A. N.; Kovalev, V. V. Tetrahedron Lett. 1996, 37, 543–546. doi:10.1016/0040-4039(95)02236-8
Return to citation in text: [1] -
Stepakov, A. V.; Molchanov, A. P.; Kostikov, R. R. Russ. J. Org. Chem. 2007, 43, 538–543. doi:10.1134/S1070428007040082
Return to citation in text: [1] [2] -
Laali, K. K.; Sarca, V. D.; Okazaki, T.; Brock, A.; Der, P. Org. Biomol. Chem. 2005, 3, 1034–1042. doi:10.1039/b416997b
Return to citation in text: [1] [2] -
Pilgrim, W. R.; Lagiere, J. Process for the preparation of 1-adamantane derivatives. U.S. Patent 5,015,758, May 14, 1991.
Return to citation in text: [1] -
Cincinelli, R.; Dallavalle, S.; Merlini, L.; Penco, S.; Pisano, C.; Carminati, P.; Giannini, G.; Vesci, L.; Gaetano, C.; Illy, B.; Zuco, V.; Supino, R.; Zunino, F. J. Med. Chem. 2003, 46, 909–912. doi:10.1021/jm025593y
Return to citation in text: [1] -
Liu, Z.; Xiang, J. Org. Process Res. Dev. 2006, 10, 285–288. doi:10.1021/op050223f
Return to citation in text: [1] -
Gelbard, G. Ind. Eng. Chem. Res. 2005, 44, 8468–8498. doi:10.1021/ie0580405
And references therein.
Return to citation in text: [1] -
Harmer, M. A.; Sun, Q. Appl. Catal., A 2001, 221, 45–62. doi:10.1016/S0926-860X(01)00794-3
Return to citation in text: [1] -
Corain, B.; Zecca, M.; Jeřábek, K. J. Mol. Catal. A: Chem. 2001, 177, 3–20. doi:10.1016/S1381-1169(01)00305-3
Return to citation in text: [1] -
Alexandratos, S. D. Ind. Eng. Chem. Res. 2009, 48, 388–398. doi:10.1021/ie801242v
Return to citation in text: [1] -
Zou, G.; Wang, N.; Wang, R. Method for preparing 2-(1-adamantyl)-4-bromophenol. Chinese Patent CN101955417A, Jan 26, 2011.
Return to citation in text: [1] -
Satyamurthy, N.; Barrio, J. R. J. Org. Chem. 1983, 48, 4394–4396. doi:10.1021/jo00171a050
Return to citation in text: [1] -
Charpentier, B.; Bernardon, J.-M.; Eustache, J.; Millois, C.; Martin, B.; Michel, S.; Shroot, B. J. Med. Chem. 1995, 38, 4993–5006. doi:10.1021/jm00026a006
Return to citation in text: [1] [2] -
Aigami, K.; Inamoto, Y.; Takaishi, N.; Hattori, K.; Takatsuki, A.; Tamura, G. J. Med. Chem. 1975, 18, 713–721. doi:10.1021/jm00241a015
Return to citation in text: [1] [2] -
Brenna, E.; Fuganti, C.; Fronza, G.; Gatti, F. G.; Sala, F.; Serra, S. Tetrahedron 2007, 63, 2351–2356. doi:10.1016/j.tet.2006.12.038
Return to citation in text: [1]
| 1. | Shroot, B.; Eustache, J.; Bernardon, J.-M. Benzonaphthalene derivatives and compositions. U.S. Patent 4,717,720, Jan 5, 1988. |
| 2. | Dawson, M. I.; Harris, D. L.; Liu, G.; Hobbs, P. D.; Lange, C. W.; Jong, L.; Bruey-Sedano, N.; James, S. Y.; Zhang, X.-k.; Peterson, V. J.; Leid, M.; Farhana, L.; Rishi, A. K.; Fontana, J. A. J. Med. Chem. 2004, 47, 3518–3536. doi:10.1021/jm030524k |
| 3. | Cincinelli, R.; Dallavalle, S.; Nannei, R.; Carella, S.; de Zani, D.; Merlini, L.; Penco, S.; Garattini, E.; Giannini, G.; Pisano, C.; Vesci, L.; Carminati, P.; Zuco, V.; Zanchi, C.; Zunino, F. J. Med. Chem. 2005, 48, 4931–4946. doi:10.1021/jm049440h |
| 4. | Lorenzo, P.; Alvarez, R.; Ortiz, M. A.; Alvarez, S.; Piedrafita, F. J.; de Lera, Á. R. J. Med. Chem. 2008, 51, 5431–5440. doi:10.1021/jm800285f |
| 5. | Pérez-Rodríguez, S.; Ortiz, M. A.; Pereira, R.; Rodríguez-Barrios, F.; de Lera, Á. R.; Piedrafita, F. J. Eur. J. Med. Chem. 2009, 44, 2434–2446. doi:10.1016/j.ejmech.2009.01.011 |
| 6. | Dawson, M. I.; Xia, Z.; Jiang, T.; Ye, M.; Fontana, J. A.; Farhana, L.; Patel, B.; Xue, L. P.; Bhuiyan, M.; Pellicciari, R.; Macchiarulo, A.; Nuti, R.; Zhang, X.-K.; Han, Y.-H.; Tautz, L.; Hobbs, P. D.; Jong, L.; Waleh, N.; Chao, W.; Feng, G.-S.; Pang, Y.; Su, Y. J. Med. Chem. 2008, 51, 5650–5662. doi:10.1021/jm800456k |
| 7. | Cincinelli, R.; Dallavalle, S.; Nannei, R.; Merlini, L.; Penco, S.; Giannini, G.; Pisano, C.; Vesci, L.; Ferrara, F. F.; Zuco, V.; Zanchi, C.; Zunino, F. Bioorg. Med. Chem. 2007, 15, 4863–4875. doi:10.1016/j.bmc.2007.04.057 |
| 8. | Dawson, M. I.; Xia, Z.; Liu, G.; Fontana, J. A.; Farhana, L.; Patel, B. B.; Arumugarajah, S.; Bhuiyan, M.; Zhang, X.-K.; Han, Y.-H.; Stallcup, W. B.; Fukushi, J.-i.; Mustelin, T.; Tautz, L.; Su, Y.; Harris, D. L.; Waleh, N.; Hobbs, P. D.; Jong, L.; Chao, W.-r.; Schiff, L. J.; Sani, B. P. J. Med. Chem. 2007, 50, 2622–2639. doi:10.1021/jm0613323 |
| 9. | Tribulovich, V. G.; Garabadzhiu, A. V.; Kalvin’sh, I. Pharm. Chem. J. 2011, 45, 241–244. doi:10.1007/s11094-011-0605-z |
| 18. | Stepakov, A. V.; Molchanov, A. P.; Kostikov, R. R. Russ. J. Org. Chem. 2007, 43, 538–543. doi:10.1134/S1070428007040082 |
| 29. | Charpentier, B.; Bernardon, J.-M.; Eustache, J.; Millois, C.; Martin, B.; Michel, S.; Shroot, B. J. Med. Chem. 1995, 38, 4993–5006. doi:10.1021/jm00026a006 |
| 20. | Pilgrim, W. R.; Lagiere, J. Process for the preparation of 1-adamantane derivatives. U.S. Patent 5,015,758, May 14, 1991. |
| 21. | Cincinelli, R.; Dallavalle, S.; Merlini, L.; Penco, S.; Pisano, C.; Carminati, P.; Giannini, G.; Vesci, L.; Gaetano, C.; Illy, B.; Zuco, V.; Supino, R.; Zunino, F. J. Med. Chem. 2003, 46, 909–912. doi:10.1021/jm025593y |
| 22. | Liu, Z.; Xiang, J. Org. Process Res. Dev. 2006, 10, 285–288. doi:10.1021/op050223f |
| 1. | Shroot, B.; Eustache, J.; Bernardon, J.-M. Benzonaphthalene derivatives and compositions. U.S. Patent 4,717,720, Jan 5, 1988. |
| 2. | Dawson, M. I.; Harris, D. L.; Liu, G.; Hobbs, P. D.; Lange, C. W.; Jong, L.; Bruey-Sedano, N.; James, S. Y.; Zhang, X.-k.; Peterson, V. J.; Leid, M.; Farhana, L.; Rishi, A. K.; Fontana, J. A. J. Med. Chem. 2004, 47, 3518–3536. doi:10.1021/jm030524k |
| 3. | Cincinelli, R.; Dallavalle, S.; Nannei, R.; Carella, S.; de Zani, D.; Merlini, L.; Penco, S.; Garattini, E.; Giannini, G.; Pisano, C.; Vesci, L.; Carminati, P.; Zuco, V.; Zanchi, C.; Zunino, F. J. Med. Chem. 2005, 48, 4931–4946. doi:10.1021/jm049440h |
| 4. | Lorenzo, P.; Alvarez, R.; Ortiz, M. A.; Alvarez, S.; Piedrafita, F. J.; de Lera, Á. R. J. Med. Chem. 2008, 51, 5431–5440. doi:10.1021/jm800285f |
| 5. | Pérez-Rodríguez, S.; Ortiz, M. A.; Pereira, R.; Rodríguez-Barrios, F.; de Lera, Á. R.; Piedrafita, F. J. Eur. J. Med. Chem. 2009, 44, 2434–2446. doi:10.1016/j.ejmech.2009.01.011 |
| 6. | Dawson, M. I.; Xia, Z.; Jiang, T.; Ye, M.; Fontana, J. A.; Farhana, L.; Patel, B.; Xue, L. P.; Bhuiyan, M.; Pellicciari, R.; Macchiarulo, A.; Nuti, R.; Zhang, X.-K.; Han, Y.-H.; Tautz, L.; Hobbs, P. D.; Jong, L.; Waleh, N.; Chao, W.; Feng, G.-S.; Pang, Y.; Su, Y. J. Med. Chem. 2008, 51, 5650–5662. doi:10.1021/jm800456k |
| 7. | Cincinelli, R.; Dallavalle, S.; Nannei, R.; Merlini, L.; Penco, S.; Giannini, G.; Pisano, C.; Vesci, L.; Ferrara, F. F.; Zuco, V.; Zanchi, C.; Zunino, F. Bioorg. Med. Chem. 2007, 15, 4863–4875. doi:10.1016/j.bmc.2007.04.057 |
| 8. | Dawson, M. I.; Xia, Z.; Liu, G.; Fontana, J. A.; Farhana, L.; Patel, B. B.; Arumugarajah, S.; Bhuiyan, M.; Zhang, X.-K.; Han, Y.-H.; Stallcup, W. B.; Fukushi, J.-i.; Mustelin, T.; Tautz, L.; Su, Y.; Harris, D. L.; Waleh, N.; Hobbs, P. D.; Jong, L.; Chao, W.-r.; Schiff, L. J.; Sani, B. P. J. Med. Chem. 2007, 50, 2622–2639. doi:10.1021/jm0613323 |
| 9. | Tribulovich, V. G.; Garabadzhiu, A. V.; Kalvin’sh, I. Pharm. Chem. J. 2011, 45, 241–244. doi:10.1007/s11094-011-0605-z |
| 15. | Olah, G. A.; Török, B.; Shamma, T.; Török, M.; Prakash, G. K. S. Catal. Lett. 1996, 42, 5–13. doi:10.1007/BF00814460 |
| 16. | Prakash, G. K. S.; Yan, P.; Tórók, B.; Bucsi, I.; Tanaka, M.; Olah, G. A. Catal. Lett. 2003, 85, 1–6. doi:10.1023/A:1022133227407 |
| 17. | Shokova, E. A.; Khomich, A. N.; Kovalev, V. V. Tetrahedron Lett. 1996, 37, 543–546. doi:10.1016/0040-4039(95)02236-8 |
| 18. | Stepakov, A. V.; Molchanov, A. P.; Kostikov, R. R. Russ. J. Org. Chem. 2007, 43, 538–543. doi:10.1134/S1070428007040082 |
| 19. | Laali, K. K.; Sarca, V. D.; Okazaki, T.; Brock, A.; Der, P. Org. Biomol. Chem. 2005, 3, 1034–1042. doi:10.1039/b416997b |
| 30. | Aigami, K.; Inamoto, Y.; Takaishi, N.; Hattori, K.; Takatsuki, A.; Tamura, G. J. Med. Chem. 1975, 18, 713–721. doi:10.1021/jm00241a015 |
| 10. | Alexander, J. B.; Schrock, R. R.; Davis, W. M.; Hultzsch, K. C.; Hoveyda, A. H.; Houser, J. H. Organometallics 2000, 19, 3700–3715. doi:10.1021/om000336h |
| 11. | Gademann, K.; Chavez, D. E.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2002, 41, 3059–3061. doi:10.1002/1521-3773(20020816)41:16<3059::AID-ANIE3059>3.0.CO;2-I |
| 12. | Golisz, S. R.; Bercaw, J. E. Macromolecules 2009, 42, 8751–8762. doi:10.1021/ma901659q |
| 13. | Waltman, A. W.; Grubbs, R. H. Organometallics 2004, 23, 3105–3107. doi:10.1021/om049727c |
| 14. | Watanabe, T.; Ishida, Y.; Matsuo, T.; Kawaguchi, H. Dalton Trans. 2010, 39, 484–491. doi:10.1039/b911082h |
| 31. | Brenna, E.; Fuganti, C.; Fronza, G.; Gatti, F. G.; Sala, F.; Serra, S. Tetrahedron 2007, 63, 2351–2356. doi:10.1016/j.tet.2006.12.038 |
| 27. | Zou, G.; Wang, N.; Wang, R. Method for preparing 2-(1-adamantyl)-4-bromophenol. Chinese Patent CN101955417A, Jan 26, 2011. |
| 29. | Charpentier, B.; Bernardon, J.-M.; Eustache, J.; Millois, C.; Martin, B.; Michel, S.; Shroot, B. J. Med. Chem. 1995, 38, 4993–5006. doi:10.1021/jm00026a006 |
| 15. | Olah, G. A.; Török, B.; Shamma, T.; Török, M.; Prakash, G. K. S. Catal. Lett. 1996, 42, 5–13. doi:10.1007/BF00814460 |
| 30. | Aigami, K.; Inamoto, Y.; Takaishi, N.; Hattori, K.; Takatsuki, A.; Tamura, G. J. Med. Chem. 1975, 18, 713–721. doi:10.1021/jm00241a015 |
| 23. |
Gelbard, G. Ind. Eng. Chem. Res. 2005, 44, 8468–8498. doi:10.1021/ie0580405
And references therein. |
| 24. | Harmer, M. A.; Sun, Q. Appl. Catal., A 2001, 221, 45–62. doi:10.1016/S0926-860X(01)00794-3 |
| 25. | Corain, B.; Zecca, M.; Jeřábek, K. J. Mol. Catal. A: Chem. 2001, 177, 3–20. doi:10.1016/S1381-1169(01)00305-3 |
| 26. | Alexandratos, S. D. Ind. Eng. Chem. Res. 2009, 48, 388–398. doi:10.1021/ie801242v |
| 19. | Laali, K. K.; Sarca, V. D.; Okazaki, T.; Brock, A.; Der, P. Org. Biomol. Chem. 2005, 3, 1034–1042. doi:10.1039/b416997b |
| 28. | Satyamurthy, N.; Barrio, J. R. J. Org. Chem. 1983, 48, 4394–4396. doi:10.1021/jo00171a050 |
© 2012 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)