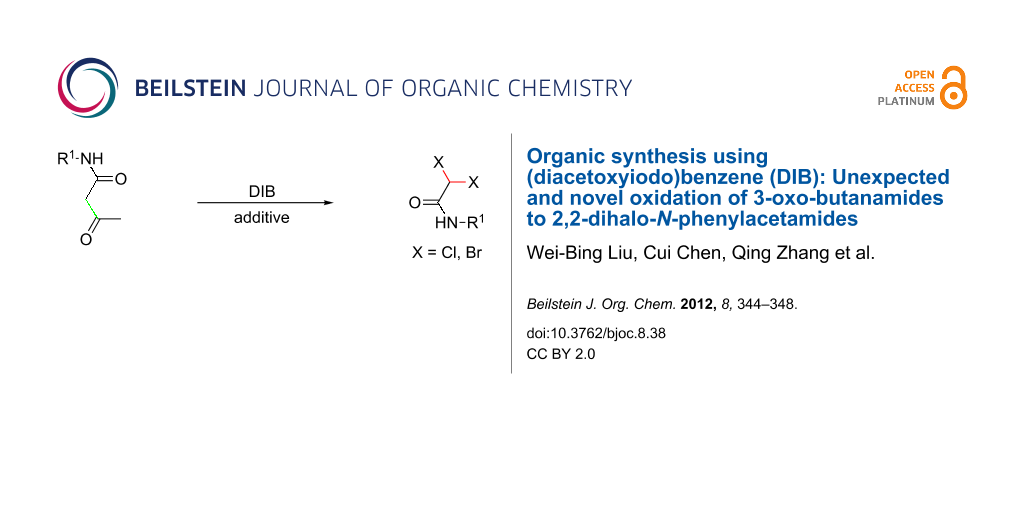
Abstract
A novel and reliable method for the direct preparation of 2,2-dihalo-N-phenylacetamides is reported. The key transformation involves the cleavage of a carbon–carbon bond in the presence of DIB and a Lewis acid as the halogen source, and thus this method significantly expands the value of DIB as a unique and powerful tool in chemical synthesis. This protocol not only adds a new aspect to reactions that use other hypervalent iodine reagents but also provides a wide space for the synthesis of disubstituted acetamides.

Graphical Abstract
Introduction
Hypervalent iodine(III) reagents [1-18] have received much attention, as reflected by the plethora of publications and reviews [19-23]. This is due to their low toxicity, ready availability, easy handling, clean transformation, and reactivity, which is similar to heavy-metal-based oxidants, including harmful elements, such as Pb(IV), Hg(II), and Tl(III), as well as transition metal-catalyzed processes [24-30]. Recently, we reported an efficient acetoxylation approach to synthesize 1-carbamoyl-2-oxopropyl acetate derivatives by using (diacetoxyiodo)benzene (DIB) (Scheme 1) [31].
![[1860-5397-8-38-i1]](/bjoc/content/inline/1860-5397-8-38-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 1-carbamoyl-2-oxopropyl acetates.
Scheme 1: Synthesis of 1-carbamoyl-2-oxopropyl acetates.
During the course of conditional optimization to synthesize 1-carbamoyl-2-oxopropyl acetate derivatives, we surprisingly found that almost none of the desired acetoxylation product was obtained, but 2,2-dichloro-N-phenylacetamide was provided as the major product, upon addition of Lewis acids such as FeCl3, ZnCl2 and CuCl2 in the reaction system. Based on this result, we developed a simple and efficient approach to the synthesis of 2,2-dihalo-N-phenylacetamides, on which we report herein (Scheme 2). To the best of our knowledge, there are several reports on chlorination and bromination reactions with PhI(OAc)2 and a halogen source such as TMSBr, lithium halide or pyridinium halide [32-34]. Also, there are several reports on the synthesis of difunctionalized acetamide derivatives [35-38], but this report is the first to describe the synthesis of 2,2-dihalo-N-phenylacetamides through an oxidative process with PhI(OAc)2 and Lewis acids as the halogen source.
![[1860-5397-8-38-i2]](/bjoc/content/inline/1860-5397-8-38-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2,2-dihalo-N-phenylacetamides.
Scheme 2: Synthesis of 2,2-dihalo-N-phenylacetamides.
Results and Discussion
Initially, we used 3-oxo-N-phenylbutanamide (1a) as the model substrate to optimize the reaction conditions in different solvents, temperatures and amounts of DIB (Table 1). The best result was obtained in dioxane in the presence of 1.3 equiv of DIB and 1.5 equiv of zinc(II) chloride at room temperature for one hour (Table 1, entry 11). For this transformation, FeCl3 and ZnCl2 were suitable Lewis acids (Table 1, entry 2 and entry 3), and dioxane and DMF were practical solvents among the various solvents examined (Table 1, entry 3 and entry 8). It is noteworthy that no product 2a was obtained when the reaction was carried out without the addition of Lewis acids (Table 1, entry 1) or without DIB (Table 1, entry 5).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-38-i7.svg?max-width=637&scale=1.0)
|
||||
| entry | solvent | additive (1.5 equiv) | time (h) | yield (%)b |
|---|---|---|---|---|
| 1 | dioxane | none | 1 | – |
| 2 | dioxane | FeCl3 | 1 | 78 |
| 3 | dioxane | ZnCl2 | 1 | 81 |
| 4c | dioxane | ZnCl2 | 1 | 75 |
| 5d | dioxane | ZnCl2 | 1 | – |
| 6 | cyclohexane | ZnCl2 | 1 | 26 |
| 7 | DCE | ZnCl2 | 1 | 42 |
| 8 | DMF | ZnCl2 | 1 | 80 |
| 9 | DMSO | ZnCl2 | 1 | 46 |
| 10e | dioxane | ZnCl2 | 1 | 31 |
| 11f | dioxane | ZnCl2 | 1 | 89 |
| 12g | dioxane | ZnCl2 | 1 | 84 |
| 13f | dioxane | ZnCl2 | 0.5 | 53 |
| 14f | dioxane | ZnCl2 | 1.5 | 89 |
| 15f | dioxane | ZnCl2 | 2 | 89 |
a1a (0.25 mmol), solvent (2 mL), DIB (1.0 equiv); bGC yield; cZnCl2 (1.0 equiv); dwithout DIB; eDIB (0.5 equiv); fDIB (1.3 equiv); gDIB (2.0 equiv).
After optimizing the reaction conditions, we used a range of 3-oxo-N-phenylbutanamides to explore the substrate scope and limitations of this reaction. As shown in Scheme 3, all the reactions proceeded smoothly and gave the corresponding N-phenyl dichloroacetamides 2a–2k exclusively and in good to excellent isolated yields. It was also found that the number and the electronic properties of the substituents on the benzene ring had little effect on the reaction. For example, the reactions of 3-oxo-N-phenylbutanamide (1a), 3-oxo-N-o-tolylbutanamide (1b), N-(2,4-dimethoxyphenyl)-3-oxobutanamide (1j) and N-(4-chloro-2,5-dimethoxyphenyl)-3-oxobutanamide (1k) all led to their corresponding N-phenyl dichloroacetamides (Scheme 3, 2a, 2b, 2j, and 2k) in good isolated yields. In addition, the position of the substituents on the benzene ring also has little effect on this transformation, such that N-(2-chlorophenyl)-3-oxobutanamide (1c), N-(4-chlorophenyl)-3-oxobutanamide (1d), N-(4-methoxyphenyl)-3-oxobutanamide (1e) and N-(2-methoxyphenyl)-3-oxobutanamide (1f) could also serve as good substrates in this protocol (Scheme 3, 2c, 2d, 2e, and 2f).
![[1860-5397-8-38-i3]](/bjoc/content/inline/1860-5397-8-38-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of dichloroacetamides. Reagents and conditions: 1 (1.0 mmol), dioxane (2 mL), DIB (1.3 equiv), ZnCl2(1.5 equiv); yields % are isolated yields.
Scheme 3: Synthesis of dichloroacetamides. Reagents and conditions: 1 (1.0 mmol), dioxane (2 mL), DIB (1.3 eq...
Next, in order to expand the scope of this protocol, we employed ZnBr2 as a reagent under the same reaction conditions, and we were pleased to find that the corresponding dibromo derivatives were obtained as the products. As shown in Scheme 4, all tested substrates provided the corresponding dibromoacetamides 3a–3l in good to excellent isolated yields, which not only greatly expanded the application scope of this protocol but also provided a wide space for the synthesis of 2,2-dihalo-N-phenylacetamides.
![[1860-5397-8-38-i4]](/bjoc/content/inline/1860-5397-8-38-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of dibromoacetamides. Reagents and conditions: 1 (1.0 mmol), dioxane (2 mL), DIB (1.3 equiv), ZnBr2 (1.5 equiv); yields % are isolated yields.
Scheme 4: Synthesis of dibromoacetamides. Reagents and conditions: 1 (1.0 mmol), dioxane (2 mL), DIB (1.3 equ...
In spite of the widespread use of DIB, there is no direct precedent for DIB-mediated cleavage of C–C bonds. In particular, the application of this protocol to synthesize difunctionalized acetamides from 3-oxo-butanamides is reported here for the first time. In order to probe the mechanism of this transformation, we employed 2,2-dichloro-3-oxo-N-phenylbutanamide (1m) and 2,2-dibromo-3-oxo-N-phenylbutanamide (1n) as reactants under acidic conditions in the presence of Zn(OAc)2 (Scheme 5), and we found that the reaction can also give the corresponding product 2,2-dichloro-N-phenylacetamide (2a) and 2,2-dibromo-N-phenylacetamide (3a).
![[1860-5397-8-38-i5]](/bjoc/content/inline/1860-5397-8-38-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
On the basis of these preliminary results, a mechanistic proposal for this transformation, exemplified by the formation of 2a, is depicted in Scheme 6. Initially, the reaction involved generation of the known chlorinating agent (dichloroiodo)benzene (PhICl2) [39], followed by dichlorination of the β-keto amide of 3-oxo-N-phenylbutanamide (1a) to give intermediate 4. It is well known that Lewis acids can activate 1,3-diketones [40] to produce intermediate 5 and 6. This complexation not only increases the nucleophilicity of the methylene carbon atom, but also simultaneously increases the electrophilicity of the carbonyl carbon atom. Consequently, nucleophilic attack of the acetate ion on the carbonyl carbon atom affords intermediate 7. A subsequent carbon–carbon bond cleavage of the labile α,α-dichloro β-keto amide through a retro-Claisen condensation reaction [41] generates intermediate 8. Finally, the electrophilic attack of a proton on the carbon–carbon double bond resulted in the final product 2,2-dichloro-N-phenylacetamide (2a).
![[1860-5397-8-38-i6]](/bjoc/content/inline/1860-5397-8-38-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Tentative mechanism for the synthesis of 2,2-dihalo-N-phenylacetamides.
Scheme 6: Tentative mechanism for the synthesis of 2,2-dihalo-N-phenylacetamides.
Conclusion
In summary, we have shown an efficient and operationally simple method to synthesize 2,2-dihalo-N-phenylacetamides. The mild reaction conditions, good substrate scope and good to excellent yields make the present protocol potentially useful in organic synthesis. Moreover, it should be pointed out that this transformation includes an oxidative process involving the cleavage of a carbon–carbon bond, which significantly expands the value of DIB as a unique and powerful tool in chemical synthesis. Future studies on the application of this protocol to the synthesis of other difunctionalized acetamides and detailed investigations of the reaction mechanism are in progress.
Supporting Information
| Supporting Information File 1: Experimental details and characterization of compounds. | ||
| Format: PDF | Size: 191.1 KB | Download |
References
-
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c
Return to citation in text: [1] -
Wirth, T. Angew. Chem., Int. Ed. 2005, 44, 3656–3665. doi:10.1002/anie.200500115
Return to citation in text: [1] -
Moriarty, R. M. J. Org. Chem. 2005, 70, 2893–2903. doi:10.1021/jo050117b
Return to citation in text: [1] -
Ladziata, U.; Zhdankin, V. V. ARKIVOC 2006, (ix), 26–58.
Return to citation in text: [1] -
Zhdankin, V. V. ARKIVOC 2009, (i), 1–62.
Return to citation in text: [1] -
Dohi, T.; Kita, Y. Chem. Commun. 2009, 2073–2085. doi:10.1039/b821747e
Return to citation in text: [1] -
Yusubov, M. S.; Zhdankin, V. V. Mendeleev Commun. 2010, 20, 185–191. doi:10.1016/j.mencom.2010.06.001
Return to citation in text: [1] -
Uyanik, M.; Ishihara, K. Chem. Commun. 2009, 2086–2099. doi:10.1039/b823399c
Return to citation in text: [1] -
Ngatimin, M.; Lupton, D. W. Aust. J. Chem. 2010, 63, 653–658. doi:10.1071/CH09625
Return to citation in text: [1] -
Yusubov, M. S.; Nemykin, V. N.; Zhdankin, V. V. Tetrahedron 2010, 66, 5745–5752. doi:10.1016/j.tet.2010.04.046
Return to citation in text: [1] -
Satam, V.; Harad, A.; Rajule, R.; Pati, H. Tetrahedron 2010, 66, 7659–7706. doi:10.1016/j.tet.2010.07.014
Return to citation in text: [1] -
Uyanik, M.; Ishihara, K. Aldrichimica Acta 2010, 43, 83–91.
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Synthesis 2011, 517–538. doi:10.1055/s-0030-1258328
Return to citation in text: [1] -
Desjardins, S.; Andrez, J. C.; Canesi, S. Org. Lett. 2011, 13, 3406–3409. doi:10.1021/ol201149u
Return to citation in text: [1] -
Singh, F. V.; Wirth, T. Org. Lett. 2011, 13, 6504–6507. doi:10.1021/ol202800k
Return to citation in text: [1] -
Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. J. Am. Chem. Soc. 2011, 133, 16410–16413. doi:10.1021/ja207775a
Return to citation in text: [1] -
Guilbault, A.-A.; Legault, C. Y. ACS Catal. 2012, 2, 219–222. doi:10.1021/cs200612s
Return to citation in text: [1] -
Varvoglis, A. Tetrahedron 1997, 53, 1179–1255. doi:10.1016/S0040-4020(96)00970-2
Return to citation in text: [1] -
Brand, J. P.; González, D. F.; Nicolai, S.; Waser, J. Chem. Commun. 2011, 47, 102–115. doi:10.1039/c0cc02265a
Return to citation in text: [1] -
Niedermann, K.; Früh, N.; Vinogradova, E.; Wiehn, M. S.; Moreno, A.; Togni, A. Angew. Chem., Int. Ed. 2011, 50, 1059–1163. doi:10.1002/anie.201006021
Return to citation in text: [1] -
Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2010, 49, 2175–2177. doi:10.1002/anie.200907352
Return to citation in text: [1] -
Koller, R.; Stanek, K.; Stolz, D.; Aardoom, R.; Niedermann, K.; Togni, A. Angew. Chem., Int. Ed. 2009, 48, 4332–4336. doi:10.1002/anie.200900974
Return to citation in text: [1] -
Ochiai, M.; Miyamoto, K. Eur. J. Org. Chem. 2008, 4229–4239. doi:10.1002/ejoc.200800416
Return to citation in text: [1] -
Richardson, R. D.; Wirth, T. Angew. Chem., Int. Ed. 2006, 45, 4402–4404. doi:10.1002/anie.200601817
Return to citation in text: [1] -
Kita, Y.; Tohma, H.; Yakura, T. Trends Org. Chem. 1992, 3, 113–128.
Return to citation in text: [1] -
Kita, Y.; Takada, T.; Tohma, H. Pure Appl. Chem. 1996, 68, 627–630. doi:10.1351/pac199668030627
Return to citation in text: [1] -
Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523–2584. doi:10.1021/cr010003+
Return to citation in text: [1] -
Tohma, H.; Kita, Y. J. Synth. Org. Chem., Jpn. 2004, 62, 116–127.
Return to citation in text: [1] -
Liu, W.-B.; Chen, C.; Zhang, Q.; Zhu, Z.-B. Beilstein J. Org. Chem. 2011, 7, 1436–1440. doi:10.3762/bjoc.7.167
Return to citation in text: [1] -
Evans, P. A.; Brandt, T. A. J. Org. Chem. 1997, 62, 5321–5326. doi:10.1021/jo970525i
Return to citation in text: [1] -
Ngatimin, M.; Gartshore, C. J.; Kindler, J. P.; Naidu, S.; Lupton, D. W. Tetrahedron Lett. 2009, 50, 6008–6011. doi:10.1016/j.tetlet.2009.08.038
Return to citation in text: [1] -
Hamamoto, H.; Hattori, S.; Takemaru, K.; Miki, Y. Synlett 2011, 1563–1566. doi:10.1055/s-0030-1260791
Return to citation in text: [1] -
Pasquato, L.; Santoni, G.; Modena, G. Eur. J. Org. Chem. 2001, 3457–3460. doi:10.1002/1099-0690(200109)2001:18<3457::AID-EJOC3457>3.0.CO;2-C
Return to citation in text: [1] -
Porzelle, A.; Woodrow, M. D.; Tomkinson, N. C. O. Org. Lett. 2010, 12, 1492–1495. doi:10.1021/ol100196a
Return to citation in text: [1] -
Fujiu, T.; Izumi, K.; Sekiguchi, S. Bull. Chem. Soc. Jpn. 1985, 58, 1055–1056. doi:10.1246/bcsj.58.1055
Return to citation in text: [1] -
Katagiri, N.; Niwa, R.; Furuya, Y.; Kato, T. Chem. Pharm. Bull. 1983, 31, 1833–1841. doi:10.1248/cpb.31.1833
Return to citation in text: [1] -
Paquette, L. A., Ed. Encyclopedia of Reagents for Organic Synthesis; Wiley: New York, 1995; Vol. 6, p 3984.
Return to citation in text: [1] -
Christoffers, J. Chem. Commun. 1997, 943–944. doi:10.1039/a700838d
Return to citation in text: [1] -
Biswas, S.; Maiti, S.; Jana, U. Eur. J. Org. Chem. 2010, 2861–2866. doi:10.1002/ejoc.201000128
Return to citation in text: [1]
| 1. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c |
| 2. | Wirth, T. Angew. Chem., Int. Ed. 2005, 44, 3656–3665. doi:10.1002/anie.200500115 |
| 3. | Moriarty, R. M. J. Org. Chem. 2005, 70, 2893–2903. doi:10.1021/jo050117b |
| 4. | Ladziata, U.; Zhdankin, V. V. ARKIVOC 2006, (ix), 26–58. |
| 5. | Zhdankin, V. V. ARKIVOC 2009, (i), 1–62. |
| 6. | Dohi, T.; Kita, Y. Chem. Commun. 2009, 2073–2085. doi:10.1039/b821747e |
| 7. | Yusubov, M. S.; Zhdankin, V. V. Mendeleev Commun. 2010, 20, 185–191. doi:10.1016/j.mencom.2010.06.001 |
| 8. | Uyanik, M.; Ishihara, K. Chem. Commun. 2009, 2086–2099. doi:10.1039/b823399c |
| 9. | Ngatimin, M.; Lupton, D. W. Aust. J. Chem. 2010, 63, 653–658. doi:10.1071/CH09625 |
| 10. | Yusubov, M. S.; Nemykin, V. N.; Zhdankin, V. V. Tetrahedron 2010, 66, 5745–5752. doi:10.1016/j.tet.2010.04.046 |
| 11. | Satam, V.; Harad, A.; Rajule, R.; Pati, H. Tetrahedron 2010, 66, 7659–7706. doi:10.1016/j.tet.2010.07.014 |
| 12. | Uyanik, M.; Ishihara, K. Aldrichimica Acta 2010, 43, 83–91. |
| 13. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 14. | Merritt, E. A.; Olofsson, B. Synthesis 2011, 517–538. doi:10.1055/s-0030-1258328 |
| 15. | Desjardins, S.; Andrez, J. C.; Canesi, S. Org. Lett. 2011, 13, 3406–3409. doi:10.1021/ol201149u |
| 16. | Singh, F. V.; Wirth, T. Org. Lett. 2011, 13, 6504–6507. doi:10.1021/ol202800k |
| 17. | Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. J. Am. Chem. Soc. 2011, 133, 16410–16413. doi:10.1021/ja207775a |
| 18. | Guilbault, A.-A.; Legault, C. Y. ACS Catal. 2012, 2, 219–222. doi:10.1021/cs200612s |
| 32. | Evans, P. A.; Brandt, T. A. J. Org. Chem. 1997, 62, 5321–5326. doi:10.1021/jo970525i |
| 33. | Ngatimin, M.; Gartshore, C. J.; Kindler, J. P.; Naidu, S.; Lupton, D. W. Tetrahedron Lett. 2009, 50, 6008–6011. doi:10.1016/j.tetlet.2009.08.038 |
| 34. | Hamamoto, H.; Hattori, S.; Takemaru, K.; Miki, Y. Synlett 2011, 1563–1566. doi:10.1055/s-0030-1260791 |
| 31. | Liu, W.-B.; Chen, C.; Zhang, Q.; Zhu, Z.-B. Beilstein J. Org. Chem. 2011, 7, 1436–1440. doi:10.3762/bjoc.7.167 |
| 24. | Ochiai, M.; Miyamoto, K. Eur. J. Org. Chem. 2008, 4229–4239. doi:10.1002/ejoc.200800416 |
| 25. | Richardson, R. D.; Wirth, T. Angew. Chem., Int. Ed. 2006, 45, 4402–4404. doi:10.1002/anie.200601817 |
| 26. | Kita, Y.; Tohma, H.; Yakura, T. Trends Org. Chem. 1992, 3, 113–128. |
| 27. | Kita, Y.; Takada, T.; Tohma, H. Pure Appl. Chem. 1996, 68, 627–630. doi:10.1351/pac199668030627 |
| 28. | Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+ |
| 29. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523–2584. doi:10.1021/cr010003+ |
| 30. | Tohma, H.; Kita, Y. J. Synth. Org. Chem., Jpn. 2004, 62, 116–127. |
| 19. | Varvoglis, A. Tetrahedron 1997, 53, 1179–1255. doi:10.1016/S0040-4020(96)00970-2 |
| 20. | Brand, J. P.; González, D. F.; Nicolai, S.; Waser, J. Chem. Commun. 2011, 47, 102–115. doi:10.1039/c0cc02265a |
| 21. | Niedermann, K.; Früh, N.; Vinogradova, E.; Wiehn, M. S.; Moreno, A.; Togni, A. Angew. Chem., Int. Ed. 2011, 50, 1059–1163. doi:10.1002/anie.201006021 |
| 22. | Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2010, 49, 2175–2177. doi:10.1002/anie.200907352 |
| 23. | Koller, R.; Stanek, K.; Stolz, D.; Aardoom, R.; Niedermann, K.; Togni, A. Angew. Chem., Int. Ed. 2009, 48, 4332–4336. doi:10.1002/anie.200900974 |
| 41. | Biswas, S.; Maiti, S.; Jana, U. Eur. J. Org. Chem. 2010, 2861–2866. doi:10.1002/ejoc.201000128 |
| 39. | Paquette, L. A., Ed. Encyclopedia of Reagents for Organic Synthesis; Wiley: New York, 1995; Vol. 6, p 3984. |
| 35. | Pasquato, L.; Santoni, G.; Modena, G. Eur. J. Org. Chem. 2001, 3457–3460. doi:10.1002/1099-0690(200109)2001:18<3457::AID-EJOC3457>3.0.CO;2-C |
| 36. | Porzelle, A.; Woodrow, M. D.; Tomkinson, N. C. O. Org. Lett. 2010, 12, 1492–1495. doi:10.1021/ol100196a |
| 37. | Fujiu, T.; Izumi, K.; Sekiguchi, S. Bull. Chem. Soc. Jpn. 1985, 58, 1055–1056. doi:10.1246/bcsj.58.1055 |
| 38. | Katagiri, N.; Niwa, R.; Furuya, Y.; Kato, T. Chem. Pharm. Bull. 1983, 31, 1833–1841. doi:10.1248/cpb.31.1833 |
© 2012 Liu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)