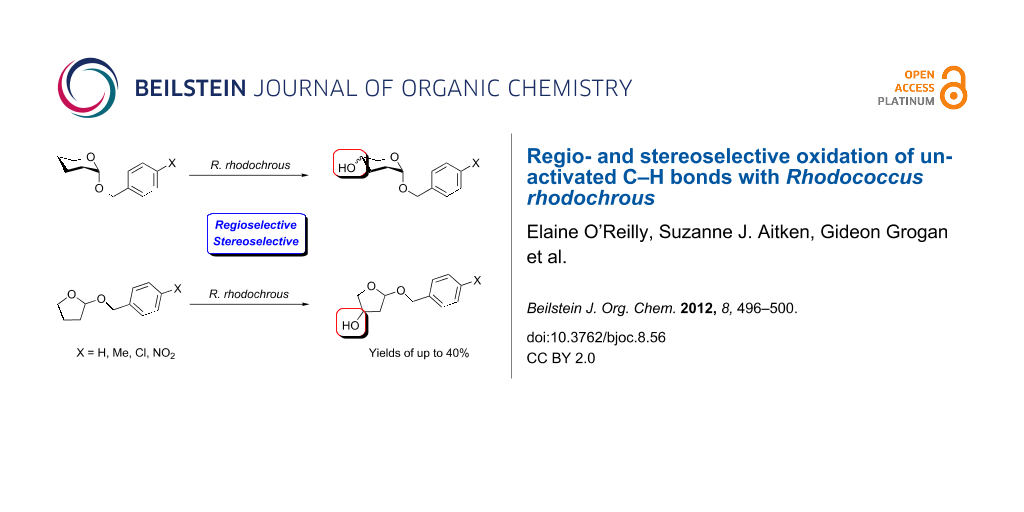
Abstract
The ability of Rhodococcus rhodochrous (NCIMB 9703) to catalyse the regio- and stereoselective hydroxylation of a range of benzyloxy-substituted heterocycles has been investigated. Incubation of 2-benzyloxytetrahydropyrans with resting cell suspensions of the organism yielded predominantly a mixture of 5-hydroxylated isomers in combined yields of up to 40%. Exposure of the corresponding 2-benzyloxytetrahydrofuran derivatives to the cell suspensions gave predominantly the 4-hydroxylated isomers in yields of up to 26%. Most interestingly, 2-(4-nitrobenzyloxy)tetrahydrofuran and 2-(4-nitrobenzyloxy)tetrahydropyran were transformed in high yields to the 4-hydroxylated and 5-hydroxylated products, respectively.

Graphical Abstract
Introduction
Selective C–H activation remains a challenge for synthetic chemists, who often rely on differences in the steric and electronic properties of bonds to achieve regioselectivity [1]. The preparative-scale generation of hydroxylated intermediates can often provide synthetically useful derivatives as well as pharmaceutically important drug metabolites and lead compounds [2-6]. Difficulties associated with chemical routes for the stereo- and regioselective preparation of such compounds has led to the exploitation of natural and recombinant whole-cell biocatalysts as tools for their preparation [7-11].
Many hydroxylating enzymes have been identified within bacteria belonging to the genus Rhodococcus. These organisms are ubiquitous in the environment and their ability to activate recalcitrant compounds and xenobiotics with an array of mono- and dioxygenases has made them the focus of research into bioremediation and biocatalyst development [12]. The soil bacterium Rhodococcus rhodochrous (also referred to as Corynebacterium sp. 7E1C, Rhodococcus rhodochrous ATCC 19067 and Gordonia rubripertinctus 7E1C) is known to oxidise a wide variety of aliphatic hydrocarbons, including n-alkanes and isoalkanes, and has been reported to catalyse the terminal hydroxylation of chloroalkanes and long-chain alkenes [13]. It has been suggested that this strain contains an alkane-inducible NADH-dependent hydroxylase that catalyses the oxidation of n-octane to 1-octanol [13,14]. In light of this, we have explored the ability of this strain to perform regio- and stereoselective oxidations of selected protected heterocyclic substrates. Protected substrates are of particular interest, as subtle changes in the protecting group have been shown to affect the regioselectivity of hydroxylation [10].
An initial substrate screen with the organism revealed that carbobenzyloxy-protected (Cbz) piperidine 1, which is known to be hydroxylated by various microorganisms [15], was oxidised selectively in the 4-position, as indicated in Scheme 1 [10].
![[1860-5397-8-56-i1]](/bjoc/content/inline/1860-5397-8-56-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Regioselective hydroxylation of Cbz-piperidine by Rhodococcus rhodochrous resting cells.
Scheme 1: Regioselective hydroxylation of Cbz-piperidine by Rhodococcus rhodochrous resting cells.
This encouraging result prompted us to challenge this organism with a variety of novel heterocyclic substrates. Substrates based on oxygen heterocycles were thought to be particularly interesting since, upon deprotection, they would yield polyfunctionalised molecules that represent valuable chiral intermediates. A number of p-substituted benzyloxytetrahydropyrans and furans were synthesised and incubated with resting cell suspensions of R. rhodochrous. Our aim in this study was to establish whether the organism is capable of catalysing the hydroxylation of this panel of compounds and to investigate whether these hydroxylations proceeded with regio-, diastereo- and enantioselectivity. We were also interested in establishing whether the oxidising capabilities of this organism were the result of cytochrome P450 activity.
Results and Discussion
Transformations of THP ether derivatives
The results of the biohydroxylations of the p-substituted 2-benzyloxypyrans 3a–d are summarised in Scheme 2.
![[1860-5397-8-56-i2]](/bjoc/content/inline/1860-5397-8-56-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Results from the incubation of THP ether derivatives with Rhodococcus rhodochrous showing the isolated yields; *an inseparable mixture of products was obtained.
Scheme 2: Results from the incubation of THP ether derivatives with Rhodococcus rhodochrous showing the isola...
The regiochemistry and relative stereochemistry of the biohydroxylated products were determined by analysis of 1H NMR coupling constants and by COSY and NOE/NOESY experiments, which identified the 5-axial and 5-equatorial hydroxylated 2-benzopyran isomers 4 and 5 as the major products.
We initially examined the regioselectivity of the biohydroxylation of tetrahydropyran (THP) derivative 3a and found that an inseparable mixture of hydroxylated products was obtained. Studies have shown that minor alterations in substrate structure can strongly affect the selectivity of Beauveria bassiana ATCC 7159 [16,17]. Interestingly, the introduction of substituents on the aryl ring of some substrates can assist in directing hydroxylation away from the substituted position and can improve regioselectivity. For example, varying the aryl ring substituents on N-arylpiperidines can direct hydroxylation away from the substituted position and improve the product yield [18]. Although we did not observe any aromatic hydroxylation of 3a during our studies, we observed a significant improvement in regioselectivity with the THPs 3b–d, in which a substituent was present on the phenyl ring.
Analysis of the products arising from the biotransformation with p-methyl 3b and the p-chloro 3c derivatives revealed that the bacterium was capable of catalysing hydroxylation selectively in the C5-position, providing the C5-axial isomers 4b and 4c and C5-equitorial hydroxylated isomers 5b and 5c in significant yields. The p-nitro substituted THP 3d was also regioselectively hydroxylated at C5 and this oxidation was considerably quicker than that of 3b and 3c, resulting in an appreciably higher yield of the C5-isomers 4d and 5d (Scheme 2). The transformations were found to be highly reproducible with the exception of the p-chloro substituted benzyloxypyran 3c, in which both hydroxylated products were not consistently isolated. In each case, no starting material was recovered, which suggests further breakdown of the THPs by the organism, although this was not investigated further.
As reported previously with other hydroxylating systems, it is often possible to modify the selectivity of microbial hydroxylation by minor alterations of the substrate structure [16,17]. Although exposure of 3a to the resting cell suspension resulted in a complex mixture of hydroxylation products, compound 6 demonstrates that insertion of one methylene unit was sufficient to improve the regioselectivity dramatically. THP 7 was isolated in excellent yield (35%) and as the sole regioisomer (Scheme 3).
![[1860-5397-8-56-i3]](/bjoc/content/inline/1860-5397-8-56-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Influence of substrate structure and linker length on the regioselectivity of hydroxylation.
Scheme 3: Influence of substrate structure and linker length on the regioselectivity of hydroxylation.
At this stage it remained unclear as to whether the gain in regioselectivity observed with 6 was due to the increased length of the ether linkage or due to its spatial orientation. NMR evidence confirmed that the ether in 3a–d sits in the axial position due to the anomeric effect. In the case of substrate 6, the ether sits in the sterically favoured equatorial position. In order to investigate this effect, substrate 8 (Scheme 3), which combines the axial positioning with the longer ether chain, was synthesised and subjected to the resting cell suspensions. A complex mixture of inseparable products was obtained. This result suggested that the length of the linker was not the most important factor influencing the regioselectivity.
Transformation of THF ether derivatives
The ability of the organism to catalyse the selective hydroxylation of the THP series 3b–d, prompted us to investigate the biotransformations of the corresponding racemic tetrahydrofuran (THF) compounds 9a–d. The results, shown in Scheme 4, were largely analogous to those from the THP series. In the case of the THF ring systems, the site of hydroxylation was again determined by NMR, although the relative stereochemistry for all of the derivatives in this series has not yet been determined. In the cases of the substrates 9a and 9b, in which the aryl substituents are hydrogen and methyl, respectively, single products were isolated and identified as the C4 hydroxylated products 10a and 10b. In the case of the p-chloro substituted 2-benzyloxytetrahydrofuran 9c, 14% of mixed hydroxylated furan derivatives were isolated along with 29% of p-chlorobenzoic acid. The isolation of the benzoic acid derivative suggests that benzylic hydroxylation takes place, leading to cleavage of the unstable acetal [19,20]; however, this was not investigated further and the oxidation products were not characterised. The most interesting transformation from this series was that of 2-(p-nitrobenzyloxy)tetrahydrofuran (9d), which was selectively oxidised to provide product 10d in substantially higher yield (26%) than the other derivatives in the series.
![[1860-5397-8-56-i4]](/bjoc/content/inline/1860-5397-8-56-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Regioselective hydroxylation following incubation of THF ether derivatives with Rhodococcus rhodochrous; *see discussion.
Scheme 4: Regioselective hydroxylation following incubation of THF ether derivatives with Rhodococcus rhodoch...
Following the apparent increase in regioselectivity induced by the insertion of a methylene unit into THP 6, the organism was challenged with the corresponding THF derivative. In this case, however, the transformation produced only a small amount of a complex mixture of hydroxylated products, which was not purified.
These transformations demonstrate a definite bias towards C5 hydroxylation on the THP series and a preference for C4 hydroxylation on the THF series.
Enantioselectivity of hydroxylation
The product resulting from the transformation of the p-nitro substituted THF derivative 9d was chosen for some initial screens with the aim of investigating the enantioselectivity of hydroxylation. The MTPA (Mosher) ester 12d was prepared on reaction of alcohol 10d with (R)-methoxytrifluorophenylacetic acid in the presence of DCC and pyridine. Only one diastereomer was clear in the 1H NMR, but examination of the 19F NMR clearly showed two fluorine resonances from which an enantiomeric excess of 93% was calculated. The S-camphanic ester 11d was prepared in a 76% yield resulting from the reaction of the alcohol 10d with S-camphanic acid chloride in the presence of pyridine. 1H NMR analysis revealed that the resulting ester was a single diastereomer, and the absolute stereochemistry of the THF portion was confirmed by X-ray crystallography and found to be 2R,4R.
The two products arising from the hydroxylation of THP derivative 3d showed high optical rotation values, and their enantioselectivity was investigated further. Mosher’s esters 13d and 14d of THP alcohols 4d and 5d, respectively, were prepared in good yield under the same conditions as those employed for alcohol 10d. The 19F NMR of Mosher’s ester 14d showed a single peak suggesting an enantiomeric excess of greater than 95%. It should be noted, however, that authentic samples of the enantiomers are not available and it is possible that the resonances may overlap. Examination of the Mosher’s ester 13d revealed two resonances in the 19F NMR with approximately equal peak area. These preliminary results suggest that the organism catalyses the axial hydroxylation of both enantiomers of 3d, providing 4d as a mixture of enantiomers.
Nature of the hydroxylating enzyme
One of the aims of this study was to investigate the nature of the hydroxylating enzyme in an attempt to establish whether the catalytic protein was a cytochrome P450. Inhibition studies with 1-aminobenzotriazole and metyrapone showed that hydroxylation was inhibited by both of these compounds. These cytochrome P450 inhibitors are known to block the activity of the enzyme by different mechanisms [10,12] and are taken to be evidence that a cytochrome P450 system operates in these transformations.
Conclusion
Rhodococcus rhodochrous has been shown to mediate the regio-, diastereo- and enantioselective hydroxylation of unactivated C–H bonds on selected THF and THP derivatives. We have demonstrated that the introduction of substituents onto the aryl ring of the THP ethers can dramatically affect the regioselectivity of hydroxylation. Most notably, exposure of the p-nitro substituted THP ether 3d to resting cells provided the C5-equatorial and C5-axial regioisomers in 18% and 21%, respectively. Similarly, the organism catalysed the regioselective hydroxylation of p-nitro substituted THF ether 10d providing the C4-hydroxylated product as the sole regioisomer.
Our investigations into the enantioselectivity of hydroxylation revealed that the p-nitro THF derivative was hydroxylated selectively, providing the 2R,4R derivative exclusively. The organism catalysed the enantioselective hydroxylation of p-nitro THP 3d, providing cis-hydroxylated 5d as the sole diastereomer. In contrast, our results indicate that the organism catalyses the axial hydroxylation of both enantiomers of 3d, providing 4d as a mixture of enantiomers. As alcohol 4d showed a strong optical rotation value, this result requires further investigation.
Future efforts will focus on the identification, cloning and heterologous expression of cytochrome P450 genes from this organism to allow for more detailed investigations. It is envisaged that these transformations could be used to produce important hydroxylated, polyfunctionalised chiral intermediates.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterisation data of synthesised, previously unknown compounds. | ||
| Format: PDF | Size: 208.7 KB | Download |
References
-
Chen, M. S.; White, M. C. Science 2007, 318, 783–787. doi:10.1126/science.1148597
Return to citation in text: [1] -
Weis, R.; Winkler, M.; Schittmayer, M.; Kambourakis, S.; Vink, M.; Rozzell, J. D.; Glieder, A. Adv. Synth. Catal. 2009, 351, 2140–2146. doi:10.1002/adsc.200900190
Return to citation in text: [1] -
Schroer, K.; Kittelmann, M.; Lütz, S. Biotechnol. Bioeng. 2010, 106, 699–706. doi:10.1002/bit.22775
Return to citation in text: [1] -
O'Reilly, E.; Köhler, V.; Flitsch, S. L.; Turner, N. J. Chem. Commun. 2011, 47, 2490–2501. doi:10.1039/C0CC03165H
Return to citation in text: [1] -
Robin, A.; Köhler, V.; Jones, A.; Ali, A.; Kelly, P. P.; O’Reilly, E.; Turner, N. J.; Flitsch, S. L. Beilstein J. Org. Chem. 2011, 7, 1494–1498. doi:10.3762/bjoc.7.173
Return to citation in text: [1] -
Robin, A.; Roberts, G. A.; Kisch, J.; Sabbadin, F.; Grogan, G.; Bruce, N.; Turner, N. J.; Flitsch, S. L. Chem. Commun. 2009, 2478–2480. doi:10.1039/b901716j
Return to citation in text: [1] -
Peters, M. W.; Meinhold, P.; Glieder, A.; Arnold, F. H. J. Am. Chem. Soc. 2003, 125, 13442–13450. doi:10.1021/ja0303790
Return to citation in text: [1] -
Vail, R. B.; Homann, M. J.; Hanna, I.; Zaks, A. J. Ind. Microbiol. Biotechnol. 2005, 32, 67–74. doi:10.1007/s10295-004-0202-1
Return to citation in text: [1] -
Çelik, A.; Flitsch, S. L.; Turner, N. J. Org. Biomol. Chem. 2005, 3, 2930–2934. doi:10.1039/b506159h
Return to citation in text: [1] -
Flitsch, S. L.; Aitken, S. J.; Chow, C. S.-Y.; Grogan, G.; Staines, A. Bioorg. Chem. 1999, 27, 81–90. doi:10.1006/bioo.1999.1135
Return to citation in text: [1] [2] [3] [4] -
Grogan, G.; Roberts, G. A.; Parsons, S.; Turner, N. J.; Flitsch, S. L. Appl. Microbiol. Biotechnol. 2002, 59, 449–454. doi:10.1007/s00253-002-1054-0
Return to citation in text: [1] -
Larkin, M. J.; Kulakov, L. A.; Allen, C. C. R. Curr. Opin. Biotechnol. 2005, 16, 282–290. doi:10.1016/j.copbio.2005.04.007
Return to citation in text: [1] [2] -
Jurtshuk, P.; Cardini, G. E.; Gibson, D. T. Crit. Rev. Microbiol. 1971, 1, 239–289. doi:10.3109/10408417109104483
Return to citation in text: [1] [2] -
Cardini, G.; Jurtshuk, P. J. Biol. Chem. 1968, 243, 6070–6072.
Return to citation in text: [1] -
Chang, D.; Feiten, H.-J.; Engesser, K.-H.; van Beilen, J. B.; Witholt, B.; Li, Z. Org. Lett. 2002, 4, 1859–1862. doi:10.1021/ol025829s
Return to citation in text: [1] -
Aitken, S. J.; Grogan, G.; Chow, C. S.-Y.; Turner, N. J.; Flitsch, S. L. J. Chem. Soc., Perkin Trans. 1 1998, 3365–3370. doi:10.1039/A805800H
Return to citation in text: [1] [2] -
Holland, H. L.; Morris, T. A.; Nava, P. J.; Zabic, M. Tetrahedron 1999, 55, 7441–7460. doi:10.1016/S0040-4020(99)00393-2
Return to citation in text: [1] [2] -
Floyd, N.; Munyeman, F.; Roberts, S. M.; Willetts, A. J. J. Chem. Soc., Perkin Trans. 1 1993, 881–882. doi:10.1039/P19930000881
Return to citation in text: [1] -
Schwaneberg, U.; Schmidt-Dannert, C.; Schmitt, J.; Schmid, R. D. Anal. Biochem. 1999, 269, 359–366. doi:10.1006/abio.1999.4047
Return to citation in text: [1] -
Cirino, P. C.; Arnold, F. H. Angew. Chem., Int. Ed. 2003, 42, 3299–3301. doi:10.1002/anie.200351434
Return to citation in text: [1]
| 1. | Chen, M. S.; White, M. C. Science 2007, 318, 783–787. doi:10.1126/science.1148597 |
| 13. | Jurtshuk, P.; Cardini, G. E.; Gibson, D. T. Crit. Rev. Microbiol. 1971, 1, 239–289. doi:10.3109/10408417109104483 |
| 12. | Larkin, M. J.; Kulakov, L. A.; Allen, C. C. R. Curr. Opin. Biotechnol. 2005, 16, 282–290. doi:10.1016/j.copbio.2005.04.007 |
| 7. | Peters, M. W.; Meinhold, P.; Glieder, A.; Arnold, F. H. J. Am. Chem. Soc. 2003, 125, 13442–13450. doi:10.1021/ja0303790 |
| 8. | Vail, R. B.; Homann, M. J.; Hanna, I.; Zaks, A. J. Ind. Microbiol. Biotechnol. 2005, 32, 67–74. doi:10.1007/s10295-004-0202-1 |
| 9. | Çelik, A.; Flitsch, S. L.; Turner, N. J. Org. Biomol. Chem. 2005, 3, 2930–2934. doi:10.1039/b506159h |
| 10. | Flitsch, S. L.; Aitken, S. J.; Chow, C. S.-Y.; Grogan, G.; Staines, A. Bioorg. Chem. 1999, 27, 81–90. doi:10.1006/bioo.1999.1135 |
| 11. | Grogan, G.; Roberts, G. A.; Parsons, S.; Turner, N. J.; Flitsch, S. L. Appl. Microbiol. Biotechnol. 2002, 59, 449–454. doi:10.1007/s00253-002-1054-0 |
| 19. | Schwaneberg, U.; Schmidt-Dannert, C.; Schmitt, J.; Schmid, R. D. Anal. Biochem. 1999, 269, 359–366. doi:10.1006/abio.1999.4047 |
| 20. | Cirino, P. C.; Arnold, F. H. Angew. Chem., Int. Ed. 2003, 42, 3299–3301. doi:10.1002/anie.200351434 |
| 2. | Weis, R.; Winkler, M.; Schittmayer, M.; Kambourakis, S.; Vink, M.; Rozzell, J. D.; Glieder, A. Adv. Synth. Catal. 2009, 351, 2140–2146. doi:10.1002/adsc.200900190 |
| 3. | Schroer, K.; Kittelmann, M.; Lütz, S. Biotechnol. Bioeng. 2010, 106, 699–706. doi:10.1002/bit.22775 |
| 4. | O'Reilly, E.; Köhler, V.; Flitsch, S. L.; Turner, N. J. Chem. Commun. 2011, 47, 2490–2501. doi:10.1039/C0CC03165H |
| 5. | Robin, A.; Köhler, V.; Jones, A.; Ali, A.; Kelly, P. P.; O’Reilly, E.; Turner, N. J.; Flitsch, S. L. Beilstein J. Org. Chem. 2011, 7, 1494–1498. doi:10.3762/bjoc.7.173 |
| 6. | Robin, A.; Roberts, G. A.; Kisch, J.; Sabbadin, F.; Grogan, G.; Bruce, N.; Turner, N. J.; Flitsch, S. L. Chem. Commun. 2009, 2478–2480. doi:10.1039/b901716j |
| 10. | Flitsch, S. L.; Aitken, S. J.; Chow, C. S.-Y.; Grogan, G.; Staines, A. Bioorg. Chem. 1999, 27, 81–90. doi:10.1006/bioo.1999.1135 |
| 12. | Larkin, M. J.; Kulakov, L. A.; Allen, C. C. R. Curr. Opin. Biotechnol. 2005, 16, 282–290. doi:10.1016/j.copbio.2005.04.007 |
| 10. | Flitsch, S. L.; Aitken, S. J.; Chow, C. S.-Y.; Grogan, G.; Staines, A. Bioorg. Chem. 1999, 27, 81–90. doi:10.1006/bioo.1999.1135 |
| 18. | Floyd, N.; Munyeman, F.; Roberts, S. M.; Willetts, A. J. J. Chem. Soc., Perkin Trans. 1 1993, 881–882. doi:10.1039/P19930000881 |
| 15. | Chang, D.; Feiten, H.-J.; Engesser, K.-H.; van Beilen, J. B.; Witholt, B.; Li, Z. Org. Lett. 2002, 4, 1859–1862. doi:10.1021/ol025829s |
| 16. | Aitken, S. J.; Grogan, G.; Chow, C. S.-Y.; Turner, N. J.; Flitsch, S. L. J. Chem. Soc., Perkin Trans. 1 1998, 3365–3370. doi:10.1039/A805800H |
| 17. | Holland, H. L.; Morris, T. A.; Nava, P. J.; Zabic, M. Tetrahedron 1999, 55, 7441–7460. doi:10.1016/S0040-4020(99)00393-2 |
| 10. | Flitsch, S. L.; Aitken, S. J.; Chow, C. S.-Y.; Grogan, G.; Staines, A. Bioorg. Chem. 1999, 27, 81–90. doi:10.1006/bioo.1999.1135 |
| 13. | Jurtshuk, P.; Cardini, G. E.; Gibson, D. T. Crit. Rev. Microbiol. 1971, 1, 239–289. doi:10.3109/10408417109104483 |
| 14. | Cardini, G.; Jurtshuk, P. J. Biol. Chem. 1968, 243, 6070–6072. |
| 16. | Aitken, S. J.; Grogan, G.; Chow, C. S.-Y.; Turner, N. J.; Flitsch, S. L. J. Chem. Soc., Perkin Trans. 1 1998, 3365–3370. doi:10.1039/A805800H |
| 17. | Holland, H. L.; Morris, T. A.; Nava, P. J.; Zabic, M. Tetrahedron 1999, 55, 7441–7460. doi:10.1016/S0040-4020(99)00393-2 |
© 2012 O’Reilly et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)